

Abstract
Naïve CD4 T cells transferred into lymphopenic mice undergo spontaneous proliferation and induce chronic inflammation in the intestine. Cellular mechanisms regulating the proliferative and inflammatory processes are not fully understood. In this study, we report that IFNγ signaling in host cells plays a major role in limiting both T cell expansion and T cell-induced intestinal inflammation. However, the role for IFNγ appears to be distinct depending on the target cells. IFNγ signaling in DCs controls T cell expansion, while IFNγ signaling in neutrophils seems to regulate both T cell expansion and inflammation. IFNγ signaling in non-hematopoietic cells may control inflammation. Therefore, our results suggest novel immunoregulatory functions for IFNγ to orchestrate colitogenic T cell responses through its distinct action on different non-T cell target cells.
Introduction
Homeostatic dysregulation is a potential cause of chronic inflammation such as autoimmunity and inflammatory bowel disease (IBD) (1). To explore mechanisms that link these two events investigators have employed a T cell-induced colitis model induced by naïve CD4 T cells transferred into immunodeficient hosts (2, 3). Transferred T cells undergo spontaneous proliferation (SP) and differentiate into effector cells in response to both self- and commensal-Ag (4). Gut Ag reactive T cells producing proinflammatory cytokines including IFNγ, IL-17, and GM-CSF are generated during this process and mediate the inflammation in the intestine. Understanding the pathways through which colitogenic effector cells are generated and their generation is regulated is thus of great importance.
Ag presenting cells (APC), especially dendritic cells (DCs), play an indispensable role in inducing CD4 T cell SP (5). APC-derived cytokines, including IL-1β, IL-12, IL-23, and IL-27, regulate T cell differentiation into different pathogenic effector cells (6, 7). One pathway that we have overlooked over the years is the role of cytokine-mediated APC activation in T cell immunity. Iwasaki and colleagues recently reported that DC activation by IL-1 is both required and sufficient to generate virus specific CD8 T cells (8). Quintana and colleagues reported that IL-27 signaling in DCs limits the generation of encephalitogenic Th1/Th17 subsets and the development of EAE (9). By contrast, we recently reported an opposing result that IL-27 signaling in APCs selectively promotes the generation of colitogenic Th17 effector cells and the development of T cell-induced colitis (10). These results strongly suggest that a cytokine signaling in APCs plays an important regulatory role in T cell immunity and T cell-mediated inflammation.
IFNγ is a pleiotropic cytokine that regulates many different cellular functions (11). Its signature roles in macrophage activation, host defense against intracellular pathogens, and Th1 type cell-associated inflammation including IBD have been extensively investigated (12). It also plays an anti-inflammatory role in limiting inflammation by attenuating tissue damages (13). Because of its pleiotropic features, our understanding the precise mechanism by which IFNγ mediates various immune functions during inflammatory responses is still incomplete.
Here, we report that IFNγ signaling in APCs plays an important role in limiting T cell SP and the subsequent development of intestinal inflammation. Wild type (WT) naïve CD4 T cells transferred into IFNγR−/− Rag−/− mice undergo uncontrolled expansion and induce acute severe intestinal inflammation even at 7 days post transfer, while IFNγR+/+ Rag−/− recipients show no signs of inflammation at the same time of analysis. Bone marrow chimeras and adoptive DC transfer experiments identified that while IFNγ signaling in DCs and in infiltrating Gr1+ neutrophils is essential to limit T cell expansion, IFNγ signaling in non-hematopoietic cells may control intestinal inflammation. Therefore, IFNγ mediates various anti-inflammatory functions by targeting different cell types during T cell-induced chronic inflammation in the intestine.
Materials and Methods
Mice
C57BL/6, B6 Ly5.1, B6 Thy1.1, B6 IFNγR−/−, B6 Rag1−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). IFNγR−/− Rag−/− mice were bred at the animal facility of the Lerner Research Institute. All animal procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee.
