

Abstract
A single–base pair resolution silkworm genetic variation map was constructed from 40 domesticated and wild silkworms, each sequenced to approximately threefold coverage, representing 99.88% of the genome. We identified ∼16 million single-nucleotide polymorphisms, many indels, and structural variations. We find that the domesticated silkworms are clearly genetically differentiated from the wild ones, but they have maintained large levels of genetic variability, suggesting a short domestication event involving a large number of individuals. We also identified signals of selection at 354 candidate genes that may have been important during domestication, some of which have enriched expression in the silk gland, midgut, and testis. These data add to our understanding of the domestication processes and may have applications in devising pest control strategies and advancing the use of silkworms as efficient bioreactors.
The domesticated silkworm, Bombyx mori, has a mid-range genome size of ∼432 Mb (1), is the model insect for the order Lepidoptera, has economically important values (e.g., silk and bioreactors production), and has been domesticated for more than 5000 years (2). Because of human selection, silkworms have evolved complete dependence on humans for survival (3), and more than 1000 inbred domesticated strains are kept worldwide (3). Archaeological and genetic evidences indicate that the domesticated silkworm originated from the Chinese wild silkworm, Bombyx mandarina, that is found throughout Asia, where modern sericulture and silkworm domestication were initiated.
The origin of the domesticated silkworm is a long-standing question that has not been settled by previous limited biochemical and molecular analyses. Two hypotheses suggested a unique domestication but disagreed on the ancestral variety. One hypothesis, based on isoenzyme polymorphism, proposed mono-voltinism as ancestral variety (voltinism represents number of generations per annum), from which bi- and multi-voltine were derived by artificial selection (4); the other proposed the reverse path considering evidence from archaeology, history, and genetics (5). An alternative hypothesis based on random amplification of polymorphic DNA indicated that the ancestral domestic silkworm strains were issued, not from a unique variety, but from mixed geographic locations and ecological types (6). These theories are conflicting, probably because they were derived from incomplete genetic information. Consequently, we present here a genome-wide detailed genetic variation map in hopes to help reconstruct the silkworm domestication history.
The data consisted of 40 samples from 29 phenotypically and geographically diverse domesticated silkworm lines [categorized by geographical regions (3): Chinese, Japanese, tropical, European lineages, and the mutant system], as well as 11 wild silkworms from various mulberry fields in China (table S1). We sequenced each genome at approximately threefold coverage, after creating single- and paired-end (PE) libraries with inserts of PEs ranging from base pairs 137 to 307 (7).
Raw short reads were mapped against the refined 432-Mb reference genome from Dazao (1) with the program SOAP (8). We pooled all reads from the 40 complete genomes and identified 15,986,559 single-nucleotide polymorphisms (SNPs) using SoapSNP (7, 9) (table S3A).The accuracy of the SNP calling was evaluated with Sequenom (San Diego, California) genotyping of a representative subset of variants in all 40 varieties, resulting in a 96.7% validation rate (7).
We then pooled separately all 29 domesticated strains and all 11 wild varieties and obtained SNP sets for each (7). The number of SNPs in the domestic versus wild varieties was 14,023,573 and 13,237,865, respectively (table S3A). To account for the different number of domestic and wild strains, we used the population-size scaled mutation rate θS to measure genetic variation (10) (table S3B). We found that θS,domesticated (0.0108) was significantly smaller than θS,wild (0.0130) [Mann-Whitney U (MWU), P = 1.10 × 10−7], which may reflect differences in effective population size and demographic history (including domestication and artificial selection). The rate of heterozygosity in domesticated strains was more than twofold lower than that of wild varieties (0.0032 versus 0.0080, respectively) (MWU, P = 3.33 × 10−6). This reduction in heterozygosity is most likely due to inbreeding or the bottleneck experienced by domesticated lines.
