

Abstract
Background
Infantile hemangioma (IH) is the most common tumor of infancy. The first-line therapy for IH is propranolol, a non-selective β-adrenergic receptor antagonist. However, mechanisms for the therapeutic effect of propranolol and regrowth of IH following cessation of treatment in some cases are not clear. We have recently shown that IH arises from multipotent stem cells. Whether IH stem cells are responsive to propranolol and are selectively targeted is unknown, and is the focus of this study.
Methods
IH stem cells were exposed to propranolol and assayed for cellular and molecular alterations. We used endothelial cells (ECs) as controls and bone marrow-mesenchymal progenitor cells (bm-MPCs) as normal stem/progenitor counterparts to determine selectivity.
Results
Our results show that propranolol significantly reduced IH stem cell growth but failed to induce caspase-3 activation. Normal bm-MPCs and mature ECs showed maintained or increased caspase-3 activation and significantly reduced cyclin-D1 levels. We further show that IH stem cells may escape apoptosis by inducing anti-apoptotic pathways.
Conclusions
This study reveals that propranolol does not induce apoptosis in IH stem cells, which is in contrast to ECs. Escape from apoptosis in IH stem cells may involve induction of anti-apoptotic pathways.
Introduction
Infantile hemangioma (IH) is a benign vascular tumor affecting 1 out of 100 newborns (1,2). IH undergoes three developmental phases: a proliferative phase, where the tumour grows rapidly and comprises undifferentiated cells during the first year of life; an involuting phase, where tumor growth slows and vessels become prominent; and an involuted phase, where fibrofatty tissue replaces much of the tumor mass (3). A unique feature only seen in IH is that the tumor follows this natural course and spontaneously regresses. Hence, most IH pose no serious threat or complications to the infant; however, in problematic cases that interfere with health and normal function due to the size or location of the tumor, patients may require immediate treatment (4). For example, obstructive IH in organs, such as eyes or airway, require immediate attention because the tumor may inhibit normal development and function of the organ to impair the infant permanently (3,5).
Current treatments for IH include surgery when necessary and use of corticosteroids, despite the severe side effects when taken for extended periods at high doses. Recently, propranolol was discovered to be an effective treatment for IH (6), with higher efficacy and minimal side effects when compared to corticosteroid use (7). Propranolol is a non-selective β-adrenergic receptor antagonist that has been widely used for complications such as angina pectoris, myocardial infarction, and hypertension. Although the mechanism of therapeutic effect of propranolol is unknown, theories suggest vasoconstriction, endothelial cell apoptosis, and inhibition of angiogenesis by modulating vascular endothelial growth factors (8–11). In fact, a number of recent studies have shown that propranolol treatment of normal endothelial cells as well as endothelial cells derived from IH specimens causes activation of caspase-3 (12,13). Caspase-3 is an important regulator of cellular apoptosis and is recognized as an indispensable death protease for apoptotic chromatin condensation and DNA fragmentation in all cell types examined (reviewed in (14)). In addition to inducing apoptosis, propranolol also decreases the expression of various cyclins in endothelial cell, thus disrupting cell cycle progression and growth (12).
A puzzling finding from a few propranolol treatment studies in patients is that some IHs regrow upon cessation of propranolol treatment (15–17). This has been attributed to early treatment withdrawal and/or a long proliferating phase of IH. Previously, we have shown that IH arises from multipotential stem cells (termed hemSCs) (18). HemSCs, isolated based on expression of stem cell antigen CD133, form glucose transporter-1 (Glut1) positive microvessels in immunodeficient mice. These Glut1-positive vessels are later replaced by human adipocytes that mimic the natural stages of human IH. Interestingly IH-derived endothelial cells are unable to produce microvessels (18). This suggests that hemSCs may be responsible for the recurrence of IH upon cessation of propranolol treatment possibly owing to the non-responsiveness of hemSCs to propranolol. In this study, we have explored this possibility by treating primary hemSCs with propranolol to determine whether propranolol induces caspase-3 activation and apoptosis as has been shown for vascular endothelial cells. We have also studied bone marrow-derived mesenchymal progenitor cells (bm-MPCs) as normal counterparts of hemSCs to determine whether changes (if any) observed in hemSCs are specific or whether the response depends on the stem/progenitor phenotype. In addition, we investigated possible signaling pathways involved upon propranolol treatment.
Results
Atypical phenotype of IH endothelium
A number of studies have investigated the effect of propranolol on IH-derived endothelial cells to offer insight into the mechanisms of therapeutic effect of propranolol (12,13,19). These studies show that propranolol causes apoptosis in IH endothelial cells by activating caspase-3 and also blocks other cellular activities including migration and tubule formation (12,13). This effect of propranolol is also exhibited by normal endothelial cells (9,12). We have shown that IH arises from multipotent stem cells (18). CD133-selected cells from human IH produce hemangioma lesions in mice. Interestingly, IH-derived endothelial cells (exhibiting mature endothelial phenotype as assessed by endothelial cell markers) fail to produce Glut1-positive microvessels (18). This suggests that the cell of interest, at least in proliferating phase IH when treatments are required, is the hemSC. Therefore, we performed immunostaining for CD133 to probe for the number of microvessels that are lined by CD133-expressing cells. Results from two proliferating IH specimens show that all microvessels within IH tissues are immunoreactive to CD133 (figure 1). This suggested that for understanding the effect of propranolol, CD133-selected hemSCs are essential.