Cell sorting and adoptive transfer
Lymph node naive CD4 T cells were obtained as previously reported (5). 1×106 naive T cells were transferred alone or in combination with sorted CD11b+ Gr1+ cells or with 1×106 MACS purified CD11c+ splenic DCs into Rag−/− mice. Mice were sacrificed to measure donor cell expansion at day 7. To measure cytokine expression cells harvested were stimulated with PMA (10ng/ml) and Ionomycin (1µM) for 4 hrs in the presence of 2 µM Monensin (Calbiochem, San Diego, CA) during the last 2 hrs. Cells were immediately fixed with 4% paraformaldehyde, permeabilized, and stained with fluorescence conjugated antibodies.
Bone marrow reconstitution
Bone marrow chimeras were generated as previously reported (5).
Lamina Propria cell Isolation
Recipients were sacrificed at the indicated time points after T-cell transfer. Colons were isolated and cleaned in HBSS. Colons were cut into small pieces and resuspended in HBSS with 0.5µM EDTA and 15µg/mL Dithiothreitol (DTT) and shaken for 15 min twice at room temperature. Colons were then resuspended in cRPMI with 400µg/mL DNase and 1mg/mL collagenase and shaken at 37°C for 90 min. Supernatant and colon was passed through a 70µM strainer and washed. Cells were resuspended in a 33% Percoll gradient and spun at room temperature for 20 min. Pellet was collected, washed and used for further experiments.
Real time PCR
Colon tissue was disrupted using a TissueLyser II (Qiagen, Valencia, CA). Total RNA was extracted using an RNeasy column (Qiagen, Valencia, CA). cDNA was subsequently obtained using a SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). Real time PCR was performed using gene specific primers and probe sets (Applied Biosystem, Foster City, CA) and ABI 7500 PCR machine (Applied Biosystem).
FACS Analysis
Cells were stained with anti-BrdU (PRB-1), anti-CD4 (RM4-5), anti-CD11c, anti-CD11b (M1/70), anti-CD25 (PC61), anti-Ly6G (Gr1), anti-CD44 (IM7), anti-CD45.1 (A20), anti-Thy1.1 (HIS51), anti-Thy1.2 (30-H12), anti-IL-17A (eBio17B7), anti-IFN-γ (XMG1.2), anti-MHCII (M5/114.15.2) (all Abs from eBioscience) and anti-GM-CSF, (140706, BD PharMingen, San Diego, CA). Cells were acquired using a FACS LSR II (BD Biosciences) and analyzed using a FlowJo software (Treestar, Ashland, OR).
Immunohistochemistry
For H&E staining colon tissue was fixed in 10% Acetic Acid/60% Methanol. Slides were stained with hematoxylin and eosin. For immunohistochemistry paraffin embedded colon sections were incubated overnight at 4°C in primary anti-CD3 (ab5690, abcam, Cambridge, MA) followed by secondary biotinylated goat anti-rabbit IgG (Jackson Immuno Research, West Grove, PA). Vectastain Elite ABC Kit and DAB (Vector Laboratories, Burlingame, CA) was used for color development, and processed for mounting using Clarifier I, Bluing Solution, Clear-Rite 3 and Mounting Media (all Richard-Allan, Kalamazoo, MI).
Data analysis
Statistical significance was determined by the Student’s t-test using the Prism 5 software (GraphPad, La Jolla, CA). A p value of <0.05 was considered statically significant.
