

SUMMARY
Interactions among ADP-ribosylation factors (ARFs), various adaptor proteins, and membrane lipids are essential for intracellular vesicle transport of a variety of cellular materials. Here we present NMR-based information on the nature of the interaction of yeast ARF1 (yARF1) and the pleckstrin homology (PH) domain of four-phosphate-adaptor protein 1 (FAPP1) as it occurs at a model membrane surface. Interactions favor a model in which FAPP1 is partially embedded in the membrane and interacts with a membrane-associated ARF1 molecule primarily through contacts between residues in switch I of ARF1 and regions near and under the solution exposed C-terminal extension of the PH domain. The ARF1 binding site on FAPP1-PH is distinct from a positively charged phosphatidylinositol-4-phosphate (PI4P) binding site. A structural model is constructed that supports coincidence detection of both activated ARF and PI4P as a mechanism facilitating FAPP1 recruitment to membranes.
INTRODUCTION
Intracellular vesicle transport, involving budding of a cargo carrying vesicle from one membrane, transport, and fusion with another membrane, is essential to cellular processes such as establishment and maintenance of organelle identity, post-translational protein modification to cisternal and transmembrane proteins, transfer and recycling of materials to and from degradative organelles, and maintenance of membrane composition (Campelo and Malhotra, 2012). ADP-ribosylation factors (ARFs1) are key components of the vesicle budding process (Donaldson and Jackson, 2011; Kahn, 2009; Mizuno-Yamasaki et al., 2012). They bind to a number of adaptor and effector proteins that mediate accumulation of vesicle cargo, influence assembly at particular regions of budding membranes, and alter metabolism of phospholipids (like phosphatidylinositols involved in cell signaling). Structures of ARF-adaptor or ARF-effector complexes, especially as they exist on membrane surfaces, are an important basis from which a detailed, molecular understanding of these processes will emerge. Four phosphate adapter protein 1 (FAPP1) is one such protein believed to play a role in regulation of vesicular cargo transport from the trans-Golgi network (TGN) to the plasma membrane (Godi et al., 2004). Here we present NMR-based information on the nature of the interaction of yeast ARF1 (yARF1) and the pleckstrin homology (PH) domain of FAPP1 as it occurs at a model membrane surface.
ARF1 is a ~20 kDa N-myristoylated GDP-GTP binding protein and prototype of the ARF family, within the RAS superfamily of regulatory GTPases. Models for its function have evolved over time with the current model involving a loose association of the GDP loaded form with a donor membrane, possibly in association with a guanine exchange factor (GEF) that promotes exchange of GDP for GTP (Cherfils and Zeghouf, 2013; East and Kahn, 2011). The GTP form of ARF1 (ARF • GTP) is tightly anchored to the membrane and may accumulate in curvature prone regions of the membrane, or actually promote curvature to facilitate budding (Ambroggio et al., 2013). This “activated” conformation also has higher affinity for effectors that generate biological output; including allosteric activation of enzymes (e.g., PI kinases, phospholipase D, cholera toxin) and recruitment to membranes of protein adaptors (e.g., FAPP1, AP-1, MINT3, COPI). There is a substantial amount of structural data on ARF1, beginning with crystal structures of non-myristoylated ARF1 • GDP (Amor et al., 1994) and a truncated non-myristoylated form of ARF1 • GTP (Δ17-ARF1 • GTP) (Goldberg, 1998). Several year ago we were able to produce a myristoylated form of yARF1 (Liu et al., 2009) and reported NMR structures of the myristoylated yARF1•GDP in solution and myristoylated yARF1•GTP associated with a model lipid bilayer membrane (bicelle) composed of mixtures of dimyristoylphosphatdylcholine (DMPC) and dihexanoylphosphatdylcholine (DHPC) (Liu et al., 2010).