Figure 1. CD133-positive cells line hemangioma vessels.
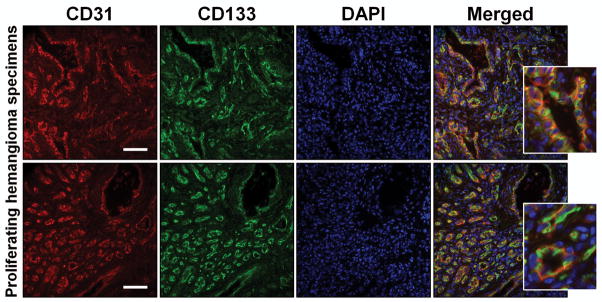
Proliferating hemangioma specimens were double-labeled for CD31 (endothelial cell marker; Red) and CD133 (stem cell antigen; Green). DAPI (blue) was used as counterstain. Staining illustrates complete co-localization of CD133 and CD31 in both proliferating hemangioma specimens (images were taken at 20X magnification; inserts illustrate high magnification; scale bar = 200μm).
β2 and β3 are the predominant β-adrenergic receptors in IH stem cells
We first assayed for β-adrenergic receptor expression in hemSCs and compared the levels to mature endothelial cells. Our results show that hemSCs express both β2 and β3 adrenergic receptors (figure 2a). β1 adrenergic receptor mRNA level, although detectable, were significantly lower. Interestingly, we found that bm-MPCs share β-adrenergic receptor profile with hemSCs. Mature endothelial cells (human microvascular endothelial cells; HDMECs) on the other hand exhibited higher level of β1 adrenergic receptor expression (figure 2a). No significant differences were found in the level of β2 or β3 adrenergic receptors between endothelial cells, hemSCs, and bm-MPCs.
Figure 2. Propranolol reduces hemSC growth.
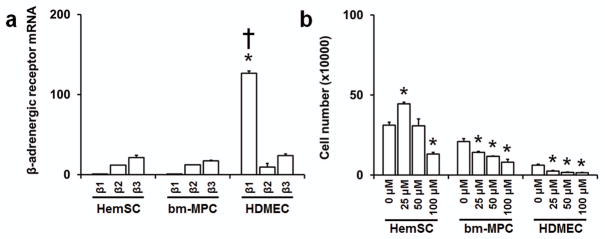
(a) mRNA levels of β-adrenergic receptors in CD133-selected hemSCs and normal bm-MPCs and HDMECs were determined by real time qRT-PCR (data normalized to β-actin; *p<0.05 compared to β1 adrenergic receptor mRNA levels in hemSCs and bm-MPCs; †p<0.05 compared to β2 and β3 adrenergic receptor mRNA levels). RNA was isolated from cells cultured in EBM2 growth media under identical conditions. The specificity of the amplification was determined by melting curve analysis (data not shown). (b) Total live cell number after 72 hours of treatment with different concentrations of propranolol. Propranolol treatment at 100 μM showed reduced number of cells as compared to control in all cell types tested (*p<0.05 compared to respective control).
Propranolol inhibits IH stem cell growth
Our next objective was to determine the effect of propranolol on the growth of hemSCs. We cultured the cells with 25, 50, 100 μM propranolol and assayed for cell number at both 24 and 72 hours. We chose these concentrations based on previous studies that have observed significant differences in IH endothelial cells (9,12). Furthermore, propranolol has been shown to have immediate effects in inhibiting cell viability at 24 hours (13). Interestingly, no significant changes to cell number were observed in any cell type at 24 hours (data not shown). At 72 hours, however, bm-MPCs and HDMECs showed significant reductions in cell number at all concentrations of propranolol when compared to control (figure 2b). In contrast, a proliferative effect was observed when hemSCs were treated with 25 μM propranolol and a reduction in total live cell number with 100 μM propranolol (figure 2b).
Reduced cell number following propranolol treatment is not due to apoptosis in IH stem cells
As the cell number decreased significantly for all cell types when cultured in 100 μM propranolol (at 72 hours), we examined whether this was due to apoptosis. Therefore, cells treated with propranolol were assayed for active caspase-3. Caspase-3 is the most frequently activated death protease and has been shown to play a role in inducing endothelial cell apoptosis upon propranolol treatment (12,13). Unexpectedly, hemSCs treated with 100 μM propranolol showed a significant reduction in the level of active caspase-3. This suggested that in hemSCs, the reduction in cell number might be due to inhibited cell growth and not apoptosis. bm-MPCs did not show a significant difference upon propranolol treatment as caspase-3 levels remained unchanged (figure 3a). On the other side of the spectrum, propranolol-treated HDMECs showed a significant increase in the level of activated caspase-3 as expected. These data demonstrate that propranolol treatment induces apoptosis in HDMECs, whereas the decrease in cell number in hemSCs and possibly bm-MPCs is mediated by a reduction in cell growth.
