

Abstract
Inhaled gene carriers must penetrate the highly viscoelastic and adhesive mucus barrier in the airway in order to overcome rapid mucociliary clearance and reach the underlying epithelium; however, even the most widely used viral gene carriers are unable to efficiently do so. We developed two polymeric gene carriers that compact plasmid DNA into small and highly stable nanoparticles with dense polyethylene glycol (PEG) surface coatings. These highly compacted, densely PEG-coated DNA nanoparticles rapidly penetrate human cystic fibrosis (CF) mucus ex vivo and mouse airway mucus ex situ. Intranasal administration of the mucus penetrating DNA nanoparticles greatly enhanced particle distribution, retention and gene transfer in the mouse lung airways compared to conventional gene carriers. Successful delivery of a full-length plasmid encoding the cystic fibrosis transmembrane conductance regulator protein was achieved in mouse lungs and airway cells, including a primary culture of mucus-covered human airway epithelium grown at air-liquid interface, without causing acute inflammation or toxicity. Highly compacted mucus penetrating DNA nanoparticles hold promise for lung gene therapy.
Keywords: airway gene therapy, mucus barrier, DNA nanoparticle, multiple particle tracking
1. Introduction
Pulmonary complications are the primary cause of morbidity and mortality associated with cystic fibrosis (CF), asthma, and other life-threatening disorders [1, 2]. A great deal of effort has aimed at development of gene therapy strategies for the airways, including clinical evaluation of a large number of viral and non-viral gene delivery systems in patients with lung diseases [3, 4]. The majority of these trials have utilized recombinant viruses, but immunogenicity, safety concerns, and inefficient gene transfer have precluded their success [3, 5]. Alternatively, non-viral gene carriers offer potential for improved safety, ease of manufacturing and scale up, possibility of repeat dosing without generating a therapy-disabling immune response, and ability to accommodate larger plasmid DNA compared to commonly tested viruses, such as adeno-associated virus. A polymeric gene delivery platform, based on a copolymer of 10 kDa polyethylene glycol (PEG) conjugated to a 30-mer poly-L-lysine (PLL) via a cysteine residue (PEG10k-CK30/DNA), was safely administered to the nares of CF patients with negligible serum or nasal inflammatory responses in a clinical trial [6], thus encouraging further development of similar strategies.
A largely overlooked barrier to efficient gene delivery to the lung airways is the highly adhesive and hyperviscoelastic mucus gel in the airways of patients, including those with CF, asthma and chronic obstructive pulmonary lung disease (COPD), that traps and prevents the access of gene carriers to the underlying epithelium. Mucus in patients with advanced obstructive lung diseases is composed of a dense mesh of mucin fibers, large macromolecules containing a high density of negatively charged glycans interspersed with periodic hydrophobic regions [7]. In the airways of CF [8, 9] and COPD [10] patients, elevated levels of bacterial and endogenous DNA, as well as actin filaments from degraded neutrophils, further contributes to the dense mucus mesh structure and increased adhesivity of the mucus gel layer. We previously estimated the average pore size in CF mucus to be 140 ± 50 nm (range: 60 – 300 nm) [11], markedly smaller than the average pore size of 340 ± 70 nm for human cervicovaginal mucus secretions from healthy, non-CF volunteers [12]. As a consequence of the elevated adhesivity and tighter mesh size, we recently found that several clinically tested viral [13] and non-viral [14, 15] gene carriers are incapable of efficiently penetrating human CF mucus.
We sought to engineer polymeric gene carriers that are sufficiently small to diffuse through the mucus mesh and enter cells via non-specific endocytic pathways, and that possess a muco-inert surface to avoid adhesion to mucus constituents. Cationic polymers possess a high density of protonable amines that facilitate efficient DNA condensation [16, 17], protect cargo DNA from enzymatic degradation [18, 19], and provide proton-buffering that may facilitate endosomal escape of cargo DNA from degradative vesicles in cells [20]. Nevertheless, gene transfer efficiency by cationic polymer-based gene carriers is strongly reduced in mucus-coated cells compared to mucus-depleted tissues [21], likely a consequence of entrapment of cationic gene carriers in the mucus gel via electrostatic association with the negatively charged mucus constituents. We have previously demonstrated that a dense coating of low MW (2 – 5 kDa) PEG markedly improved the diffusion of polymeric nanoparticles (NP) through human cervicovaginal mucus [22, 23], chronic rhinosinusitis mucus [24] and CF airway mucus [11]. PEG coatings also have shown to reduce toxicity of cationic polymers in vivo [25] and in humans [6] to clinically safe levels. Dense PEG coatings, however, interfere with DNA compaction by cationic polymers, resulting in loosely compacted larger particles [26, 27]. Larger particles with PEG surface coatings have shown inferior cellular uptake compared to smaller but similarly coated particles [28, 29]. We describe an approach to produce small, stable NP with sufficiently dense PEG coatings to allow rapid mucus penetration, and we show it is applicable to two widely used polymer systems, polyethylenimine (PEI) and poly-L-lysine (PLL). We show that mucus penetrating DNA NP mediate efficient gene transfer to airway cells in vitro and in vivo without eliciting acute toxic or inflammatory responses.
2. Materials and methods
2.1. CF mucus collection
Mucus spontaneously expectorated from CF patients ages 24 – 37, was collected at the Johns Hopkins Adult Cystic Fibrosis Program. Mucus collection was performed under informed consent on a protocol approved by the Johns Hopkins Medicine Institutional Review Board. Samples were acquired from the weekly CF outpatient clinic, placed immediately on ice, and studied the same day. The total number of individual samples used for the present study was 7.
2.2. Polymer preparation
Methoxy PEG N-hydroxysuccinimide (mPEG-NHS, 5 kDa, Sigma-Aldrich, St. Louis, MO) was conjugated to 25 kDa branched PEI (Sigma-Aldrich) to yield PEG5k-PEI copolymer. Briefly, PEI was dissolved in ultrapure distilled water and pH was adjusted to 7.5–8. Approximately 50 molar excess of mPEG-NHS was added to PEI solution and allowed to react overnight. After the reaction, the polymer solution was extensively dialyzed against ultrapure distilled water and lyophilized. Nuclear magnetic resonance (NMR) was used to confirm a PEG:PEI ratio to be ~37 (Fig. S1). 1H NMR (500 MHz, D2O): δ 2.48–3.20 (br, CH2CH2NH), 3.62–3.72 (br, CH2CH2O). The lyophilized polymers were dissolved in ultrapure distilled water and pH was adjusted to ~7–7.5.
A 30-mer PLL (K30) was synthesized by Fmoc-mediated solid-phase peptide synthesis using an automated peptide synthesizer (Symphony Quartet, Protein Technologies, Tucson, AZ). The final peptide products, including CK30 and C4K33, were synthesized by manually adding amino acids via a standard HBTU (O-(1-Benzotriazolyl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate) coupling procedure [30]. Crude peptides were purified by HPLC (Shimadzu Scientific Instruments, Columbia, MD) and molecular weights were confirmed by MALDI-TOF mass spectrometry (Voyager DE-STR, Applied Biosystems, Foster City, CA). Block copolymers of PEG and PLL, including PEG10k-CK30 and (PEG5k)4-C4K33, were prepared as previously described [31]. Briefly, 10 and 5 kDa PEG was conjugated to CK30 and C4K33, respectively, via the reaction between sulfhydryl group of cysteine residue and maleimide group of PEG. Completion of the reaction was verified using a 4-PDS assay for unreacted sulfhydryls [31]. The chemical structures of copolymers are depicted in Fig. S2.