PH domains are ~120 residues in length and found within hundreds of different human proteins, including FAPP1. They are important in providing spatial targeting information through direct binding to different PIPs (notably PI4P, PI4,5P2 and PI3,4,5P3) or protein partners; including G protein βγ dimers (Touhara et al., 1994; Wang et al., 1994), protein kinase C (Yao et al., 1994), and GTPases (e.g., Rac1) (Snyder et al., 2003) or ARFs (Godi et al., 2004). FAPP1 is a 33 kDa protein that contains a 110 residue N-terminal PH domain. The FAPP1-PH domain specifically interacts with both the GTP-bound form of ARF1 and phosphatidylinositolphosphates (PIPs); negatively charged lipids that are concentrated in the trans-Golgi membranes. Specificity has been demonstrated for phosphatidylinositol-4-phosphates (PI4Ps), consistent with their enrichment at the TGN. The ARF1 and PIP binding sites are believed to be non-overlapping (He et al., 2011). Thus, the PH domain provides for the possibility of coincidence detection of two signals in one highly localized space, one proteinaceous and one lipid; both may be required before sufficient affinity of FAPP1 for a specific membrane is achieved and consequent downstream signaling occurs. PIP binding may also facilitate FAPP1-ARF1 interactions as ARF too has a hypothesized PIP binding site (Randazzo, 1997; Terui et al., 1994). With both proteins concentrating in PIP rich membrane regions protein-protein interactions would be promoted. This idea of coincidence detection of two or more inputs providing specificity to GTPase actions on a membrane is rapidly emerging as an important concept in modeling cell signaling (Haucke, 2005; Lemmon, 2007; Malaby et al., 2013; Richardson et al., 2012; Wieffer et al., 2012).
Both crystal and NMR structures of the FAPP1-PH domain exist (He et al., 2011; Lenoir et al., 2010). These display similar structures with a 7 stranded beta barrel capped by an alpha helix near the C-terminus and having an extended loop between strands 1 and 2 near the N-terminus. There have also been studies of the interaction of FAPP1-PH with ARF1 and with PI4P analogs using NMR chemical shift perturbations of FAPP1 cross-peaks in 15N-1H HSQC (heteronuclear single quantum coherence) spectra to identify binding sites. While the sites for ARF1 and PI4P interaction were clearly indicated to be different, considerable overlap in chemical shift perturbation existed. The FAPP1 resonance perturbation studies were done with a form of ARF1 locked in its GTP form by a Q71L mutation, but missing parts needed for membrane association, namely its myristoylated N-terminal helix. While the Q71L mutant is likely to display a GTP-like structure, the absence of an interaction with a native membrane environment could clearly alter protein-protein associations. Previous studies with PI4P were also done primarily with versions of PI4P carrying no fatty acids or short chain fatty acids on PI4P’s glycerol backbone. When micelle solubilized versions of PI4P were used, the detergent was dodecylphosphorylcholine, a detergent which we have found to disrupt the native structure of ARF1 (unpublished data). Hence, there is value and pursuing further studies in the presence of a phospholipid based interaction surface. Chemical shifts in (HSQC spectra of human ARF1 (hARF1) on adding FAPP1-PH were also noted in one of the previous studies (He et al., 2011), but with resonance assignments only available for the GDP form of hARF1 (Seidel et al., 2004), a specific interaction surface on ARF was not identified.
Here we present data on the interaction of a full-length myristoylated form of S. cerevisiae ARF1 (yARF1) with FAPP1-PH on the surface of a DMPC/DHPC model membrane bicelle, as well as the interaction of long chain, bicelle incorporated, PI4P with FAPP1-PH. FAPP1-PH used in this study is a human protein and previous interaction studies have been carried out with hARF1. However, it is the yARF1 construct that has allowed efficient production of myristoylated ARF1 and membrane associated studies (Liu et al., 2010). Moreover, NMR resonance assignments are available for the GTP-bound form of yARF1, allowing identification of FAPP1 interaction surfaces on ARF1 in a membrane-like environment. The yARF1 sequence is 78% identical to hARF1, high but still raises some concern about conservation of interactions described here. We attempt to address this concern by comparison of chemical shift perturbations on FAPP1 on adding myr-yARF1•GTPγS in bicelles with those on FAPP1 on adding Δ17-hARF1•GTPγS in solution. We also use paramagnetic perturbations from a nitroxide spin-labeled analog of yARF1, observations that are more directly distance dependent (Battiste and Wagner, 2000), to support binding sites on FAPP1 identified by chemical shift data. The unpaired electron on the nitroxide provides long range paramagnetic spin relaxation enhancement (PRE) of nuclear spin sites that drops off in a 1/r6 manner. These enhancements have been used effectively in many other protein-protein association studies (Clore and Iwahara, 2009; Clore et al., 2007) and add significantly to confidence in the chemical shift perturbation data presented.
Our experiments result in several key findings. Most notably, the presence of a membrane mimetic does have an effect on the nature of FAPP1-PH interactions. While similar to those observed in solution, the ARF1 binding site is shifted slightly away from the hypothesized membrane insertion end of FAPP1-PH and the PIP binding site is shifted toward that end. This is consistent with insertion of both PIP and FAPP1-PH into the membrane surface. Identification of the FAPP1-PH binding sites on ARF1 for the first time allows the construction of a structural model for the ARF1-FAPP1-PIP complex and assessment of the possible biological importance of this ternary complex.