Figure 3. Propranolol reduces active caspase-3 level in hemSCs.
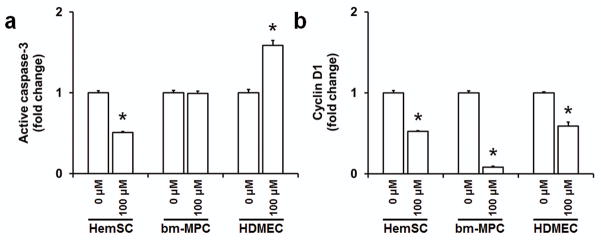
Levels of activated caspase-3 (a) and cyclin-D1 (b) in hemSCs, bm-MPCs and HDMECs following 100 μM propranolol treatment for 72 hours (*p<0.05 compared to respective 0 μM treatment/control).
Propranolol halts cell cycle progression but does not induce apoptosis in hemSCs
We examined the effect of propranolol on cyclin-D1 level. Cyclin-D1 is a key regulator in the progression from G1/S phase and has recently been shown to be maintained in G2 phase (20). Recent studies have shown that propranolol reduces cyclin-D1 in a time-dependent manner in endothelial cells (9). Surprised by previous experimental findings, we wanted to determine if propranolol reduces cyclin-D1 in hemSCs. Our results do show significantly reduced cyclin-D1 in hemSCs upon propranolol treatment (figure 3b). Similarly, bm-MPCs and HDMECs cyclin-D1 levels were also significantly reduced (figure 3b). The greatest change in cyclin-D1 was seen in bm-MPCs which may explain the reduced cell number seen earlier. These results suggest that propranolol inhibits cell cycle progression in all cell types by decreasing level of cyclin-D1.
Propranolol induces anti-apoptotic pathways in hemSCs
In order to understand the possible mechanism by which propranolol induces apoptosis in endothelial cells but not hemSCs, we used quantitative RT-PCR to profile for genes important in the central mechanisms of cellular death. We used Human Cell Death Pathway Finder PCR Array (Qiagen; see Methods for details) which comprises 84 key genes important for cell survival and apoptosis. HemSCs, bm-MPCs, and HDMECs were cultured in normal growth media or in media containing different concentrations of propranolol for 72 hours. Using this PCR-based array, we found that propranolol significantly induces various anti-apoptotic pathways in hemSCs and normal bm-MPCs (figure 4a). These included Akt (also known as protein kinase B; induced 31.2x in hemSCs, 7.58x bm-MPC), Bcl2 (295.9 x in hemSCs, 143.2x bm-MPC)/Bcl2A (23.47x in hemSC, 10.97x in bmMPC), and insulin-like growth factor receptor-1 (IGFR-1, 53.68x in hemSC, 9.29x in bm-MPC). In contrast, we did not observe any alteration of these anti-apoptotic pathways in HDMECs (figure 4a).
Figure 4. Propranolol induces anti-apoptotic signaling pathways in hemSCs.

Propranolol induced expression of anti-apoptotic genes (a), TNF superfamily signaling pathway (b) and interferon γ (c) in hemSCs and bm-MPCs but not HDMECs (mRNA levels were measured using SABiosciences RT2 Cell Death Pathway Finder and normalized to β-actin and GAPDH; Red dashed lines in panels a and b, highlight 100 μM propranolol groups; graphs shown in a–c are representative of multiple PCR arrays).
TNF- α, but not IFNγ, reduced cell growth in IH stem cells
In addition to inducing anti-apoptotic pathways, we observed that propranolol treatment significantly increased tumor necrosis factor (TNF) pathway members and interferon γ (IFNγ) in hemSCs and to a lesser degree in bm-MPCs (figures 4b and 4c). This led us to question whether TNF or IFN signaling mediates changes we have observed in hemSCs upon propranolol challenge. We first determined the effect of exogenous TNF-α treatment in hemSCs. Interestingly, there was a significant cell number reduction in hemSCs upon exposure to 50 and 100 ng/mL TNF-α (figure 5a). We then investigated the effect of TNF-α on activated caspase-3 level in hemSCs. If propranolol is acting through TNF-α pathway, then we would expect no increase in caspase-3 activity. Indeed, our results show that TNF-α treated hemSCs did not change active caspase-3 level when compared with control (figure 5b). We then tested the possible involvement of IFNγ by exposing hemSCs and bm-MPCs to exogenous IFNγ. Our studies show no significant effect on hemSC or bm-MPC growth at 24 or 72 hours (figure 5c and 5d). This suggests that TNF-α, but not IFNγ, have anti-proliferating effect in vitro and may be involved in propranolol’s action.