Results and Discussion
Lack of host IFNγR signaling during CD4 T cell SP accelerates T cell-mediated intestinal inflammation
Host IFNγ signaling controls the expansion, contraction, and differentiation of CD8 T cells following immunization and during lymphopenia-induced proliferation (14, 15). To determine whether it also regulates naïve CD4 T cell SP and the subsequent development of intestinal inflammation, we transferred WT naïve polyclonal CD4 T cells into Rag−/− recipients that express or lack IFNγR. Unlike Rag−/− mice that develop chronic intestinal inflammation and weight loss starting ~6 weeks post transfer (16), IFNγR−/− Rag−/− recipients developed acute weight loss (Fig 1A) and severe colonic inflammation 7 days post transfer, at which Rag−/− recipients exhibited no signs of inflammation (Fig 1B). Of note, IFNγR−/− Rag−/− recipients do not show any signs of intestinal inflammation prior to transfer (not shown). We observed a dramatic increase of T cell recovery in all tested lymphoid and intestinal tissues of IFNγR−/− Rag−/− mice (Fig 1C). Enhanced T cell expansion was noticeable in circulating donor CD4 T cells. Starting at 5 days after transfer, the proportion of circulating CD4 T cells dramatically increased between 6 and 7 days of transfer in IFNγR−/− conditions, while T cells only gradually increased in IFNγR+/+ conditions (Fig 1D). Effector T cells producing IFNγ, IL-17A and GM-CSF are the chief inflammatory cells mediating the diseases (3, 17). Consistent with severe inflammation, the absolute numbers of cells producing IFNγ, IL-17A, or GM-CSF were significantly greater in both mesenteric LN (mLN) and the lamina propria (LP) of IFNγR−/− Rag−/− recipients, although the frequency of these cells was similar between the groups (Fig 1E and not shown). T cells producing IL-10 remained low in both groups (not shown). Colon expression of IL-1β and IL-23p19 was significantly higher in IFNγR−/− recipients, while IL-10 and IL-6 expression was comparable (not shown). Transfer of IFNγR−/− CD4 T cells into Rag−/− mice did not result in elevated T cell immunity (Fig 1F), indicating that IFNγ signaling in T cells play little role in this process. Therefore, IFNγ signaling in host cells directly limits SP and T cell-induced inflammation in the intestine.
Figure 1. Rag−/− mice deficient in IFNγR develop severe colon inflammation following naïve CD4 T cell transfer.

FACS purified naive Thy1.1+ CD4 T cells were transferred into Rag−/− or IFNγR−/− Rag−/− recipients. (A) Weight loss after T cell transfer. (B) The colon tissues were harvested 7 days post transfer and H&E stained. 20x magnification. (C) Absolute numbers of total donor CD4 T cells in the indicated tissues were enumerated 7 days post transfer by FACS analysis. (D) The recipients were daily bled, and circulating donor T cells were examined. (E) Cells from the indicated tissues were harvested 7 days post transfer, ex vivo stimulated with PMA/Ionomycin, and stained for intracellular cytokine expression. Absolute numbers of IFNγ, IL-17A and GM-CSF producing CD4 T cells were determined by FACS analysis. (F) CD4 T cells isolated from Thy1.1 WT or IFNγR−/− mice were transferred into Rag−/− recipients. The absolute numbers of the donor T cells was examined 7 days post transfer. All experiments were repeated 2-3 times and similar results were observed. Each symbol represents individually tested animals. *, p<0.05; ***, p<0.001.
IFNγ signaling in hematopoietic cells controls T cell expansion
To explore mechanisms underlying IFNγ-mediated regulation of CD4 T cell immunity, we examined the target cells of IFNγ responsible for the enhanced T cell expansion. Bone marrow (BM) chimeras were generated (Rag−/− BM → IFNγR−/− Rag−/−, W → K and IFNγR−/− Rag−/− BM → Rag−/−, K → W), and naïve CD4 T cells were transferred into the mice. Enhanced expansion of CD4 T cells was only found when BM derived cells lack IFNγR (K → W) (Fig 2), while the level of T cell expansion of the W → K group was similar to that of Rag−/− mice (dotted line). Interestingly, elevated T cell expansion was only observed in the lymphoid tissues of K → W groups (Fig 2), while the accumulation of proliferating cells in the LP was similar to that of Rag−/− mice (dotted line). Consistent with this finding, severe inflammation seen in IFNγR−/− Rag−/− recipients did not develop in K → W groups (Supp Fig S1). Therefore, IFNγ signaling in hematopoietic cells in the lymphoid tissues may control T cell expansion, IFNγ signaling in non-hematopoietic cells especially on the target LP tissues may regulate T cell expansion and the subsequent development of inflammation in the intestine (18).