RESULTS
FAPP1 interaction surfaces from PI4P induced shifts
Previous structural investigations of the interaction of FAPP1-PH with PIPs were conducted in solution using micelle imbedded or soluble versions of PIPs having short alkyl chains or just the PIP headgroup. Hence, we felt it appropriate to repeat these studies using a PI4P with longer acyl chains (PI4P isolated from brain) and the bilayer-like membrane model (a bicelle) that was used in our the structural characterization of myr-yARF1•GTPγS. These interactions were monitored using perturbations of HSQC/TROSY (transverse relaxation optimized spectroscopy) peaks from 15N-labeled FAPP1-PH on adding PI4P as well as perturbation of 31P resonances of PI4P on adding FAPP1-PH. In the latter experiments the phosphate monoester peak from PI4P in DMPC/DHPC bicelles is well resolved at 4ppm in the absence of FAPP1-PH (Figure 1B). Interaction with FAPP1-PH induces a substantial downfield shift as the protein concentration is increased in the presence of 2mM PI4P, nearing saturation at a concentration of 3.5 mM FAPP1-PH. The gradual shift with significant broadening at intermediate levels is indicative of rapid to intermediate exchange between bound and free forms on a sub-ms time scale. Complete saturation of PI4P with FAPP1-PH could not be achieved because of indications of precipitation at 3.5mM, but non-linearity in the titration suggests a dissociation constant in the 0.2–0.3 mM range. The phosphate diester peak from PI4P is obscured by the abundant peaks from phosphatidylcholine lipids in the supporting bicelle.
Figure 1.
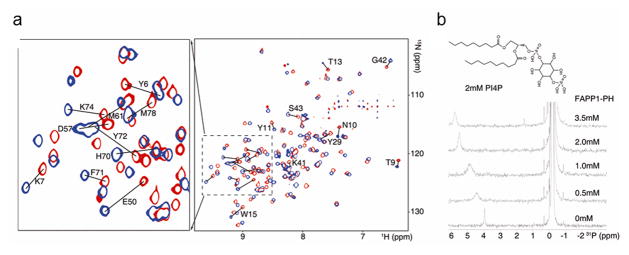
FAPP1-PH interacts with PI4P. A, Superimposed TROSY spectra of FAPP1-PH in the presence of 10% DMPC/DHPC (q=0.25) bicelle (red) and bicelle doped with 8mM PI4P (blue). Assignments are indicated for residues that undergo combined chemical shift changes greater than 0.15ppm. B, 31P spectra from serial titration of FAPP1-PH (0–3.5 mM) into 10% DMPC/DHPC (q=0.25) bicelle doped with 2mM PI4P, highlighting the migration and broadening/re-narrowing of the PI4P phosphate monoester signal.
Chemical shift perturbations of crosspeaks in the 15N-1H spectra of FAPP1-PH in the presence of PI4P-containing bicelles in comparison to pure phosphatidylcholine bicelles are shown in Figure 1A. Many peaks show substantial shift differences both from FAPP1-PH in the absence of bicelles and in the presence of bicelles not containing PI4P. This necessitated reassignment of FAPP1-PH spectra under bicelle-interacting conditions. Assignments for HSQC/TROSY peaks were achieved using a combination of backbone centered experiments on 13C, 15N labeled and 2D, 13C, 15N labeled samples (see methods section). 1H and 15N amide shifts for the bicelle containing but PI4P-free sample are generally consistent with the lipid-free shifts as reported by Lenoir et al. (BMRB deposit 16082). Outliers with >0.1ppm combined 1H and 15N shifts include residues L22, G25, H70, and S90-L95. L22 and G25 are structurally proximate to the N-terminus where our constructs differ (see methods) and S90-L95 are in a poorly structured region close to the C-terminus where constructs also differ. H70 is close to the hypothesized membrane insertion region and deviations could arise from FAPP1-PH interaction with DMPC/DHPC lipids as the published assignments were made in the absence of lipids, although pH differences could also have a significant impact on a histidine resonance. On comparing shifts in the presence of PI4P containing bicelles as compared to bicelles without PI4P the largest shifts are seen for T13, W15, Y29, E50, H70, and Y72. These residues are highlighted on the crystal structure of FAPP1-PH by coloring exposed surfaces red; surfaces of residues with lesser perturbations are colored yellow. Perturbed regions are again generally consistent with results reported previously using short, soluble PI4P, except that the perturbations of residues in the N-terminal loop are stronger (bottom of structure in Figure 2A) and the perturbations near I44 and I65-Q69 on the upper part of the molecule as depicted in Figure 2A are not as strong as previously observed. The strong interactions at the center coincide with a positive patch of residues that are likely to drive PI4P interaction (see electrostatic plot in Figure 2B). The displacement toward the bottom of the depicted structure may result from the long chain PI4P being more deeply buried in a bilayer-like membrane.