Figure 5. TNF activation leads to reduced cell growth in hemSCs.

TNF-α treatment significantly reduced cell number (a) in hemSCs after 72 hours but did not change activated caspase-3 level (b) (*p<0.05 compared to control). HemSCs treated with IFNγ showed no significant change in cell number after 24 (c) or 72 (d) hours.
Propranolol-induced altered differentiation in hemSC
While our studies were underway, a report showed that presence of propranolol in adipogenic differentiation media increased the differentiation level in hemSCs as compared to cells in adipogenic media alone (21). Higher levels of C/EBPβ and δ were found at day 4. Interestingly, when the cells were maintained in the differentiation media supplemented with propranolol for 7 days, significant cell death was observed. This is in contrast to our observations in normal growth media where a significant reduction in cell number is evident without apoptosis. Therefore, we explored the possibility that this enhanced adipogenic differentiation with propranolol is mediated through cell growth regulation. To test this idea, we treated hemSCs with mitomycin C to inhibit proliferation and tested for C/EBP expression. Our results show that C/EBPα was significantly higher when mitomycin C-treated hemSCs were exposed to adipogenic differentiation media for 4 days (figure 6a). No change was observed in PPARγ2 levels. C/EBPα is a critical transcription factor in adipogenesis and enhanced levels suggest that inhibition of cell proliferation increases the differentiation capacity of hemSCs and this may be the mechanism underlying propranolol’s effect. We then assayed for β-adrenergeic receptor expression and show here that adipogenesis is associated with significantly higher levels of all three adrenergic receptors (figure 6b). Therefore, cell death in adipogenic media supplemented with propranolol (21) might be due to increased expression of β-adrenergic receptors, which accompanies hemSC differentiation.
Figure 6. Growth inhibition enhances adipogenesis.
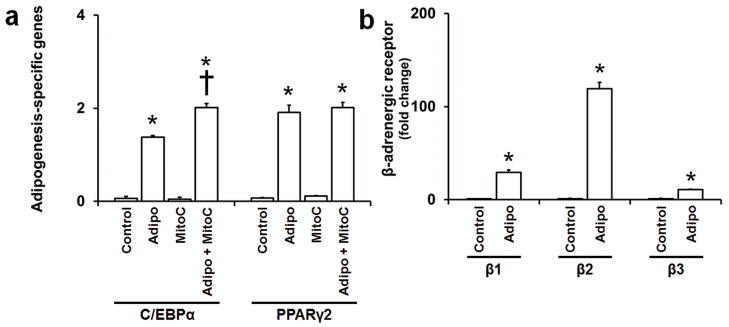
Effect of mitomycin-C treatment on adipogenic differentiation (a) in hemSCs at day 4 (Adipo = adipogenic media, MitoC = mitomycin C; *p<0.05 compared to control media; †p<0.05 compared to adipogenic differentiation media without mitomycin-C treatment). (b) Induction of β-adrenergic receptors following adipogenic differentiation in hemSCs at day 7 (Adipo = adipogenic media; *p<0.05 compared to control media).
Discussion
Propranolol is now widely used for treating IH; however, the mechanism of therapeutic effect is still unknown. In addition, some IH regrow after stopping propranolol treatment (15–17). In the present study, we have demonstrated that propranolol does not induce apoptosis in hemSCs, cells responsible for IH initiation, as seen in mature/differentiated endothelial cells. This suggests that the direct effect of propranolol in IHs may be through modulating mature endothelial cells and angiogenesis (figure 7a). The mechanism by which hemSCs and possibly normal progenitor cells (modeled here by bm-MPCs) escape apoptosis may include induction of anti-apoptotic pathways. We found Akt, Bcl2/Bcl2A, and IGFR1 to be significantly induced in hemSCs and bm-MPCs. Akt induction is of particular importance here as this pro-survival kinase counteracts caspase-3 activity (22–24). Bcl2 downregulation has been shown to increase caspase-3 in breast cancer cells (25). Furthermore, Bcl2A mediates anti-apoptotic effects of fibroblast growth factor in chrondogenic progenitor-like cell line (26). Therefore, these pathways may be involved in reducing/counteracting caspase-3 activity in hemSCs but not mature endothelial cells.
Figure 7. Proposed mechanism of therapeutic effect of propranolol.
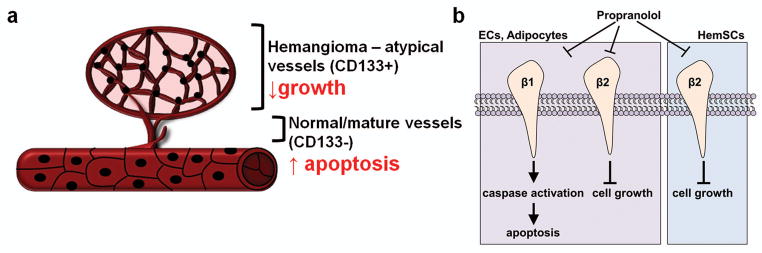
Schematic illustrating the proposed mechanism of propranolol on hemSCs and mature endothelial cells. (a) Schematic illustrating the penetrance of apoptotic effect of propranolol in hemangioma regression. (b) Propranolol leads to cell apoptosis in endothelial cells and other mature cell types including adipocytes through alteration of β1 adrenergic receptor signaling. Engagement of β2 (and possibly β3) receptors leads to cell cycle arrest and growth inhibition.