2.3. Gene carrier formulation
The pd1GL3-RL plasmid DNA was a kind gift from Professor Alexander M. Klibanov (M.I.T), pBAL and pBACH plasmid DNA were produced by Copernicus Therapeutics Inc. (Cleveland, OH), pUMVC-nt-β-gal plasmid DNA was purchased from Aldevron (Fargo, ND), and pcDNA3.1 WT-CFTR (Fig. S3A) and pEGFP WT-CFTR (Fig. S3B) plasmid DNA were designed in house. The plasmid DNA was propagated, purified and fluorescently labeled as described in Supplementary Materials and Methods.
Gene carriers were formed by the drop-wise addition of 9 – 10 volume of plasmid DNA (0.2 mg/ml) to 1 volume of a swirling polymer solution. PEI solutions were prepared at an optimized nitrogen to phosphate (N/P) ratio of 6 and at different PEI to PEG5k-PEI ratios. PLL solutions were prepared with PEG10k-CK30, (PEG5k)4-C4K33 or a blend of CK30 (25%) and (PEG5k)4-C4K33 (75%) at optimized N/P ratios. For fluorescence imaging, 20% Cy3- or Cy5-labeled DNA (i.e. 80% unlabeled DNA) was used to assemble fluorescently labeled gene carriers. The mixture of DNA and polymer solutions was incubated for 30 min at room temperature to form gene carriers, followed by syringe filtration (0.2 μm). Gene carriers were washed twice with 10 volume of ultrapure distilled water, and re-concentrated to 1 mg/ml using Amicon® Ultra Centrifugal Filters (100,000 MWCO, Millipore Corp., Billerica, MA) to remove free polymers. DNA concentration was determined via absorbance at 260 nm using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE).
2.4. Physicochemical properties and stability of gene carriers
Gene carriers were imaged using transmission electron microscopy (TEM, Hitachi H7600, Japan) to determine their morphology and size. Hydrodynamic diameter and ζ-potential were determined by dynamic light scattering and laser Doppler anemometry, respectively, using a Nanosizer ZS90 (Malvern Instruments, Southborough, MA). Haparin displacement and DNase protection assays were performed as described in Supplementary Materials and Methods.
2.5. Multiple particle tracking in CF Mucus
Cy3-labeled gene carriers were added to ~30 μl of CF mucus (3% dilution), transferred to custom-made 30 μl microwells, and equilibrated for 1 h at 37 °C prior to microscopy. Twenty second movies at 66.7 ms temporal resolution were acquired via Evolve 512 EMCCD camera (Photometrics, Tucson, AZ) equipped on an inverted epifluorescence microscope (Axio Observer D1, Zeiss; Thornwood, NY) with a 100X/1.4 NA objective. Movies were analyzed with Metamorph software (Universal Imaging, Glendale, WI) to extract x, y-coordinates of gene carrier centroids over time. Time-averaged mean square displacement (MSD) for individual gene carriers were calculated as a function of time scale (τ) [32]. CF mucus was assumed to be locally isotropic but not necessarily homogeneous; thus, 2D diffusivity is equal to 3D diffusivity [32]. Bulk transport properties were calculated by geometric ensemble-averaging of individual MSD. Due to the intrinsically autofluorescent and heterogeneous nature of CF mucus, along with limited fluorescence intensity and photostability of fluorescently labeled gene carriers, we were forced to use a fairly long exposure time (66.7 ms) to capture fluorescent snapshots of gene carriers. This, combined with the low signal-to-noise ratio, can introduce substantial static and dynamic errors into the measured MDS values [33–35]. We estimated the static error to be less than 20 nm by tracking the displacements of gene carriers immobilized at the interface of super glue and CF mucus, which simulates the signal-to-noise ratios as observed in our experimental movies. Dynamic errors are difficult to quantify and correct for, unless the shape of the data and an analytical formula for it are known, which is only the case for a few model fluids [33]. For complex viscoeleastic materials, such as a human CF mucus sample, we do not know a priori what the MSD should look like. However, dynamic error is smaller for larger time scales, since then the time interval is substantially larger than the fixed exposure time (66.7 ms) [36]. Accordingly, to minimize the effects of dynamic error, we chose to present the MSD values at a time scale of 1 s. Furthermore, based on our previous experience, this 1 s time scale is still short enough to avoid losing too many fast moving particles in the z-direction, and still provides a sufficient number of particle displacements to reduce statistical error.
2.6. Diffusion and distribution of gene carriers in mucus of mouse trachea and lung
Balb/c mice (female, 6–8 weeks), anesthetized in a perfusion chamber with 1 – 1.5% isoflurane at 1 – 1.5 L/min oxygen flow, were intranasally instilled with Cy3-labeled gene carriers. Twenty minutes later, mice were euthanized and a small piece of trachea (~5 mm) was dissected. The trachea was then cut open longitudinally, laid flat on a glass slide (mucosal side up), and sealed with a coverslip using a thin layer of super glue to minimize dehydration. Movies and images were acquired with a fluorescence microscope to qualitatively compare the diffusion and distribution of gene carriers in mucus of mouse trachea.
To investigate the distribution of gene carriers in mouse lung, mice were euthanized and the lungs were dissected 2 h after intranasal instillation of Cy3-labeled gene carriers. Lungs were then inflated with a 50:50 mixture of optimal cutting temperature compound (OCT) and 1X PBS, embedded in 100% OCT to be frozen with liquid N2, and cryosectioned at −22°C. Tissue sections were stained with ProLong® Gold antifade reagent with DAPI (Molecular Probes, Eugene, OR) and images were captured with a fluorescence microscope.
2.7. Retention of gene carriers in mouse lung and gastrointestinal tract
Anesthetized Balb/c mice (female, 6–8 weeks) were intranasally instilled with Cy5-labeled gene carriers as described above. At various time points after the administration of gene carriers, mice were euthanized and the lung and upper GI tract (i.e. esophagus and stomach) were harvested. Mice treated with saline served as control. Fluorescent images of individual lungs and upper GI tracts were then captured with a Xenogen IVIS spectrum optical imaging device (Caliper Life Sciences, Inc., Hopkinton, MA) at the exposure time of 1 s, and total photon counts (i.e. total fluorescence) were measured and subtracted with saline controls using Living Images® 2.5 software (Caliper Life Sciences, Inc.). Lung retention and upper GI localization of gene carriers over time were calculated by normalizing total photon counts at varying time points with initial photon counts.
2.8. In vivo airway gene transfer
Anesthetized Balb/c mice (female, 6–8 weeks) were intranasally instilled with gene carriers (50 μg pd1GL3-RL plasmid DNA per mouse) as described above. Mice treated with either saline or free DNA served as controls. For the comparison between mucus-penetrating PEI/DNA NP (PEI-MPP) and PEG10k-CK30/DNA NP, mice were intranasally instilled with either gene carrier at a dose of 50 μg pBAL plasmid DNA per mouse. To quantify airway gene expression, the luciferease activity on lung tissue homogenates was measured as previously described [15]; details are provided in Supplementary Materials and Methods.