Figure 2.
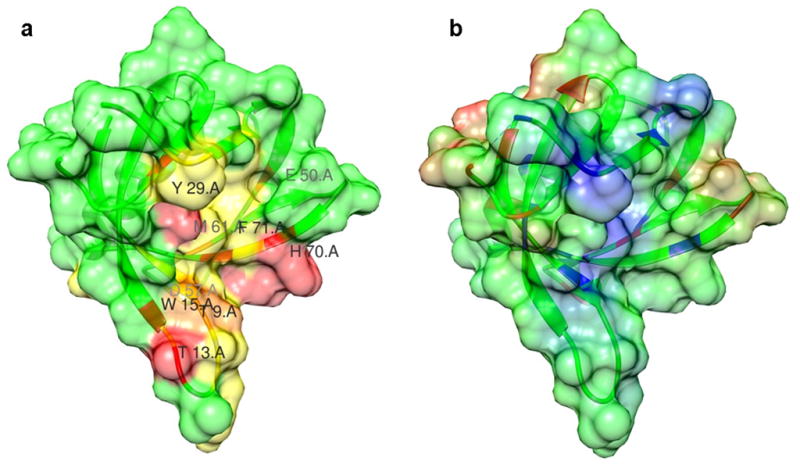
The PI4P binding interface of FAPP1-PH identified through chemical shift perturbations. A, A surface view of FAPP1-PH colored in red for residues of strong chemical shift perturbations (>0.4 ppm) upon adding PI4P and in yellow for those of weak perturbations (<0.4 and >0.2 ppm). B. The electrostatic surface of FAPP1-PH orientated in the same perspective as in A, highlighting the positively-charged pocket for PI4P binding. Positive potentials are shown in blue.
FAPP1 Interaction surfaces from ARF1 induced shifts
Given the possible concern with differences between yARF1 and hARF1 in terms of their ability to interact with FAPP1 or FAPP1-PH, it seemed prudent to compare chemical shift perturbations in HSQC spectra of FAPP1-PH using our GTPγS loaded myr-yARF1•GTPγS in bicelles to previously published data (He et al., 2011) using the soluble Δ-17-hARF1(Q71L) mutant. Because there are also differences in the FAPP1 construct used, we decided to first repeat chemical shift perturbation studies with a close analog of Δ-17-hARF1(Q71L), Δ-17-hARF1•GTPγS. Of the 14 most strongly perturbed FAPP1-PH crosspeaks that we detect, 7 were also among the 14 most strongly perturbed residues in the published data (T13, E50, V53, H54, D57, Q69, H70) (He et al., 2011). Three more of the 14 were within two residues of a strongly perturbed residue in the published work. Two of the additional perturbed resonances are near the C-terminus (A93 and C94), which could reflect differences in the C-terminal extension of FAPP1-PH used here and that used in the published work. Hence, differences in patterns of perturbation due to changes in the FAPP1-PH construct should be relatively minor and are likely to be limited to those near the C-terminus.
The experiments needed for comparison of FAPP1-PH chemical shift perturbations on adding myr-yARF1•GTPγS as compared to Δ-17-hARF1(Q71L) were conducted both with myr-yARF1•GTPγS anchored to 10% q=0.25 DMPC/DHPC bicelles and to q=0.25 DMPC/DHPC bicelles containing 5 weight % PI4P relative to other lipids. Using myr-yARF1•GTPγS anchored to bicelles in the absence of PI4P the perturbations were slightly smaller in magnitude than those seen using our soluble form of hARF1, but also included a couple of cases of crosspeaks disappearing on addition of bicelles. Disappearance can result from slower dissociation rates or slow motions in the complex, which lead to exchange broadening, or directly from resonance broadening on membrane association. Of the 11 most strongly perturbed peaks (L12, T13, E50, I51, K52, A87, L88, G89, S91, K92, and C94) 9 are among, or in close proximity of (within two residues), the moderate to strongly perturbed residues in the published data. The two additional perturbed resonances are near the C-terminus (K92, and C94). Hence, yARF1 was found to have interaction surfaces on FAPP1-PH that are conserved with those of hARF1, as expected from these functionally interchangeable proteins (Kahn et al., 1991).