We also examined whether the extrinsic apoptotic pathway is involved in propranolol-induced growth inhibition. Extrinsic pathway is primarily mediated by the TNF superfamily. We found that TNF signaling pathway genes (listed in figure 4b) are also upregulated in hemSCs and bm-MPCs upon propranolol treatment. Since it is well known that TNF signaling pathways are involved in cell death, these results were unexpected. However, recent studies show that TNF’s effect may not be limited to cell apoptosis. TNF-α causes cell cycle arrest but not death in keratinocytes (27), glioblastoma cells (28), and melanoma cell lines (29). These findings suggest two independent anti-proliferative mechanisms of TNF signaling. In hemSCs, TNF signaling may be involved in propranolol-mediated cell cycle arrest. Indeed, when we treated hemSCs to exogenous TNF-α, we noted decreased cell growth over 72 hours but no change in caspase-3 activity. Taken together with the results of anti-apoptotic gene induction, we believe propranolol may reduce hemSC growth by inducing TNF pathway and escape from caspase-3 activation and apoptosis by TNF-independent induction of anti-apoptotic genes.
Another interesting finding of this study is that hemSCs predominantly express β2 and β3 adrenergic receptors. β1 levels are almost ten-fold lower. Vascular endothelial cells, on the other hand, express significantly high level of β1 receptor compared to β2/3. These findings suggest that the differential effect of propranolol in hemSCs and endothelial cells may be due to distinct roles of β-adrenergic receptor subtypes. Communal et al. have shown that activation of β1 receptors on cardiac myocytes induces apoptosis, whereas β2 adrenergic receptor activation opposes cell death (30). Although this study involved activation of β-adrenergic receptor and not antagonism, the concept of a distinct, receptor subtype-specific role is pertinent here. Moreover, Panjala et al. have demonstrated that β1-adrenergic receptor knockout mice exhibit increased formation of degenerate capillaries in retina (31). These are interesting findings because retinal endothelial cells express β1 adrenergic receptor but not β2 (32). Also associated with acellular capillaries in the retina in knockout mice were increased level of cleaved caspase-3. Based on our data, we suggest that antagonizing β1 receptor in IH cells is associated with cell death while β2 is involved with cell cycle regulation (figure 7b). We know that hemSC predominantly express β2 and β3 receptors and show almost 10-fold lower β1 level. bm-MPCs showed a similar response to propranolol in cellular activity and molecular alterations, and share the β-adrenergic receptor profile with hemSCs.
Wong et al have recently shown that propranolol enhances adipogenesis in hemSCs (21). Specifically, presence of propranolol in adipogenic media initially caused differentiation of hemSC but significant cell death at day 7 (21), which is the typical in vitro time for full functional adipocyte differentiation. We reasoned that the initial effect of propranolol might be mediated through cell cycle disruption. To test this possibility, we treated hemSCs with mitomycin-C before exposing the cells the adipogenic differentiation media. We noted that mitomycin-C treated cells had significantly higher levels of C/EBPα induction suggesting that enhanced adipogenesis in hemSCs may be related to a change in the differentiation timeline (reduced growth and earlier differentiation). We also tested whether adipogenesis itself alters β-adrenergic receptor expression in hemSCs, thereby making cells more sensitive to propranolol and possibly explaining cell death observed in the previous study (21). Indeed, differentiation of hemSCs significantly increased the expression of all β adrenergic receptor subtypes including β1. Therefore, cell death seen in adipocytes may be associated with the increase in β1 receptor expression (29.47-fold increase as reported in our study). Increased level of β2 receptors in adipocytes may further enhance the inhibitory process by regulating cell cycle progression.
Our study represents an important step toward understanding the mechanism of propranolol action. A number of pressing questions remain. For example, is propranolol selective to IH vessels? Based on recent findings in endothelial cells (normal vs IH-derived) and our own findings in hemSCs, this seems unlikely. Alternatively, the effectiveness of propranolol in IHs may be a function of increased levels that are sustained in the capillary mass of IHs. These studies are underway in our laboratory and the findings may enhance our understanding of the mechanism of therapeutic action of propranolol and may provide more effective treatment options.