In addition to SNPs, we also identified 311,608 small insertion-deletions (indels) (table S4A), a subset of which were validated with polymerase chain reaction (7). The θS values for the indels (table S4B) were in agreement with a lower effective population size in domesticated versus wild varieties. A mate-pair relationship method (7, 11) identified 35,093 structural variants (SVs) among the 40 varieties (table S5). Over three-fourths of the SVs overlapped with transposable elements (TEs), suggesting that SV events in silkworm are probably due to TE content (12) and mobility (11). The SNPs, indels, and SVs all contributed to a comprehensive genetic variation map for the silkworm.
To elucidate the phylogeny of silkworms beyond previous studies (6, 13, 14), we used our identified SNPs to estimate a neighbor-joining tree (7) on the basis of a dissimilarity measure of genetic distance (Fig. 1A). This tree represents an average of distances among strains, so lineages cannot be directly interpreted as representing phylogenetic relationships. Instead, the distances may reflect gene flow and other population level processes related to human activities such as ancient commercial trade. The unrooted radial relationship reveals a clear split between the domesticated and wild varieties, and the domestic strains cluster into several subgroups (Fig. 1A).
Fig. 1.
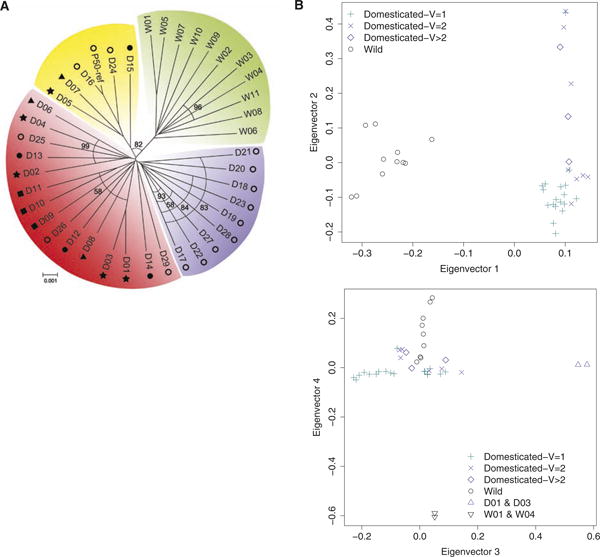
Silkworm phylogeny and population structure from PCA. (A) A neighbor-joining tree from genomic SNPs, bootstrapped with 1000 replicates (bootstrap values less than 100 are shown on arcs; those equal to 100 are not shown): green for all wild varieties; others are domesticated strains separated into three groups (purple, red, and yellow). Domesticated strains are denoted by a combination of symbols representing silkworm systems (hollow circles, Chinese; stars, Japanese; triangles, tropical; squares, European; filled circles, mutant system) and sample IDs (D01 to D29 and P50-ref for the reference genome of Dazao). Wild varieties are indicated by their IDs (W01 to W11). Scale bar, frequencies of base-pair differences. (B) PCA results of the first four statistically significant components (Tracy-Wisdom, P < 0.05). (Top) The first eigenvector separates domesticated and wild varieties, and the second divides the domesticated strains into subgroups. (Bottom) The third eigenvector separates the high-silk production Japanese domesticated strains D01 and D03 from the other domesticated strains, and the fourth separates the wild varieties W01 and W04 from the other wild varieties.
A principal components analysis (PCA) (7) classified the first four eigenvectors as significant (table S6; Tracy-Widom, P < 0.05). The first eigenvector clearly separates the domesticated and wild varieties, whereas the second eigenvector divides the domesticated strains into subgroups correlated with voltinism (Fig. 1B, top). The third principal component separates D01 and D03 (which are high–silk producing Japanese domesticated strains) from the other domesticated strains, whereas the fourth separates W01 and W04 from the other wild varieties (Fig. 1B, bottom). Results of population structure analysis (7) (fig. S3) confirmed the results of the PCA, as well as the neighbor-joining analysis. The clear genetic separation between domesticated and wild varieties suggests a unique domestication event and relatively little subsequent gene flow between the two groups.