Figure 2. IFNγ signaling in hematopoietic cells is essential to control CD4 T cell expansion.
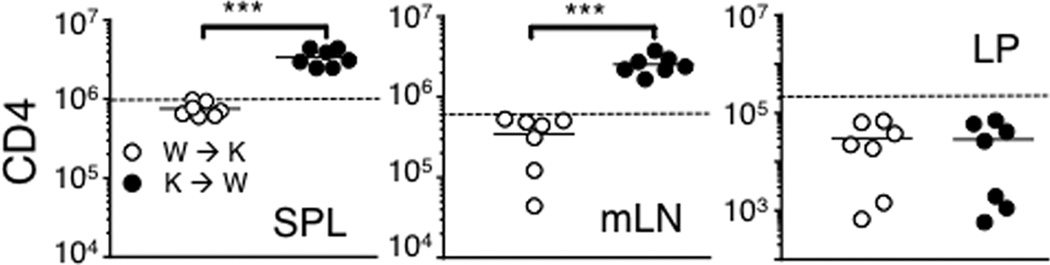
Lethally irradiated IFNγR−/− Rag−/− and Rag−/− mice were reconstituted with BM cells from Rag−/− and IFNγR−/− Rag−/− donor mice, respectively (Rag−/− BM → IFNγR−/− Rag−/−, W → K and IFNγR−/− Rag−/− BM → Rag−/−, K → W). FACS purified naive Thy1.1+ CD4 T cells were transferred into the reconstituted recipients after 6 weeks of BM reconstitution. The T cell recipients were sacrificed 7 days post transfer, and cells from the indicated tissues were stained for Thy1.1 and CD4. Absolute numbers of donor cells were calculated. T cell recovery of unmanipulated Rag−/− recipients is shown as dotted line. All experiments were repeated twice and similar results were observed. Each symbol represents individually tested animals. ***, p<0.001.
IFNγ signaling in CD11c+ DCs is sufficient to control T cell expansion
CD11c+ DCs play a central role in inducing CD4 T cell SP (5). Interestingly, the numbers of CD11c+ DCs in the mLN were substantially greater in IFNγR−/− Rag−/− recipients (Fig 3A). DC apoptosis is a key process that balances immunity and tolerance and the defects in DC apoptosis can lead to the development of autoimmunity (19). We thus examined if IFNγ signaling in DC plays a role in DC survival and T cell immunity. MHCII−/− Rag−/− mice were used as recipients. Without MHCII molecules, T cells transferred remain undivided (5). Splenic DCs isolated from WT or IFNγR−/− mice were cotransferred into these recipients so that T cell expansion can be induced by the transferred MHCII+ DCs. WT DCs transferred significantly enhanced T cell expansion (Fig 3B). Importantly, IFNγR−/− DCs further enhanced the expansion (Fig 3B). Therefore, the lack of IFNγ signaling specifically in DCs is sufficient to enhance T cell expansion. The total numbers of transferred IFNγR−/− DCs were found significantly greater than that of WT DCs (Fig 3C), suggesting that IFNγ may regulate DC homeostasis by inducing apoptosis. Without IFNγR, DC apoptosis measured by Annexin-V staining was significantly reduced (Fig 3D). Moreover, expression of anti-apoptotic molecule, bcl2 was enhanced (Fig 3D). IL-21 and GM-CSF exhibit opposing roles in conventional DC apoptosis (20). Interestingly, we found that IL-21 expression was significantly lower, while GM-CSF expression was substantially higher in IFNγR−/− Rag−/− recipients (Supp Fig S2). Therefore, IFNγ appears to regulate DC apoptosis both by directly acting on DCs and indirectly by inducing factors controlling it.
Figure 3. Prolonged IFNγR−/− DC survival promotes CD4 T cell expansion.