Methods
IH Specimens and Immunostaining
We obtained paraffin-embedded IH specimens from the Department of Pathology Archives at the London Health Sciences Centre (LHSC, London Ontario, Canada). The proliferating phase was confirmed by histological analysis. Tissue sections were deparaffinized, hydrated, and subjected to antigen retrieval using Tris/EDTA buffer (10 mM Trizma-base, 1 mM EDTA, 0.05% Tween-20, pH 9.0) in 2100 Retriever (Electron Microscopy Sciences, Hatfield, PA). To perform double staining, we incubated slides with mouse anti-human CD31 (1:50; M0823, Dako Canada, Mississauga, ON) and rabbit anti-human CD133 antibody (1:100; ab19898, Abcam, Cambridge, MA) for 1 hour at room temperature. Fluorescein- or texas red-conjugated secondary antibodies (Vector Laboratories, Burlington, ON) were used for detection. Slides were counterstained with DAPI (Vector Laboratories). Images were taken using the Olympus BX-51 microscope (Olympus Canada In., Richmond Hill, ON) equipped with a Spot Pursuit digital camera (SPOT Imaging Solutions, Sterling Heights, MI).
IH Cell Isolation and Culture
Proliferating IH-derived CD133+ cells (hemSCs) were kindly provided by Dr. Joyce Bischoff (Children’s Hospital Boston, Boston, MA). We have previously shown full characterization of hemSCs and culture conditions (18). Freshly isolated bone marrow-mesenchymal progenitor cells (bm-MPCs; isolated from bone marrow mononuclear preparations; 2M-125B, Lonza Inc., Walkersville, MD) were used as normal stem/progenitor controls. Neonatal human dermal microvascular endothelial cells (HDMECs; CC-2516, Lonza Inc) were also used as controls. Previous studies have compared normal endothelial cells with IH-derived mature endothelial cells and have shown striking similarity in propranolol response (9,12,13). All cells were cultured on fibronectin-coated (FN; 1 μg/cm2, FC010-10, Millipore, Temecula, CA) plates in Endothelial Basal Media-2 (EBM2; Lonza Inc.) supplemented with 20% FBS (Lonza Inc.) and EGM-2 SingleQuots (CC-4176, Lonza Inc.) and 1X antibiotic antimycotic media (PSF; Life Technologies, Burlington, ON). All cells were cultured under identical conditions and experiments were performed with a minimum of 2 biological replicates (different IH cell preparations) and 3 technical replicates. All studies were conducted following approval by the Research Ethics Board at The University of Western Ontario, London, Ontario, Canada.
Cell growth assay
To determine the effect of propranolol on cell growth, we plated each cell type at 5000 cell/cm2 in complete EBM2 growth media (described above). After 24 hours, media was removed and cells were exposed to 25, 50, or 100 μM (R)-(+)-propranolol hydrochloride (Propranolol; 0624, R&D Systems, Minneapolis, MN) in fresh EBM2 media. Total number of live cells was determined at 24 or 72 hours using Scepter 2.0 Automated Cell Counter (Millipore) with appropriate histogram gating setup (33,34). For some experiments, hemSCs and bm-MPCs were treated with interferon-γ (IFNγ; 285-IF-100, R&D systems) or tumor necrosis factor-α (TNF-α; 210-TA-010/CF, R&D systems) for up to 72 hours.
RNA isolation, mRNA profiling & Quantitative RT-PCR
RNA was isolated using RNeasy Micro Plus Kit (Qiagen, Mississauga, ON). Total amount of RNA was measured using Qubit RNA Broad Range Assay in a Qubit Fluorometer (Life Technologies). cDNA was then synthesized using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). We performed gene expression analyses using using RT2 Human Cell Death Pathway Finder PCR arrays (PAHS-212Z; Qiagen) in Bio-Rad CFX Connect (Bio-Rad). Data was analyzed by CFX Manager Software using normalized (ΔΔCT) method with two housekeeping genes (β-actin and GAPDH were both used for normalization after empirically determining the expression for stability in our treatment groups).
β-adrenergic receptor and adipogenesis-specific gene expression was also assessed by qRT-PCR. Reactions consisted of 10 μL ssoFast Evagreen (1725200, Bio-Rad), 2 μL of both forward and reverse primers (at a 10 μM concentration), 2 μL cDNA, and 6 μL of H2O. Adipogenesis was assessed by C/EBPα (QT00203357, Qiagen) and PPARγ2 (sequence shown in (35)). β1 (QT00204309, Qiagen), β2 (QT00200011, Qiagen), and β3 adrenergic receptor (QT00200004, Qiagen) expressions were measured similarly. Target gene mRNA data was normalized to β-actin (QT01680476, Qiagen). All reactions were performed for 40 cycles using the following temperature profiles: 95°C for 2 minutes (initial denaturation); and 55°C for 12 seconds (annealing and extension). Data was analyzed using relative quantity (ΔCT).
Caspase-3 and Cyclin-D1 Measurements
Total proteins from the cultured cells were extracted using Cell Extraction Buffer (Life Technologies) with complete protease inhibitor cocktail (Roche Diagnostics, Laval, Quebec). Proteins were measured by BCA Protein Assay Reagent (Pierce BCA Protein Assay Kit, Thermo Scientific, Rockford, IL) and equal amounts were used for active caspase-3 and cyclin-D1 measurements. To measure activated caspase-3, caspase-3 (active) Human ELISA kit (Life Technologies) was used. Data were collected using Thermo Scientific Multiskan FC Microplate Photometer (Thermo Scientific), measuring absorbance at 450nm. Cyclin-D1 level was measured similarly using PathScan Total Cyclin-D1 Sandwich ELISA kit (Cell Signaling Technology Inc., Danvers, MA).