One puzzling observation is that, although domesticated strains are clearly genetically differentiated from the wild ones, they still harbor ∼83% of the variation observed in the wild varieties. This suggests that the population-size bottleneck at domestication only reduced genetic variability mildly (7); that is, a large number of individuals must have been selected for initial domestication or else domestication occurred simultaneously in many places. To quantify this observation, we fit a simple coalescence-based genetic bottleneck model to the SNP frequency spectrum (7). The estimated model suggests that the domestication event lead to a 90% reduction in effective population size during the initial bottleneck (fig. S2). Additionally, we observed no excess of low-frequency variants in the domesticated varieties compared with the wild varieties, suggesting that there has not been obvious population growth since the domestication event and that the domestic lines probably have had a generally stable effective population size.
Our measure of pairwise linkage disequilibrium (LD) (7) showed that LD decays rapidly in silkworms, with correlation coefficient r2 decreasing to half of its maximum value at distances of ∼46 and 7 base pairs for the domesticated and wild varieties, respectively (fig. S1). The fast decay of LD implies that regions affected by selective sweeps are probably relatively small. To detect regions with significant (Z test, P < 0.005) signatures of selective sweep, we measured SNP variability and frequency spectrum following a genome-wide sliding window strategy (7) (Fig. 2A). Though the significance of our Z tests (7) cannot be interpreted literally due to correlations in LD and shared ancestral history between the two populations, the Z tests suggest differences in frequency spectra and amounts of variability between the two groups. We termed the candidate regions genomic regions of selective signals (GROSS).
Fig. 2.
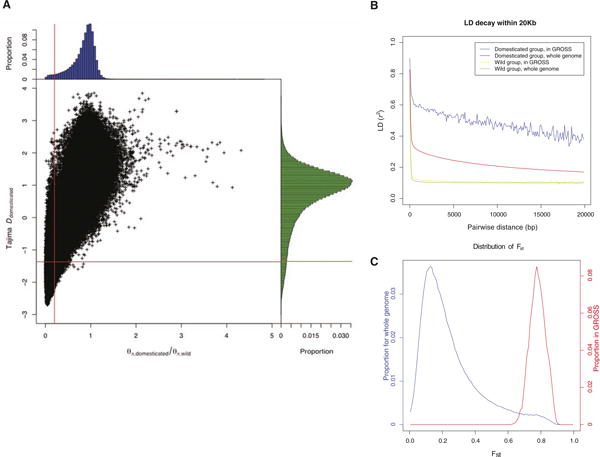
GROSS. (A) Two-dimensional distribution for θπ,domesticated/θπ,wild and Tajima's D for domesticated silkworms. 5-kb windows, data points of which locate to the left of the vertical red line (corresponding to Z test P < 0.005) and below the horizontal red line (also Z test P < 0.005), were picked out as building blocks of GROSS. (B) LD in GROSS. For domesticated silkworms, LD decays much more slowly in GROSS than in the whole genome, whereas for wild varieties, no obvious change in the pattern was observed. (C) Distribution of divergence between domesticated and wild groups in GROSS versus the whole genome (Fst) (7).
We identified a total of 1041 GROSS (7), covering 12.5 Mb (2.9%) of the genome, which may reflect genomic footprints left by artificial selection during domestication. A region affected by selective sweep typically has an elevated level of LD (15, 16), and in our GROSS, the level of LD among SNP pairs less than 20-kb apart was 2.3 times higher than genome average (Fig. 2B), consistent with the hypothesis that selection is affecting these regions. In all these regions, divergence levels (7) between the domesticated and wild groups were also elevated (Fig. 2C), confirming the differentiation of the two subpopulations.