To examine the airway distribution of transgene expression, Balb/c mice were intranasally dosed with either saline or PEI-MPP carrying pUMVC-nt-β-gal plasmid DNA (250 μg DNA per mouse). Lungs were harvested 24 h after the administration, inflated with a 50:50 mixture of OCT and 1X PBS, embedded in 100% OCT to be frozen with liquid N2, and cryosectioned at −22°C. Immunohistochemical staining of tissue sections was performed as previously reported [31].
To test whether PEI-MPP mediate the expression of mature CFTR protein, Balb/c mice were intranasally dosed with PEI-MPP carrying CFTR plasmid DNA (50 μg DNA per mouse). Depending on the type of plasmid DNA, mice were euthanized at 2 or 7 day post-administration of PEI-MPP. Lungs were harvested and CFTR protein expression was determined by western blotting as described in Supplementary Materials and Methods.
For the multi-dose study, mice were intranasally dosed once or twice with PEI-MPP (50 μg DNA per mouse) carrying pBACH plasmid DNA (mCherry) or pBAL plasmid DNA (luciferase). These two plasmids are identical except for the reporter coding sequences. Dosing regimen is shown in Fig. 3E.
Figure 3.

In vivo gene transfer to airways of Balb/c mice following (A – D) a single dose or (E) two doses of intranasal NP administration. (A) Mice were dosed with saline or gene carriers carrying pd1GL3-RL plasmid DNA, and the luciferase activity was measured 24 h after the administration (n = 5). The differences are statistically significant between the conditions indicated with asterisks (*) (** p < 0.01; *** p < 0.001). Immunohistochemical staining in lungs of mice dosed with (B) saline or (C) PEI-MPP carrying pUMVC1-nt-β-gal plasmid DNA (nuclear targeted β-galactosidase). Nova red stain (reddish brown; marked by arrows) represents β-galactosidase expression and hematoxylin stain (purplish blue) represents cell nuclei. (D) A western blot showing mature C band of wild-type CFTR protein (wtCFTR) expression (marked by arrows) in tissue taken from the lung airways of a mouse dosed with saline or PEI-MPP carrying pcDNA 3.1 WT-CFTR plasmid DNA. Mature CFTR expression mediated by PEI-MPP resulted in enhanced expression of the C band. (E) Repeated administration of PEI-MPP to airways of Balb/c mice (n = 5 – 7). Dosing regimen is shown in the inset. Briefly, mice were dosed once or twice with PEI-MPP (50 μg DNA per mouse) carrying pBACH plasmid DNA (mCherry; grey arrows) or pBAL plasmid DNA (luciferase; black arrows); the two plasmids are identical except for the reporter coding sequences. Mice in Group I and II were treated with a single dose of pBACH/MPP (negative control) and pBAL/MPP, respectively. Mice in Group III and IV were initially dosed with pBACH/MPP and subsequently with pBAL/MPP with intervals of 2 and 4 weeks, respectively, to avoid potential accumulation of luciferase expression by consecutive doses of pBAL/MPP. Luciferase activity was measured 1 week after the last administration in each group. The differences in the luciferase activities are statistically significant between the conditions indicated with asterisks (*) (*** p < 0.001).
2.9. In vitro CFTR protein expression
In vitro CFTR transgene expression was determined as described in Supplementary Materials and Methods.
2.10. Whole-cell patch clamp
Chinese hamster ovary (CHO) cells cultured in Ham’s F-12, supplemented with 10% FBS and 1% penicillin/streptomycin, were treated with PEI-MPP carrying pEGFP WT-CFTR plasmid DNA for 72 h. Whole-cell patch clamp recordings were performed as previously described [37]; details are provided in Supplementary Materials and Methods.
2.11. Airway inflammation and in vitro cytotoxicity induced by gene carriers
In vivo lung inflammation was assessed by examining lung tissues and BALF; details are provided in Supplementary Materials and Methods. In vitro cytotoxicity of gene carriers was assessed by conventional MTT (3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazonlium bromide) assay; details are provided in Supplementary Materials and Methods.
2.12. Statistical analysis
Statistically significant differences between two groups were analyzed with a two-tailed Student’s t test, assuming unequal variances. Multiple comparisons were performed using one-way analysis of variance (ANOVA) followed by post hoc test using SPSS 18.0 software (SPSS Inc. Chicago, IL). Statistical differences in luciferase activities were determined by analyzing the variances of logarithmically transformed data. P values less than 0.05 were considered significant.
3. Results
To effectively shield the highly positive surface charge of nanoparticulate gene carriers based on PEI (uncoated PEI/DNA NP; PEI-UCP hereafter), we first attempted to conjugate multiple 5 kDa PEG molecules to 25 kDa branched PEI molecules at a high PEG to PEI ratio of 50. The reaction yielded PEG5k-PEI copolymers with an average PEG to PEI ratio of 37, which is substantially higher than previously reported (< 20) [26, 38, 39]. We found that surface charge of PEG-coated DNA NP (conventionally coated PEI/DNA NP; PEI-CCP hereafter), as indicated by ζ-potential, was significantly reduced to near neutral (~ 3.5 mV) compared to PEI-UCP (~ 35.0 mV) (Table 1), indicating the presence of neutrally charged PEG on the PEG-CCP surface. However, the average hydrodynamic diameter of PEI-CCP was ~15% larger than that of PEI-UCP (Table 1). Transmission electron micrographs revealed that PEI-UCP were dense spheres (Fig. S4A), while PEI-CCP were generally larger and more diffuse (Fig. S4B). We hypothesized that the larger size of PEI-CCP was due to loose DNA compaction as a result of reduced PEI positive charges upon PEG conjugation, and/or steric hindrance by PEG chains that interfere with electrostatic interactions between DNA and PEI. Thus, we investigated whether inclusion of free PEI, mixed together with PEG5k-PEI at different PEI:PEG5k-PEI ratios (0%, 50%, 75% and 100% PEG5k-PEI), could reduce particle size while effectively shielding particle surface charge. We found that gene carriers formulated with a mixture of 25% PEI and 75% PEG5k-PEI (mucus-penetrating PEI/DNA NP; PEI-MPP hereafter) were: (i) highly compacted (Fig. S4C) similar to PEI-UCP (Fig. S4A), with hydrodynamic diameters ~ 50 nm (Fig. S5A and Table 1), and (ii) near neutral in surface charge comparable to PEI-CCP (~3.0 mV; Fig. S5B and Table 1). As expected from the reduced number of amines and steric hindrance by PEG chains, PEI-CCP exhibited lower DNA compaction stability in presence of anionic heparin than PEI-UCP (Figs. S4D and S4E). However, PEI-MPP showed an improved stability compared to PEI-CCP (Figs. S4E and S4F). Both PEI-CCP and PEI-MPP protected cargo DNA against DNase challenge as efficiently as the uncoated PEI-UCP (Figs. S4G – S4I).
Table 1.