Adipogenic Differentiation
To induce adipogenic differentiation, hemSCs were seeded at a density of 40,000 cells/cm2 in StemPro Adipogenesis differentiation media (Adipo media; Life Technologies). Control media consisted of Dulbecco’s Modified Eagle Medium supplemented with 10% FBS. Media was changed every other day. RNA was isolated from cells after 7 days to perform qRT-PCR for β-adrenergic receptor expression. To determine whether cell growth/proliferation may alter adipogenesis, we pretreated hemSCs with 10 μg/mL mitomycin C (MitoC; Sigma Aldrich, Oakville, ON) for 2 hours. Cells were then washed, resuspended, and plated at 40,000 cells/cm2 in adipogenesis media. RNA was isolated at day 4 to assay for C/EBPα and PPARγ2 levels (transcription factors essential for adipogenic differentiation (33–35)).
Statistical Analysis
The data were expressed as means ± SEM. Where appropriate, analysis of variance (ANOVA) followed by two-tailed student’s unpaired t-tests were performed. P values < 0.05 were considered statistically significant.
Acknowledgments
Statement of financial support: This study was funded by the Canadian Institutes of Health Research (ZK; MOP 97783). ZK is a recipient of a New Investigator Award from the Heart and Stroke Foundation of Canada (Great-West Life and London Life New Investigator Award).
Footnotes
Disclosure: No conflicts to declare.
Category of study: basic science.
References
- 1.Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–22. doi: 10.1097/00006534-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Krol A, MacArthur CJ. Congenital hemangiomas: rapidly involuting and noninvoluting congenital hemangiomas. Arch Facial Plast Surg. 2005;7:307–11. doi: 10.1001/archfaci.7.5.307. [DOI] [PubMed] [Google Scholar]
- 3.Kleiman A, Keats EC, Chan NG, Khan ZA. Evolution of hemangioma endothelium. Exp Mol Pathol. 2012;93:264–72. doi: 10.1016/j.yexmp.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 4.Frieden IJ, Eichenfield LF, Esterly NB, Geronemus R, Mallory SB. Guidelines of care for hemangiomas of infancy. American Academy of Dermatology Guidelines/Outcomes Committee. J Am Acad Dermatol. 1997;37:631–7. doi: 10.1016/s0190-9622(97)70183-x. [DOI] [PubMed] [Google Scholar]
- 5.Durr ML, Meyer AK, Huoh KC, Frieden IJ, Rosbe KW. Airway hemangiomas in PHACE syndrome. Laryngoscope. 2012;122:2323–9. doi: 10.1002/lary.23475. [DOI] [PubMed] [Google Scholar]
- 6.Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taieb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–51. doi: 10.1056/NEJMc0708819. [DOI] [PubMed] [Google Scholar]
- 7.Price CJ, Lattouf C, Baum B, et al. Propranolol vs corticosteroids for infantile hemangiomas: a multicenter retrospective analysis. Arch Dermatol. 2011;147:1371–6. doi: 10.1001/archdermatol.2011.203. [DOI] [PubMed] [Google Scholar]
- 8.Richter GT, Friedman AB. Hemangiomas and vascular malformations: current theory and management. Int J Pediatr. 2012;2012:645678. doi: 10.1155/2012/645678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamy S, Lachambre MP, Lord-Dufour S, Beliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53:200–8. doi: 10.1016/j.vph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Bingham MM, Saltzman B, Vo NJ, Perkins JA. Propranolol reduces infantile hemangioma volume and vessel density. Otolaryngol Head Neck Surg. 2012;147:338–44. doi: 10.1177/0194599812451570. [DOI] [PubMed] [Google Scholar]
- 11.Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas. Pediatrics. 2011;128:e259–66. doi: 10.1542/peds.2010-0029. [DOI] [PubMed] [Google Scholar]
- 12.Stiles J, Amaya C, Pham R, et al. Propranolol treatment of infantile hemangioma endothelial cells: A molecular analysis. Exp Ther Med. 2012;4:594–604. doi: 10.3892/etm.2012.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji Y, Li K, Xiao X, Zheng S, Xu T, Chen S. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J Pediatr Surg. 2012;47:2216–23. doi: 10.1016/j.jpedsurg.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 15.Xiao Q, Li Q, Zhang B, Yu W. Propranolol therapy of infantile hemangiomas: efficacy, adverse effects, and recurrence. Pediatr Surg Int. 2013;29:575–81. doi: 10.1007/s00383-013-3283-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Georgountzou A, Karavitakis E, Klimentopoulou A, Xaidara A, Kakourou T. Propranolol treatment for severe infantile hemangiomas: a single-centre 3-year experience. Acta Paediatr. 2012;101:e469–74. doi: 10.1111/j.1651-2227.2012.02783.x. [DOI] [PubMed] [Google Scholar]