B. mori has experienced intense artificial selection, represents a completely domesticated insect (3), and has become totally dependent on humans for survival. Artificial selection has also enhanced important economic traits such as cocoon size, growth rate, and digestion efficiency (3). Moreover, compared to its wild ancestor B. mandarina, B. mori has gained some representative behavioral characteristics (such as tolerance to human proximity and handling, as well as extensive crowding) and lost other traits (such as flight, predators, and diseases avoidance). However, to date no genes have been identified as domestication genes under artificial selection. Within GROSS, we identified 354 protein-coding genes that represent good candidates for domestication genes (table S9). Their Gene Ontology annotation (17) showed the most representation in the categories of “binding” and “catalytic” in molecular function, as well as “metabolic” and “cellular” in biological process (fig. S4).
Considering published expression profiles performed on different tissues in fifth-instar day 3 of Dazao with genome-wide microarray (18), we found that 159 of our GROSS genes exhibit differential expression. Of these, 4, 32, and 54 genes are enriched in tissues of silk gland, midgut, and testis, respectively (fig. S5). Among the genes enriched in the silk gland is silk gland factor-1 (Sgf-1), a homolog of a Drosophila melanogaster Fkh gene. Sgf-1 regulates the transcription of the B. mori glue protein-encoding sericin-1 gene and of three fibroin genes encoding fibroin light chain, fibroin heavy chain, and fhx/P25 (19, 20). Another silk gland-enriched gene, BGIBMGA005127, homologous to the Drosophila sage gene, was overexpressed fourfold in a high-silk strain compared with Dazao (fig. S6). In Drosophila, the products of Fkh and sage genes cooperate to regulate the transcription of the glue genes SG1 and SG2, which are crucial for the synthesis and secretion of glue proteins (21, 22). Additionally, midgut- and testis-enriched genes suggest that genes involved in energy metabolism and reproduction have also been under artificial selection during domestication (7). Specifically, we identified three likely candidates for artificial selection: (i) NM_001130902 is homologous to paramyosin protein in Drosophila and may be related to flight (23). (ii) NM_001043506 is homologous to fattyacyl desaturase (desat1) in Drosophila, which is related to courtship behaviors, because mutations in desat1 can change the pattern of sex pheromones production and discrimination (24). Finally, (iii) BGIBMGA000972 is homologous to tyrosine-protein kinase Btk29A in Drosophila, which is involved in male genitalia development (25).
In sericulture, silkworms are typically categorized by their geographic origins (3). Voltinism, which results from adaptation to ecological conditions, and geographic systems have been central to previous studies of silkworm origin and domestication (4–6). Our findings indicate that a unique domestication event occurred and, although voltinism correlates with genetic distances, major genetically cohesive strains cannot be identified on the basis of voltinism. We observed no correlation between longitudes of the sample origins and any of the principal components, but we did find a significant correlation between the latitudes and eigenvectors 2 and 4 in the PCA (table S7). Although this correlation might be due to isolation by distance, this result also agrees with previous studies suggesting that climate affects silkworm biology (2).
The silkworm data reported here represent a large body of genome sequences for a lepidopteran species and offer a source of near-relatives in this clade for comparative genomic analysis. We further proposed a set of candidate domestication genes that, in addition to being putatively under artificial selection, also show higher expression levels in tissues important for silkworm economic traits. Because a proportion of the GROSS genes were probably important in domestication, functional verification of these candidate genes may enable a comprehensive understanding of the differences of biological characteristics between B. mori and B. mandarina. Moreover, domesticated silkworms have been used as bioreactors (26, 27), and such an effort may provide useful clues to help improve the capacity and capability of silkworms to produce foreign proteins (26). These findings may also aid in the understanding of how to enhance traits of interest in other organisms in an environmentally safe manner and, because the wild silkworm is a destructive pest, allow new approaches for pest control.