Diffusion of Gene Carriers in CF Mucus.
| Gene Carriers | Hydrodynamic Diameter (nm) a | ζ - potential (mV) b | MSDw/<MSD> c |
|---|---|---|---|
| PEI-UCP | 52 ± 1.0 | 34 ± 1.0 | 7,700 |
| PEI-CCP | 66 ± 5.0 | 3.5 ± 0.7 | 1,800 |
| PEI-MPP | 56 ± 2.5 | 3.0 ± 0.5 | 110 |
| PLL-CCP | 64 ± 8.0 | 0.5 ± 2.0 | 15,000 |
| PLL-MPP | 58 ± 6.5 | 3.0 ± 1.0 | 110 |
Measured by dynamic light scattering. Error values indicate standard deviations of three independent measurements (mean ± SD).
Measured at pH 7.4. Error values indicate standard deviations of three independent measurements (mean ± SD).
MSDw is the theoretical diffusivity of particles in water calculated from the Stokes-Einstein equation. <MSD> is measured at time scale of 1 s. The MSDw/<MSD> ratio indicates by what multiple the average particle movement rate in CF mucus is slower than in pure water.
To visualize the diffusion of gene carriers in human mucus, we formulated DNA NP compacting fluorescently labeled DNA. We quantified the translational motion of hundreds of PEI-UCP in CF mucus using multiple particle tracking (MPT). As expected from their positive surface charge, motions of PEI-UCP were strongly hindered in mucus freshly expectorated by CF patients, as was evident by their highly constrained, non-Brownian time-lapse traces (Fig. 1A). The geometric averaged mean square displacement (<MSD>) of PEI-UCP in four different mucus samples was 4.4 × 10−3 μm2 at a time scale (τ) of 1 s (Fig. 1B), which is ~7,700-fold lower than the same sized NP in water at τ = 1 s (Table 1). We next quantified the diffusion of gene carriers formulated with PEG5k-PEI (PEI-CCP) in CF mucus. We found that the movements of PEI-CCP were strongly hindered in CF mucus, similar to uncoated PEI-UCP, with <MSD> 1,800-fold lower in mucus compared to in water (Fig. 1B and Table 1). This suggests that DNA compacted with PEG5k-PEI alone, the conventional method used today, does not provide a muco-inert surface coating of gene carriers for efficient diffusion in CF mucus. In contrast, PEI-MPP displayed time-lapse trajectories that spanned markedly greater distances (Fig. 1A). PEI-MPP moved with an <MSD> only 100-fold smaller than their theoretical MSD in water (Table 1). Unlike PEI-UCP and PEI-CCP, the distribution of individual <MSD> for PEI-MPP was bimodal, with only a small fraction of trapped gene carriers and a majority population that rapidly penetrated CF mucus (bottom panel in Fig. 1C).
Figure 1.

Transport of gene carriers in CF mucus. (A, D) Representative trajectories of gene carriers in CF mucus during 20 s movies. The mean square displacement (MSD) of individual traces shown are within one standard deviation of the ensemble-averaged geometric MSD (<MSD>). (B, E) <MSD> of gene carriers as a function of time scale (τ). (C, F) Distribution of the logarithmic MSD of individual gene carriers at τ = 1 s. Data represents 4 and 3 independent experiments for PEI-based and PLL-based gene carriers, respectively, with an average of n > 100 gene carriers per experiment.
We next investigated whether this approach can be applied to another widely used cationic polymer. We previously found that PLL-based DNA NP with a single segment of PEG (5 or 10 kDa) conjugated to PLL, the latter of which is identical to DNA NP developed and clinically tested by Copernicus Therapeutics, Inc. (i.e. PEG10k-CK30/DNA), were unable to efficiently penetrate CF mucus (e.g. ~7,000-fold slower in CF mucus than in water) [14]. To achieve a high density PEG coating, we synthesized a copolymer composed of 4 branched arms of 5 kDa PEG conjugated to PLL via 4 cysteine residues ((PEG5k)4-C4K33), and formulated DNA NP by compacting fluorescently labeled plasmid DNA with (PEG5k)4-C4K33 (PLL-CCP hereafter). Despite the improved PEG density, the movements of PLL-CCP were largely hindered in CF mucus, as reflected by highly constrained time-lapse traces (Fig. 1D). However, DNA NP formulated using a blend of PLL and (PEG5k)4-C4K33 (PLL-MPP hereafter) traveled a markedly greater distance in CF mucus than conventionally coated PLL-CCP (Fig. 1D). Quantitatively, PLL-MPP exhibited <MSD> at least two orders of magnitude higher than PLL-CCP at most time scales monitored (Fig. 1E), and possessed a uniformly higher individual MSD at τ = 1 s (Fig. 1F).
We next investigated the diffusion and distribution of gene carriers in mouse airways. Following intranasal administration of PEI-UCP and PEI-MPP, a significant fraction of PEI-MPP underwent rapid diffusion in the mouse trachea (Video S1), whereas a majority of PEI-UCP were completely immobilized and aggregated into large clusters (Video S2). One hour following inhalation into mouse lungs, PEI-MPP uniformly distributed along the airway epithelium throughout the lungs, whereas PEI-UCP were found aggregated and sparsely distributed in the lung airways (Figs. 2A, 2B and Fig. S6). Inhaled PEI-UCP and PEI-MPP were found in both mouse lungs and the upper GI tract immediately after administration, as expected since a portion of the administered dose is swallowed. Over 70% of PEI-MPP initially deposited in mouse lungs were retained in the airways at 6 h post-administration, whereas the concentration of PEI-UCP sharply dropped (to ~30% of the initial value) at 2 h post-administration, suggesting PEI-UCP were rapidly eliminated from the mouse lungs by mucociliary clearance (MCC) (Figs. 2C – 2E). To confirm that reduced airway retention of PEI-UCP could be attributed to MCC, we measured the concentration of gene carriers in the upper GI tract over time. We found that the concentration of PEI-UCP in the upper GI tract sharply increased at 2 h post-administration, whereas that of PEI-MPP gradually decreased over time (Fig. 2F).
Figure 2.

Lung airway distribution and retention of PEI-UCP and PEI-MPP following pulmonary administration. Distribution of (A) PEI-UCP and (B) PEI-MPP in large and small airways of mouse lungs. Red represents gene carriers and blue represents DAPI staining of lung tissues. The scale bars indicate 50 μm. (C – F) Retention of PEI-UCP and PEI-MPP in mouse lungs (n = 5). Representative fluorescent images of mouse lungs harvested at varying times after the intranasal administration of (C) PEI-UCP and (D) PEI-MPP. (E) Retention of gene carriers in the mouse lungs over time. (F) Localization of gene carriers in upper gastrointestinal (GI) tract over time. Total fluorescence (photons/s) at varying time points was normalized by initial fluorescence. The error bars indicate standard error of the mean. The differences in the % tissue retention/localization are statistically significant between the conditions indicated with asterisks (*) (* p < 0.05; *** p < 0.001).