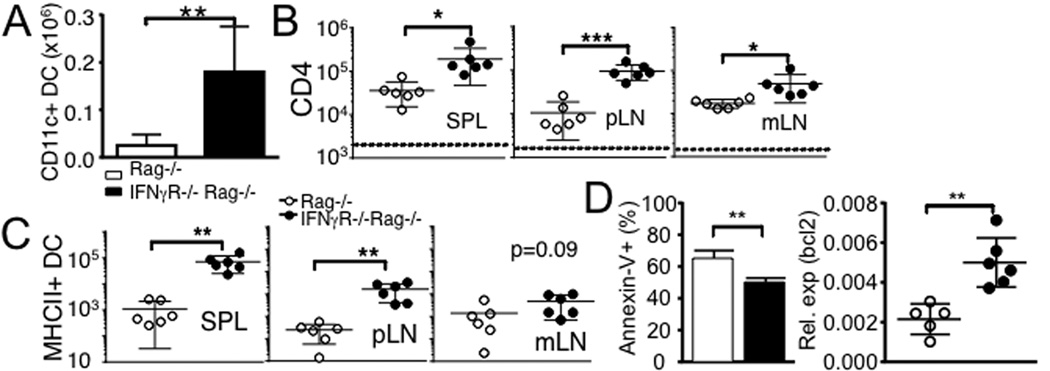
(A) The absolute numbers of CD11c+ DCs in the colon was calculated 7 days post transfer. Data are the mean ± SD of 5-6 individually tested mice from two separate experiments. (B) MHCII−/− Rag−/− mice received FACS purified Ly5.1+ naive CD4 T cells together with splenic CD11c+ DCs isolated from WT (○) or IFNγR−/− (●) mice. Donor CD4 T cell recovery was determined by FACS 7 days post transfer. (C) Absolute numbers of adoptively transferred WT and IFNγR−/− MHCII+ CD11c+ DCs were counted at day 7. All the experiments were repeated twice and similar results were observed. (D) Annexin-V staining and bcl2 expression of purified mLN DCs were examined. Data are the mean ± SD of 5-6 individually tested mice. *, p<0.05; **, p<0.01; ***, p<0.001.
Gr1+ neutrophils in response to IFNγ signaling regulate T cell expansion and intestinal inflammation
IFNγ plays an important role in attenuating tissue damage associated with chronic inflammation (13), and one pathway to achieve this is through inhibition of tissue infiltration by neutrophils. IFNγ-deficient mice exhibit dysregulated expansion of CD11b+ Gr1+ neutrophils (21, 22). Moreover, CD11b+ cells in response to IFNγ limit CD8 T cell expansion (14). Consistent with these findings, elevated accumulation of CD11b+ Gr1+ neutrophils in IFNγR−/− Rag−/− recipients was noticed in the intestinal lamina propria (Fig 4A). Interestingly, depleting neutrophils with anti-Gr1 mAb prevented severe pathology in the colon (Fig 4B), suggesting that neutrophils may directly be involved in tissue damage and inflammation, or alternatively, they may do so by controlling T cell responses. In fact, depleting neutrophils significantly reduced T cell accumulation in the lamina propria (Fig 4C).
Figure 4. Gr1+ cells in response to IFNγ regulate CD4 T cells expansion and intestinal inflammation in IFNγR−/− Rag−/− mice by iNOS-dependent mechanism.

(A) Immunohistochemistry analysis of the Gr1+ cells in the colon. Absolute numbers of Gr1+ CD11b+ cells in the colon were determined 7 days post naïve CD4 T cell transfer. Data shown represent the mean ± SD of 6 individually tested mice from two independent experiments. (B and C) IFNγR−/− Rag−/− recipients of naïve CD4 T cells were treated with RB6-8C5 anti-Gr1 mAb (150 µg) at days -1, 2, and 5 of T cell transfer. H&E staining of the colon tissues and the absolute numbers of CD4 T cells in the colon were determined at 7 days post transfer. (D) Thy1.1 naive CD4 T cells were transferred into Rag−/− recipients together with Gr1+ cells harvested from WT or IFNγR−/− mice. Donor T cells recovery was measured at day 7. (E) iNOS expression in the colon was examined by real time PCR at 7 days post CD4 transfer. (F) Naive CD4 T cells were transferred into Rag−/− recipients together with Gr1+ cells harvested from WT or iNOS−/− mice. Donor T cells recovery was measured at day 7. Data are the mean ± SD of 4-9 individually tested animals from 2-3 independent experiments. *, p<0.05; **, p<0.01; ***, p<0.001.