- 17.Bagazgoitia L, Hernandez-Martin A, Torrelo A. Recurrence of infantile hemangiomas treated with propranolol. Pediatr Dermatol. 2011;28:658–62. doi: 10.1111/j.1525-1470.2011.01644.x. [DOI] [PubMed] [Google Scholar]
- 18.Khan ZA, Boscolo E, Picard A, et al. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118:2592–9. doi: 10.1172/JCI33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji Y, Chen S, Li K, Xiao X, Zheng S, Xu T. The role of beta-adrenergic receptor signaling in the proliferation of hemangioma-derived endothelial cells. Cell Div. 2013;8:1. doi: 10.1186/1747-1028-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15:158–63. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- 21.Wong A, Hardy KL, Kitajewski AM, Shawber CJ, Kitajewski JK, Wu JK. Propranolol accelerates adipogenesis in hemangioma stem cells and causes apoptosis of hemangioma endothelial cells. Plast Reconstr Surg. 2012;130:1012–21. doi: 10.1097/PRS.0b013e318267d3db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kermer P, Klocker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 In vivo. J Neurosci. 2000;20:2–8. [PubMed] [Google Scholar]
- 23.Shimoke K, Chiba H. Nerve growth factor prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced cell death via the Akt pathway by suppressing caspase-3-like activity using PC12 cells: relevance to therapeutical application for Parkinson’s disease. J Neurosci Res. 2001;63:402–9. doi: 10.1002/1097-4547(20010301)63:5<402::AID-JNR1035>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Barber AJ, Nakamura M, Wolpert EB, et al. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276:32814–21. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- 25.Mohan S, Abdelwahab SI, Kamalidehghan B, et al. Involvement of NF-kappaB and Bcl2/Bax signaling pathways in the apoptosis of MCF7 cells induced by a xanthone compound Pyranocycloartobiloxanthone A. Phytomedicine. 2012;19:1007–15. doi: 10.1016/j.phymed.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Kim HR, Heo YM, Jeong KI, et al. FGF-2 inhibits TNF-alpha mediated apoptosis through upregulation of Bcl2-A1 and Bcl-xL in ATDC5 cells. BMB Rep. 2012;45:287–92. doi: 10.5483/bmbrep.2012.45.5.287. [DOI] [PubMed] [Google Scholar]
- 27.Basile JR, Eichten A, Zacny V, Munger K. NF-kappaB-mediated induction of p21(Cip1/Waf1) by tumor necrosis factor alpha induces growth arrest and cytoprotection in normal human keratinocytes. Mol Cancer Res. 2003;1:262–70. [PubMed] [Google Scholar]
- 28.Cheng K, Sawamura Y, Sakuma S, et al. Antiproliferative effect of tumor necrosis factor-alpha on human glioblastoma cells linked with cell cycle arrest in G1 phase. Neurol Med Chir (Tokyo) 1994;34:274–8. doi: 10.2176/nmc.34.274. [DOI] [PubMed] [Google Scholar]
- 29.Hattori T, Hayashi H, Chiba T, Onozaki K. Activation of two distinct anti-proliferative pathways, apoptosis and p38 MAP kinase-dependent cell cycle arrest, by tumor necrosis factor in human melanoma cell line A375. Eur Cytokine Netw. 2001;12:244–52. [PubMed] [Google Scholar]
- 30.Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)-and beta(2)-adrenergic receptors on cardiac myocyte apoptosis : role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 31.Panjala SR, Jiang Y, Kern TS, Thomas SA, Steinle JJ. Increased tumor necrosis factor-alpha, cleaved caspase 3 levels and insulin receptor substrate-1 phosphorylation in the beta(1)-adrenergic receptor knockout mouse. Mol Vis. 2011;17:1822–8. [PMC free article] [PubMed] [Google Scholar]
- 32.Steinle JJ, Booz GW, Meininger CJ, Day JN, Granger HJ. Beta 3-adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem. 2003;278:20681–6. doi: 10.1074/jbc.M300368200. [DOI] [PubMed] [Google Scholar]
- 33.Keats E, Khan ZA. Unique responses of stem cell-derived vascular endothelial and mesenchymal cells to high levels of glucose. PLoS One. 2012;7:e38752. doi: 10.1371/journal.pone.0038752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleiman A, Keats EC, Chan NG, Khan ZA. Elevated IGF2 prevents leptin induction and terminal adipocyte differentiation in hemangioma stem cells. Exp Mol Pathol. 2013;94:126–36. doi: 10.1016/j.yexmp.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 35.Roach EE, Chakrabarti R, Park NI, et al. Intrinsic regulation of hemangioma involution by platelet-derived growth factor. Cell Death Dis. 2012;3:e328. doi: 10.1038/cddis.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]