To investigate airway gene transfer in vivo, we intranasally dosed mice with either luciferase plasmid DNA alone, or plasmid DNA compacted into PEI-UCP or PEI-MPP (Fig. 3A). The average luciferase activities of DNA alone, PEI-UCP and PEI-MPP were 8-, 47-, and 230-fold higher than that of saline control, respectively, with PEI-MPP statistically superior to the other groups (p < 0.001). The average luciferase activity mediated by PEI-MPP was also significantly higher than that achieved by PEG10k-CK30/DNA NP (p < 0.01) (Fig. S7), a polymeric gene delivery platform recently shown to mediate partial CFTR reconstitution in a clinical trial [6]. This is in good agreement with a previous finding that gene transfer efficiency of PEG10k-CK30/DNA NP were comparable to conventional PEI/DNA NP (uncoated PEI-UCP) [31]. Importantly, immunohistochemical staining for nuclear localized β-galactosidase revealed the PEI-MPP induced uniform transgene expression primarily in airway epithelial cells; no gene expression was detected in saline controls (Figs. 3B and 3C). We also confirmed the expression of mature wild-type CFTR proteins in the lungs of mice that were intranasally dosed with PEI-MPP carrying the full-length CFTR gene in plasmid DNA (Fig. 3D). In addition, repeated dosing of PEI-MPP with intervals of 2 and 4 weeks did not reduce gene transfer efficiency in mouse lungs (Fig. 3E).
We next investigated whether mucus penetrating DNA NP are capable of mediating expression of functional CFTR protein in human primary airway epithelial cells grown at the air-liquid interface, and in other cell lines. The fully glycosylated C band of CFTR was detectable without NP treatment in a primary culture of human airway epithelial cells, which is consistent with the endogenous expression of wild-type CFTR protein in normal airway epithelium. The level of detectable C band increased when the cells were treated with PEI-MPP carrying wild-type CFTR plasmid DNA, suggesting the expression of the transfected CFTR gene in this primary culture (Fig. 4A). Likewise, a dose-dependent increase in C band was observed in a cystic fibrosis bronchial epithelial (CFBE41o-) cell line stably expressing wild-type CFTR when treated with varying doses of PEI-MPP carrying wild-type CFTR plasmid DNA (Fig. 4B and Fig. S8). B band but not C band was detected in CFBE41o- cells expressing only endogenous ΔF508 CFTR, which is indicative of immature ΔF508 CFTR residing primarily in the endoplasmic reticulum [40]. However, a fully glycosylated C band was detected when these cells were treated with PEI-MPP carrying wild-type CFTR plasmid DNA (Fig. 4C). To determine whether the expressed CFTR protein by PEI-MPP carrying CFTR plasmid DNA are functional, we conducted a whole-cell patch clamp experiment. Despite being treated with PEI-MPP carrying CFTR plasmid DNA encoding a fusion protein of green fluorescent protein (GFP) and wild-type CFTR, no current was observed in GFP-positive CHO cells devoid of cAMP (Fig. 4D), which is as expected since the CFTR is cAMP-activated chloride channel [41]. However, significant chloride currents were evident in the presence of both cAMP and a CFTR activator, forskolin (Figs. 4D, 4E and 4G), and the currents were effectively inhibited by a subsequent treatment with the CFTR inhibitor-172 (Fig. 4F) [42]. These results suggest that the cells treated with PEI-MPP produced functional cAMP-activated CFTR protein, which is consistent with what has been observed for wild-type CFTR [41].
Figure 4.
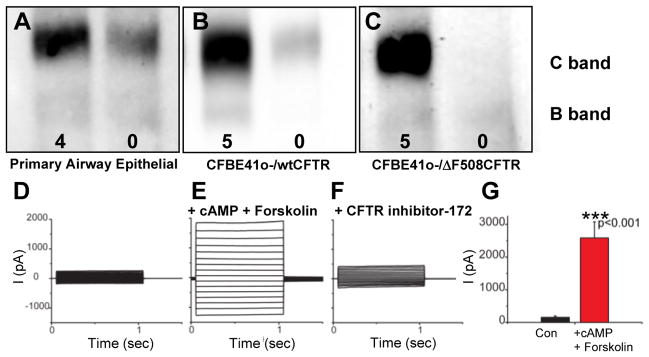
Expression of mature CFTR protein in cells transfected with PEI-MPP carrying CFTR plasmid DNA. (A – C) CFTR protein expression in cells transfected with PEI-MPP carrying pcDNA 3.1 WT-CFTR plasmid DNA (wild-type CFTR). Representative western blots of cultures of (A) primary human airway epithelial cells, (B) CFBE41o- cells stably expressing wild-type CFTR (wtCFTR), and (C) CFBE41o- cells stably expressing ΔF508 CFTR. In each panel, the left lane represents cells treated for 72 h with PEI-MPP carrying pcDNA3.1 WT-CFTR plasmid DNA, whereas the right lane represents untreated controls. Numbers in each panel represent PEI-MPP dose in terms of μg pcDNA3.1 WT-CFTR plasmid DNA. C and B bands exhibit mature, fully glycosylated and immature CFTR protein, respectively. (D – F) Function of expressed CFTR protein in CHO cells treated with PEI-MPP. Representative whole-cell currents from GFP-positive CHO cells transfected with NP carrying pEGFP WT-CFTR plasmid DNA (GFP-wtCFTR) for 72 h (D) before and (E) after bath applications of both 100 μM cAMP and 20 μM forskolin, and (F) after the subsequent addition of 10 μM CFTR inhibitor-172. (G) Current amplitude (I) at 125 mV was significantly increased (*** p < 0.001, n = 5) in PEI-MPP treated cells after the addition of both cAMP and forskolin. The error bars indicate standard error of the mean.
Despite their wide use as gene carrier platforms, several groups have reported on the toxicity of PEI [26, 39] and PLL [43]. We thus investigated whether the heavily PEG-coated PEI-MPP may cause inflammation in the mouse lung. PEI-UCP were found to cause an elevated immune cell infiltration compared to saline control (Figs. 5A and 5B). However, no increase in immune cell infiltration was observed in the lungs of mice administered PEI-MPP compared to saline control (Fig. 5C), suggesting PEI-MPP did not trigger pulmonary inflammation. Total cell counts and the concentration of TNF-α in bronchoalveolar lavage fluid (BALF) were also measured in mice dosed with PEI-UCP and PEI-MPP (Fig. 5D). PEI-UCP induced a significant increase in the total cell counts at 24 h post administration (p < 0.05), a hallmark of pulmonary inflammation, whereas the total cell counts for PEI-MPP was comparable to that for saline controls. Differences in the concentration of TNF-α in BALF were not statistically significant for all three conditions, suggesting neither PEI-UCP nor PEI-MPP induced TNF-α release 24 h after administration. We also evaluated cytotoxicity of gene carriers in both human bronchial (Fig. 5E) and alveolar (Fig. 5F) epithelial cell lines in vitro. Consistent with the in vivo toxicity study, PEI-MPP did not induce cytotoxicity at any dose tested, regardless of cell type, 48 h post-transfection, whereas PEI-UCP exhibited dose-dependent increase in cytotoxicity in both cell lines.
Figure 5.

In vivo lung inflammation and in vitro cytotoxicity induced by gene carriers. (A – D) Balb/c mice were intranasally dosed with gene carriers (PEI-UCP or PEI-MPP; 50 μg pd1GL3-RL plasmid DNA per mouse) and lung inflammation was assessed 24 h post-administration. Saline-treated mice served as controls. Hematoxylin and eosin (H & E) staining of paraffin sections of lung tissues from Balb/c mice intranasally dosed with (A) saline, (B) PEI-UCP and (C) PEI-MPP. Inflamed regions are marked by arrows. (D) Total cell counts and the concentration of TNF-α in BALF. The error bars indicate standard error of the mean. The differences are statistically significant between the conditions indicated with asterisks (*) (p < 0.05). (E, F) In vitro cytotoxicity of gene carriers to human lung cell lines. Human airway epithelial cell lines, (E) BEAS-2B and (F) A549, were transfected with varying concentrations of PEI-UCP or PEI-MPP carrying pd1GL3-RL plasmid DNA (luciferase; 0.2, 1 and 5 μg DNA per well of 96-well plates) and cell viability was measured by MTT assay at 48 h post-transfection. Asterisks (*) indicate statistically significant differences in the cell viability (* p < 0.05).