To directly examine if IFNγ signaling in neutrophils modulate T cell responses Gr1+ cells isolated from WT or IFNγR−/− mice were transferred into Rag−/− mice with CD4 T cells, and T cell expansion was determined. As shown in Fig 4D, IFNγR−/− neutrophils significantly enhanced T cell expansion compared to that of WT neutrophils. Neutrophils activated by IFNγ express inducible NO synthase (iNOS) and release NO, which can inhibit T cell proliferation (23). Indeed, iNOS expression in the LP was almost absent in IFNγR−/− Rag−/− recipients (Fig 4E). Furthermore, transfer of iNOS−/− neutrophils also resulted in enhanced T cell expansion (Fig 4F). Therefore, neutrophils stimulated by IFNγ may control T cell expansion via iNOS-dependent mechanism.
Our results reveal cell type-specific immunoregulatory pathways through which IFNγ controls CD4 T cell immunity. By controlling DC survival, IFNγ enhances T cell expansion. However, whether DC apoptosis seen in the current study is a direct effect of IFNγ (24), or IFNγ regulates other factors promoting or preventing DC apoptosis such as IL-21, GM-CSF, or type I IFN remains to be examined (25). Apart from the role for IFNγ on DCs, we also found that IFNγ acts on neutrophils to control T cell responses. IFNγ decreases KC expression and/or selectin expression, both of which are critical for neutrophil trafficking (26, 27). The lack of IFNγ signaling in recruited neutrophils may impair iNOS expression, resulting in uncontrolled T cell expansion. However, we cannot exclude the possibility that neutrophils may release factors directly damaging the tissues as depleting neutrophils prevents acute inflammation that seen in IFNγR−/− Rag−/− recipients. Alternatively, IFNγ signaling in non-hematopoietic cells in the intestine may control intestinal inflammation possibly through β-catenin signaling pathways (28). Future investigation should focus on the role of IFNγ signaling in non-T target cells in other inflammatory conditions.
Supplementary Material
Acknowledgements
We thank Sohee Kim and Jennifer Powers for technical assistance and cell sorting, respectively. We also thank Drs. Serpil Erzurum and Robert Fairchild for providing iNOS−/− mice and anti-Gr1 mAb, respectively, and Nina Vologh for immunohistochemistry.
Supported by NIH grant AI074932.