Supplementary Material
Acknowledgments
We thank two anonymous referees, L. Goodman, L. Bolund, and K. Kristiansen for providing valuable comments. This work was supported by the Ministry of Science and Technology of China (grants 2005CB121000, 2007CB815700, 2006AA10A117, 2006AA10A118, 2006AA02Z177, and 2006AA10A121), the Ministry of Education of China (Program for Changjiang Scholars and Innovative Research Team in University, grant IRT0750), Chongqing Municipal Government, the 111 Project (grant B07045), the National Natural Science Foundation of China (grants 30725008, 30890032, and 90608010), the International Science and Technology Cooperation Project (grant 0806), the Chinese Academy of Science (grant GJHZ0701-6), the Danish Platform for Integrative Biology, the Ole Rømer grant from the Danish Natural Science Research Council, and the Solexa project (grant 272-07-0196). Raw genome data are deposited in the National Center for Biotechnology Information/Short Read archive database with accession number SRA009208; silkworm genetic variations, GROSS information, and microarray data can be found in http://silkworm.swu.edu.cn/silkdb/resequencing.html.
Footnotes
Supporting Online Material: www.sciencemag.org/cgi/content/full/1176620/DC1, Materials and Methods, SOM Text, Figs. S1 to S7, Tables S1 to S10, References
References and Notes
- 1.The International Silkworm Genome Consortium. Insect Biochem Mol Biol. 2008;38:1036. [Google Scholar]
- 2.Xiang Z, Huang J, Xia J, Lu C. Biology of Sericulture. China Forestry Publishing House; Beijing: 2005. [Google Scholar]
- 3.Goldsmith MR, Shimada T, Abe H. Annu Rev Entomol. 2005;50:71. doi: 10.1146/annurev.ento.50.071803.130456. [DOI] [PubMed] [Google Scholar]
- 4.Yoshitake N. J Sericult Sci Japan. 1967;37:83. [Google Scholar]
- 5.Jiang Y. Agric Archaeol. 1987;14:316. [Google Scholar]
- 6.Lu C, Yu H, Xiang Z. Agric Sci China. 2002;1:349. [Google Scholar]
- 7.Materials and methods are available as supporting material on Science Online
- 8.Li R, Li Y, Kristiansen K, Wang J. Bioinformatics. 2008;24:713. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 9.Li R, et al. Genome Res. 2009;19:1124. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watterson GA. Theor Popul Biol. 1975;7:256. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, et al. Nature. 2008;456:60. [Google Scholar]
- 12.Biology Analysis Group et al. Science. 2004;306:1937. [Google Scholar]
- 13.Xia Q, Zhou Z, Lu C, Xiang Z. Acta Entomol Sinica. 1998;41:32. [Google Scholar]
- 14.Li M, et al. Genome. 2005;48:802. doi: 10.1139/g05-053. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen R. Annu Rev Genet. 2005;39:197. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 16.Slatkin M. Nat Rev Genet. 2008;9:477. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye J, et al. Nucleic Acids Res. 2006;34:W293. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Q, et al. Genome Biol. 2007;8:R162. doi: 10.1186/gb-2007-8-8-r162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horard B, Julien E, Nony P, Garel A, Couble P. Mol Cell Biol. 1997;17:1572. doi: 10.1128/mcb.17.3.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mach V, et al. J Biol Chem. 1995;270:9340. doi: 10.1074/jbc.270.16.9340. [DOI] [PubMed] [Google Scholar]
- 21.Abrams EW, Mihoulides WK, Andrew DJ. Development. 2006;133:3517. doi: 10.1242/dev.02525. [DOI] [PubMed] [Google Scholar]
- 22.Li TR, White KP. Dev Cell. 2003;5:59. doi: 10.1016/s1534-5807(03)00192-8. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, et al. Proc Natl Acad Sci USA. 2005;102:10522. [Google Scholar]
- 24.Marcillac F, Grosjean Y, Ferveur JF. Proc Biol Sci. 2005;272:303. doi: 10.1098/rspb.2004.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba K, et al. Mol Cell Biol. 1999;19:4405. doi: 10.1128/mcb.19.6.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda S. Annu Rev Entomol. 1989;34:351. doi: 10.1146/annurev.en.34.010189.002031. [DOI] [PubMed] [Google Scholar]
- 27.Maeda S, et al. Nature. 1985;315:592. doi: 10.1038/315592a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.