4. Discussion
Uniform expression of functional genes throughout the airway epithelium is thought to be essential to improved lung gene therapy for several diseases, including to correct phenotypical defects in the airways of CF patients [44]. However, this remains a formidable challenge due to numerous biological barriers, including the tenacious mucus layer that lines the airways and excludes conventional NP and gene carriers [8, 9, 13, 45]. By developing a formulation process that allows highly dense PEG coatings on small, stable gene nanocarriers, using widely used cationic polymers for proof-of-concept, we engineered gene delivery platforms that rapidly penetrate the most viscoelastic airway mucus gel obtained from CF patients by spontaneous expectoration. This is particularly significant given our recent finding that adeno-associated virus-5 and adenovirus do not penetrate CF mucus efficiently [13]. Highly compacted mucus- penetrating DNA NP protected cargo DNA against enzymatic degradation, did not induce significant acute lung inflammation or toxicity, and mediated a significant improvement in gene transfer to lungs of mice compared to conventional cationic gene carriers and to the only polymeric carrier tested in human CF gene therapy clinical trials (PEG10k-CK30/DNA NP) [6]. Highly compacted mucus-penetrating DNA NP carrying wild-type CFTR plasmid DNA mediated enhanced expression of wild-type CFTR in mouse lungs, human primary airway epithelial cells grown at air-liquid interface, and various cell lines in a dose-dependent manner. Enhanced in vivo airway gene transfer observed with these systems is most likely attributed to the improved mucus penetration, which led to more uniform distribution and prolonged retention in the lung by avoiding MCC.
CF mucus possesses high concentrations of negatively charged macromolecules, including mucins, DNA and actin filaments, which can interact with cationic polymers commonly used to condense plasmid DNA into NP. Based on our previous finding, which showed that as little as a 40% reduction of PEG coverage of latex NP led to a more than two orders of magnitude decrease in their average diffusion rate in human cervicovaginal mucus [22], we hypothesized that a dense surface PEG coverage is essential to effectively minimize mucoadhesion of DNA NP. However, we found that a high degree of PEG conjugation to cationic polymers led to less efficient DNA compaction and larger particle sizes, in good agreement with previous findings [26, 27]. This is likely due to fewer positive charges available for compacting DNA and/or steric interference from PEG, and poor DNA compaction may lead to inferior protection of cargo DNA [27]. Furthermore, loose compaction may result in an insufficient surface coverage of PEG, in accordance with the poor mucus penetration by conventionally PEG-coated PEI-CCP and PLL-CCP. A high degree of PEG conjugation may also reduce the buffering capacity of gene carriers [46] and, thus, gene transfer efficiency. Here, we circumvented the problems of this conventional single polymer approach by using a mixture of free and PEG-conjugated cationic polymers, which led to an effective surface shielding while retaining the efficient compaction capacity of cationic polymers. As shown with mucus penetrating PEI-MPP, this approach yielded significantly smaller carrier size and stronger compaction stability than PEI-CCP without increasing surface charge. This is most likely because cationic PEI is localized in the core of PEI-MPP (to compact the anionic DNA), while the more neutral PEG5k-PEI is primarily localized to the surface, rendering particles muco-inert. Likewise, a similar inclusion of free PLL resulted in a muco-inert coating of PLL-MPP, as evidenced by their markedly greater diffusion in CF mucus compared to conventionally coated PLL-CCP.
Cationic polymers, such as PEI and PLL, are known to evoke inflammatory and cytotoxic responses [43]. However, DNA NP formulated by condensing plasmid DNA with PEG-conjugated PLL polymers have exhibited minimal toxicity in the mouse lung [47], and have been well tolerated by CF patients [6]. Similarly, PEI-MPP did not trigger detectable acute pulmonary inflammation or cytotoxicity to airway epithelial cells in this study. Cell death induced by cytotoxic effects of therapeutic NP may cause release of cell debris, which can further thicken CF mucus. A recent report showed that PEG5k-PEI copolymers induced cytotoxicity in vitro to a level comparable to PEI polymers [46]. However, the PEG:PEI ratio tested in their study was only 4, which is ~9-fold lower than that in the present study (i.e. PEG:PEI = 37:1). Thus, the low toxicity of PEI-MPP may be attributed to the high PEG substitution achieved. Extensive removal of free PEI following NP formation by the ultrafiltration/concentration process may also have contributed to the reduced toxicity of PEI-MPP, since uncomplexed PEI has been implicated in toxicity observed with PEI-based gene carriers [48–50]. The approach taken here should also be compatible with lower MW and biodegradable polycations, which may further reduce chances of in vivo toxicity [51].
Sustained expression of functional CFTR genes is required to cure CF [52]. Recent innovations in plasmid design allowed long-term transgene expression; CFTR expression was extended to more than 6 months after a single administration to mouse lung using CpG-free ubiquitin C promoter instead of conventional CMV promoter [53]. Nevertheless, transgene expression fades along with the life span of airway epithelial cells, thus necessitating repeated dosing for life-long transgene expression. The use of most viral gene carriers remains problematic due to immunogenicity, which limits their repeated administration [5]. Recently, lentivirus was repeatedly administered to mouse lung without eliciting significant immune responses [54]; however, its potential oncogenic property should be further elucidated [55]. Here, we confirmed that non-viral PEI-MPP carrying CpG-free plasmid DNA can be repeatedly instilled to mouse lungs at dosing intervals of 2 to 4 weeks without diminishing their ability to mediate transgene expression. This is in contrast to a recent finding that airway transgene expression mediated by repeated administrations of PEI-based gene carriers at varying intervals was significantly reduced compared to a single-dose treatment [56]. PEI-MPP carrying optimized plasmids hold promise to maintain long-term expression of functional proteins in patients upon repeated dosing.
We described synthetic gene carrier platforms capable of penetrating human CF mucus. Highly compacted mucus-penetrating gene carriers possess a highly dense PEG coating that allows them to penetrate mucus barriers, achieve uniform airway distribution and prolonged lung retention, and enhance gene transfer to mouse lung while eliciting negligible acute toxic and inflammatory responses.