References
- 1.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 2.Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, Grisham MB. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. American journal of physiology. Gastrointestinal and liver physiology. 2009;296:G135–G146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Do JS, Visperas A, Dong C, Baldwin WM, 3rd, Min B. Cutting edge: Generation of colitogenic Th17 CD4 T cells is enhanced by IL-17+ gammadelta T cells. J Immunol. 2011;186:4546–4550. doi: 10.4049/jimmunol.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Do JS, Foucras G, Kamada N, Schenk AF, Shaw M, Nunez G, Paul WE, Min B. Both exogenous commensal and endogenous self antigens stimulate T cell proliferation under lymphopenic conditions. Cellular immunology. 2012;272:117–123. doi: 10.1016/j.cellimm.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Do JS, Min B. Differential requirements of MHC and of DCs for endogenous proliferation of different T-cell subsets in vivo. Proc Natl Acad Sci U S A. 2009;106:20394–20398. doi: 10.1073/pnas.0909954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 8.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol. 2013;14:246–253. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, Kuchroo VK, Robson SC, Quintana FJ. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14:1054–1063. doi: 10.1038/ni.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visperas A, Do JS, Bulek K, Li X, Min B. IL-27, targeting antigen-presenting cells, promotes Th17 differentiation and colitis in mice. Mucosal immunology. 2013 doi: 10.1038/mi.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferongamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 12.Singh UP, Singh S, Iqbal N, Weaver CT, McGhee JR, Lillard JW., Jr IFN-gamma-inducible chemokines enhance adaptive immunity and colitis. J Interferon Cytokine Res. 2003;23:591–600. doi: 10.1089/107999003322485099. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sercan O, Hammerling GJ, Arnold B, Schuler T. Innate immune cells contribute to the IFN-gamma-dependent regulation of antigen-specific CD8+ T cell homeostasis. J Immunol. 2006;176:735–739. doi: 10.4049/jimmunol.176.2.735. [DOI] [PubMed] [Google Scholar]
- 15.Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. 2010;184:2855–2862. doi: 10.4049/jimmunol.0902708. [DOI] [PubMed] [Google Scholar]
- 16.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 17.Griseri T, McKenzie BS, Schiering C, Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. 2012;37:1116–1129. doi: 10.1016/j.immuni.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paul G, Marchelletta RR, McCole DF, Barrett KE. Interferon-gamma alters downstream signaling originating from epidermal growth factor receptor in intestinal epithelial cells: functional consequences for ion transport. J Biol Chem. 2012;287:2144–2155. doi: 10.1074/jbc.M111.318139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kushwah R, Hu J. Dendritic cell apoptosis: regulation of tolerance versus immunity. J Immunol. 2010;185:795–802. doi: 10.4049/jimmunol.1000325. [DOI] [PubMed] [Google Scholar]
- 20.Wan CK, Oh J, Li P, West EE, Wong EA, Andraski AB, Spolski R, Yu ZX, He J, Kelsall BL, Leonard WJ. The cytokines IL-21 and GM-CSF have opposing regulatory roles in the apoptosis of conventional dendritic cells. Immunity. 2013;38:514–527. doi: 10.1016/j.immuni.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savarin C, Stohlman SA, Hinton DR, Ransohoff RM, Cua DJ, Bergmann CC. IFN-gamma protects from lethal IL-17 mediated viral encephalomyelitis independent of neutrophils. Journal of neuroinflammation. 2012;9:104. doi: 10.1186/1742-2094-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Veen RC. Nitric oxide and T helper cell immunity. International immunopharmacology. 2001;1:1491–1500. doi: 10.1016/s1567-5769(01)00093-5. [DOI] [PubMed] [Google Scholar]
- 24.Koshiji M, Adachi Y, Sogo S, Taketani S, Oyaizu N, Than S, Inaba M, Phawa S, Hioki K, Ikehara S. Apoptosis of colorectal adenocarcinoma (COLO 201) by tumour necrosis factor-alpha (TNF-alpha) and/or interferon-gamma (IFN-gamma), resulting from down-modulation of Bcl-2 expression. Clinical and experimental immunology. 1998;111:211–218. doi: 10.1046/j.1365-2249.1998.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattei F, Bracci L, Tough DF, Belardelli F, Schiavoni G. Type I IFN regulate DC turnover in vivo. Eur J Immunol. 2009;39:1807–1818. doi: 10.1002/eji.200939233. [DOI] [PubMed] [Google Scholar]
- 26.Melrose J, Tsurushita N, Liu G, Berg EL. IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells. J Immunol. 1998;161:2457–2464. [PubMed] [Google Scholar]
- 27.Ohmori Y, Hamilton TA. IFN-gamma selectively inhibits lipopolysaccharide-inducible JE/monocyte chemoattractant protein-1 and KC/GRO/melanoma growth-stimulating activity gene expression in mouse peritoneal macrophages. J Immunol. 1994;153:2204–2212. [PubMed] [Google Scholar]
- 28.Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, Skerra A, Li L, Parkos CA, Nusrat A. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 2010;32:392–402. doi: 10.1016/j.immuni.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.