Supplementary Material
Acknowledgments
Funding was provided by the National Institutes of Health (R01 EB003558, P01 HL51811 and F32 HL103137) and the Cystic Fibrosis foundation (HANES07XX0). We are grateful to Meghan Ramsay and Sharon Watts at the Johns Hopkins Adult Cystic Fibrosis Center for CF mucus collection. We also thank Tao Yu and Benjamin Schuster for their help with NMR measurements and MPT analysis, respectively. The mucus penetrating particle technology described in this publication is being developed by Kala Pharmaceuticals. Dr. Hanes is a co-founder of Kala Pharmaceuticals, Inc., and serves as a consultant. Dr. Hanes owns company stock, which is subject to certain restrictions under University policy.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Papiris S, Kotanidou A, Malagari K, Roussos C. Clinical review: severe asthma. Crit Care. 2002;6:30–44. doi: 10.1186/cc1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flume PA. Pulmonary complications of cystic fibrosis. Respir Care. 2009;54:618–627. doi: 10.4187/aarc0443. [DOI] [PubMed] [Google Scholar]
- 3.Griesenbach U, Alton EW. Gene transfer to the lung: lessons learned from more than 2 decades of CF gene therapy. Adv Drug Deliv Rev. 2009;61:128–139. doi: 10.1016/j.addr.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Podolska K, Stachurska A, Hajdukiewicz K, Malecki M. Gene therapy prospects--intranasal delivery of therapeutic genes. Adv Clin Exp Med. 2012;21:525–534. [PubMed] [Google Scholar]
- 5.Sinn PL, Burnight ER, McCray PB., Jr Progress and prospects: prospects of repeated pulmonary administration of viral vectors. Gene Ther. 2009;16:1059–1065. doi: 10.1038/gt.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konstan MW, Davis PB, Wagener JS, Hilliard KA, Stern RC, Milgram LJ, Kowalczyk TH, Hyatt SL, Fink TL, Gedeon CR, Oette SM, Payne JM, Muhammad O, Ziady AG, Moen RC, Cooper MJ. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther. 2004;15:1255–1269. doi: 10.1089/hum.2004.15.1255. [DOI] [PubMed] [Google Scholar]
- 7.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders NN, De Smedt SC, Van Rompaey E, Simoens P, De Baets F, Demeester J. Cystic fibrosis sputum: a barrier to the transport of nanospheres. Am J Respir Crit Care Med. 2000;162:1905–1911. doi: 10.1164/ajrccm.162.5.9909009. [DOI] [PubMed] [Google Scholar]
- 10.Sheils CA, Kas J, Travassos W, Allen PG, Janmey PA, Wohl ME, Stossel TP. Actin filaments mediate DNA fiber formation in chronic inflammatory airway disease. Am J Pathol. 1996;148:919–927. [PMC free article] [PubMed] [Google Scholar]
- 11.Suk JS, Lai SK, Wang YY, Ensign LM, Zeitlin PL, Boyle MP, Hanes J. The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials. 2009;30:2591–2597. doi: 10.1016/j.biomaterials.2008.12.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai SK, Wang YY, Hida K, Cone R, Hanes J. Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses. Proc Natl Acad Sci U S A. 2010;107:598–603. doi: 10.1073/pnas.0911748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hida K, Lai SK, Suk JS, Won SY, Boyle MP, Hanes J. Common gene therapy viral vectors do not efficiently penetrate sputum from cystic fibrosis patients. PLoS One. 2011;6:e19919. doi: 10.1371/journal.pone.0019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boylan NJ, Suk JS, Lai SK, Jelinek R, Boyle MP, Cooper MJ, Hanes J. Highly compacted DNA nanoparticles with low MW PEG coatings: In vitro, ex vivo and in vivo evaluation. J Control Release. 2011;157:72–79. doi: 10.1016/j.jconrel.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suk JS, Boylan NJ, Trehan K, Tang BC, Schneider CS, Lin JM, Boyle MP, Zeitlin PL, Lai SK, Cooper MJ, Hanes J. N-acetylcysteine enhances cystic fibrosis sputum penetration and airway gene transfer by highly compacted DNA nanoparticles. Mol Ther. 2011;19:1981–1989. doi: 10.1038/mt.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun X, Zhang N. Cationic polymer optimization for efficient gene delivery. Mini Rev Med Chem. 2010;10:108–125. doi: 10.2174/138955710791185109. [DOI] [PubMed] [Google Scholar]
- 17.Dunlap DD, Maggi A, Soria MR, Monaco L. Nanoscopic structure of DNA condensed for gene delivery. Nucleic Acids Res. 1997;25:3095–3101. doi: 10.1093/nar/25.15.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kukowska-Latallo JF, Raczka E, Quintana A, Chen C, Rymaszewski M, Baker JR., Jr Intravascular and endobronchial DNA delivery to murine lung tissue using a novel, nonviral vector. Hum Gene Ther. 2000;11:1385–1395. doi: 10.1089/10430340050057468. [DOI] [PubMed] [Google Scholar]
- 19.Ferrari S, Pettenazzo A, Garbati N, Zacchello F, Behr JP, Scarpa M. Polyethylenimine shows properties of interest for cystic fibrosis gene therapy. Biochim Biophys Acta. 1999;1447:219–225. doi: 10.1016/s0167-4781(99)00153-0. [DOI] [PubMed] [Google Scholar]
- 20.Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 21.Ferrari S, Kitson C, Farley R, Steel R, Marriott C, Parkins DA, Scarpa M, Wainwright B, Evans MJ, Colledge WH, Geddes DM, Alton EW. Mucus altering agents as adjuncts for nonviral gene transfer to airway epithelium. Gene Ther. 2001;8:1380–1386. doi: 10.1038/sj.gt.3301525. [DOI] [PubMed] [Google Scholar]
- 22.Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed Engl. 2008;47:9726–9729. doi: 10.1002/anie.200803526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A. 2007;104:1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai SK, Suk JS, Pace A, Wang YY, Yang M, Mert O, Chen J, Kim J, Hanes J. Drug carrier nanoparticles that penetrate human chronic rhinosinusitis mucus. Biomaterials. 2011;32:6285–6290. doi: 10.1016/j.biomaterials.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beyerle A, Braun A, Banerjee A, Ercal N, Eickelberg O, Kissel TH, Stoeger T. Inflammatory responses to pulmonary application of PEI-based siRNA nanocarriers in mice. Biomaterials. 2011;32:8694–8701. doi: 10.1016/j.biomaterials.2011.07.082. [DOI] [PubMed] [Google Scholar]
- 26.Petersen H, Fechner PM, Martin AL, Kunath K, Stolnik S, Roberts CJ, Fischer D, Davies MC, Kissel T. Polyethylenimine-graft-poly(ethylene glycol) copolymers: influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjug Chem. 2002;13:845–854. doi: 10.1021/bc025529v. [DOI] [PubMed] [Google Scholar]
- 27.Kichler A, Chillon M, Leborgne C, Danos O, Frisch B. Intranasal gene delivery with a polyethylenimine-PEG conjugate. J Control Release. 2002;81:379–388. doi: 10.1016/s0168-3659(02)00080-9. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y, Xie J, Tong YW, Wang CH. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J Control Release. 2007;118:7–17. doi: 10.1016/j.jconrel.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Pamujula S, Hazari S, Bolden G, Graves RA, Chinta DD, Dash S, Kishore V, Mandal TK. Cellular delivery of PEGylated PLGA nanoparticles. J Pharm Pharmacol. 2012;64:61–67. doi: 10.1111/j.2042-7158.2011.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amblard M, Fehrentz JA, Martinez J, Subra G. Methods and protocols of modern solid phase peptide synthesis. Mol Biotechnol. 2006;33:239–254. doi: 10.1385/MB:33:3:239. [DOI] [PubMed] [Google Scholar]
- 31.Ziady AG, Gedeon CR, Miller T, Quan W, Payne JM, Hyatt SL, Fink TL, Muhammad O, Oette S, Kowalczyk T, Pasumarthy MK, Moen RC, Cooper MJ, Davis PB. Transfection of airway epithelium by stable PEGylated poly-L-lysine DNA nanoparticles in vivo. Mol Ther. 2003;8:936–947. doi: 10.1016/j.ymthe.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Suh J, Dawson M, Hanes J. Real-time multiple-particle tracking: applications to drug and gene delivery. Adv Drug Deliv Rev. 2005;57:63–78. doi: 10.1016/j.addr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Savin T, Doyle PS. Static and dynamic errors in particle tracking microrheology. Biophys J. 2005;88:623–638. doi: 10.1529/biophysj.104.042457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruthardt N, Lamb DC, Brauchle C. Single-particle tracking as a quantitative microscopy-based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol Ther. 2011;19:1199–1211. doi: 10.1038/mt.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michalet X, Berglund AJ. Optimal diffusion coefficient estimation in single-particle tracking. Phys Rev E Stat Nonlin Soft Matter Phys. 2012;85:061916. doi: 10.1103/PhysRevE.85.061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crocker JC, Hoffman BD. Multiple-particle tracking and two-point microrheology in cells. Method Cell Biol. 2007;83:141–178. doi: 10.1016/S0091-679X(07)83007-X. [DOI] [PubMed] [Google Scholar]
- 37.Liang LH, Woodward OM, Chen ZH, Cotter R, Guggino WB. A novel role of protein tyrosine kinase2 in mediating chloride secretion in human airway epithelial cells. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malek A, Czubayko F, Aigner A. PEG grafting of polyethylenimine (PEI) exerts different effects on DNA transfection and siRNA-induced gene targeting efficacy. J Drug Target. 2008;16:124–139. doi: 10.1080/10611860701849058. [DOI] [PubMed] [Google Scholar]
- 39.Merkel OM, Urbanics R, Bedocs P, Rozsnyay Z, Rosivall L, Toth M, Kissel T, Szebeni J. In vitro and in vivo complement activation and related anaphylactic effects associated with polyethylenimine and polyethylenimine-graft-poly(ethylene glycol) block copolymers. Biomaterials. 2011;32:4936–4942. doi: 10.1016/j.biomaterials.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 40.Cebotaru L, Vij N, Ciobanu I, Wright J, Flotte T, Guggino WB. Cystic fibrosis transmembrane regulator missing the first four transmembrane segments increases wild type and DeltaF508 processing. J Biol Chem. 2008;283:21926–21933. doi: 10.1074/jbc.M709156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fuller CM, Benos DJ. CFTR! Am J Physiol. 1992;263:C267–286. doi: 10.1152/ajpcell.1992.263.2.C267. [DOI] [PubMed] [Google Scholar]
- 42.Verkman AS, Lukacs GL, Galietta LJ. CFTR chloride channel drug discovery--inhibitors as antidiarrheals and activators for therapy of cystic fibrosis. Curr Pharm Des. 2006;12:2235–2247. doi: 10.2174/138161206777585148. [DOI] [PubMed] [Google Scholar]
- 43.Hunter AC, Moghimi SM. Cationic carriers of genetic material and cell death: a mitochondrial tale. Biochim Biophys Acta. 2010;1797:1203–1209. doi: 10.1016/j.bbabio.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 44.Pringle IA, Hyde SC, Gill DR. Non-viral vectors in cystic fibrosis gene therapy: recent developments and future prospects. Expert Opin Biol Ther. 2009;9:991–1003. doi: 10.1517/14712590903055029. [DOI] [PubMed] [Google Scholar]
- 45.Sanders N, Rudolph C, Braeckmans K, De Smedt SC, Demeester J. Extracellular barriers in respiratory gene therapy. Adv Drug Deliv Rev. 2009;61:115–127. doi: 10.1016/j.addr.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beyerle A, Merkel O, Stoeger T, Kissel T. PEGylation affects cytotoxicity and cell-compatibility of poly(ethylene imine) for lung application: structure-function relationships. Toxicol Appl Pharmacol. 2010;242:146–154. doi: 10.1016/j.taap.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Ziady AG, Gedeon CR, Muhammad O, Stillwell V, Oette SM, Fink TL, Quan W, Kowalczyk TH, Hyatt SL, Payne J, Peischl A, Seng JE, Moen RC, Cooper MJ, Davis PB. Minimal toxicity of stabilized compacted DNA nanoparticles in the murine lung. Mol Ther. 2003;8:948–956. doi: 10.1016/j.ymthe.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Erbacher P, Bettinger T, Brion E, Coll JL, Plank C, Behr JP, Remy JS. Genuine DNA/polyethylenimine (PEI) complexes improve transfection properties and cell survival. J Drug Target. 2004;12:223–236. doi: 10.1080/10611860410001723487. [DOI] [PubMed] [Google Scholar]
- 49.Davies LA, McLachlan G, Sumner-Jones SG, Ferguson D, Baker A, Tennant P, Gordon C, Vrettou C, Baker E, Zhu J, Alton EW, Collie DD, Porteous DJ, Hyde SC, Gill DR. Enhanced lung gene expression after aerosol delivery of concentrated pDNA/PEI complexes. Mol Ther. 2008;16:1283–1290. doi: 10.1038/mt.2008.96. [DOI] [PubMed] [Google Scholar]
- 50.Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J Gene Med. 2004;6:1102–1111. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- 51.Jere D, Jiang HL, Arote R, Kim YK, Choi YJ, Cho MH, Akaike T, Cho CS. Degradable polyethylenimines as DNA and small interfering RNA carriers. Expert Opin Drug Deliv. 2009;6:827–834. doi: 10.1517/17425240903029183. [DOI] [PubMed] [Google Scholar]
- 52.Davies JC, Alton EW. Gene therapy for cystic fibrosis. Proc Am Thorac Soc. 2010;7:408–414. doi: 10.1513/pats.201004-029AW. [DOI] [PubMed] [Google Scholar]
- 53.Hyde SC, Pringle IA, Abdullah S, Lawton AE, Davies LA, Varathalingam A, Nunez-Alonso G, Green AM, Bazzani RP, Sumner-Jones SG, Chan M, Li H, Yew NS, Cheng SH, Boyd AC, Davies JC, Griesenbach U, Porteous DJ, Sheppard DN, Munkonge FM, Alton EW, Gill DR. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat Biotechnol. 2008;26:549–551. doi: 10.1038/nbt1399. [DOI] [PubMed] [Google Scholar]
- 54.Sinn PL, Anthony RM, McCray PB., Jr Genetic therapies for cystic fibrosis lung disease. Hum Mol Genet. 2011;20:R79–86. doi: 10.1093/hmg/ddr104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griesenbach U, Alton EW. Current status and future directions of gene and cell therapy for cystic fibrosis. BioDrugs. 2011;25:77–88. doi: 10.2165/11586960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 56.Davies LA, Hyde SC, Nunez-Alonso G, Bazzani RP, Harding-Smith R, Pringle IA, Lawton AE, Abdullah S, Roberts TC, McCormick D, Sumner-Jones SG, Gill DR. The use of CpG-free plasmids to mediate persistent gene expression following repeated aerosol delivery of pDNA/PEI complexes. Biomaterials. 2012;33:5618–5627. doi: 10.1016/j.biomaterials.2012.04.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.