For bicelles with PI4P incorporated, the set of shifted or broadened crosspeaks were similar to that obtained using bicelles without PI4P, though perturbations were noticeably stronger (~50% larger shifts and more crosspeaks disappear, see supplement). Of the 10 most strongly perturbed crosspeaks (T13, A47, E50, K52, V86, G89, S91, K92, C94, L95) 7 were seen in the absence of PI4P. Perturbations for two more simply couldn’t be quantified in the absence of PI4P because of peak overlap (V86 and L95). The one additional perturbation seen in the presence of PI4P is for A47, a residue close to the strongly perturbed E50. The presence of PI4P in the bicelle clearly enhances interactions with ARF1. This may simply be a result of enhanced binding of FAPP1-PH to the bicelle or possibly more optimal positioning for better interaction with membrane bound ARF1. The most notable differences from the published data using Δ-17-hARF1(Q71L) are the reduction of interactions with the N-terminal loop (N10-W15 of FAPP1-PH) where only T13 and L11 disappear or are weakly perturbed, and the lack of interaction in the Q69-Y72 region. The reduction of interactions in the N-terminal loop in the bicelle anchored system may not be surprising as this loop has been suggested to be a membrane insertion motif and may not be available for interaction with the globular cytosolic domain of ARF1. The loss of interaction in the Q69-Y72 region along with reduction of interaction with the N-terminal loop may be significant in that these are regions where PI4P interactions with FAPP1-PH are observed.
Chemical shift perturbations from TROSY-HSQC spectra for FAPP1 in the presence of bicelles containing PI4P plus myr-yARF1•GTPγS at a concentration of 1.6 mM are shown in Figure 3A. Perturbed residues are color coded in the surface depiction of the FAPP1-PH molecule in Figure 3B; most strongly perturbed (shifted or disappeared) are red (T13,E50,K52,V86,G89,S94,L95) and least perturbed are yellow (A47,V48,E80,R83,L88,S91,K92). The perturbed resonances include a weakly perturbed region near the top of the molecule a more strongly perturbed region in and under the C-terminal extension (red strand overlaying the red region to the left). It may be that the C-terminal extension changes conformation on interaction with ARF1 making the underlying region more accessible. Note that, except for T13 and E50, perturbed resonances do not include those perturbed by PI4P. The T13 perturbation is weaker than in the absence of PI4P and E50 may be subject to perturbations propagated through other structural rearrangements as it is frequently involved with electrostatic interactions with K92 of the C-terminal extension, a poorly ordered part of FAPP1 in many structures. The model shown in the Figure 3B is positioned as in Figure 2B but rotated ~180° about the vertical axis so that comparison of PI4P and ARF1 interaction surfaces can be made.
Figure 3.
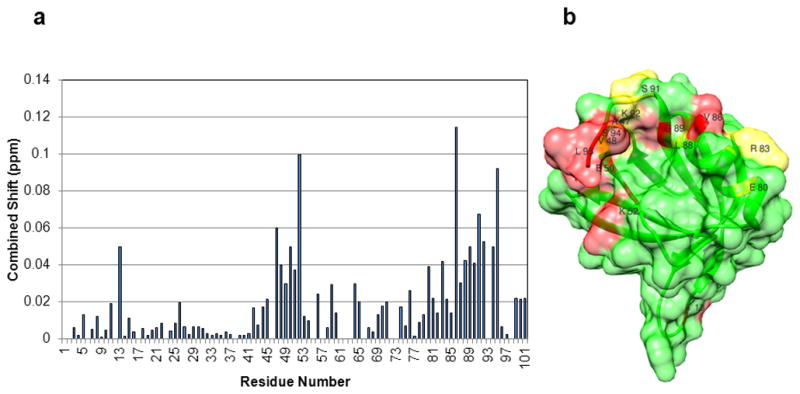
The ARF1 binding interface of FAPP1-PH identified through chemical shift perturbations. A, Residue specific chemical shift perturbations in TROSY spectra of FAPP1-PH in the presence of myr-yARF1•GTPγS in 10% DMPC/DHPC (q=0.25), 0.5% PI4P bicelles. Resonances that disappear have been set to 0.05 ppm. B, The ARF1 binding surface of FAPP1-PH colored in red for residues showing strong chemical shift perturbations upon adding ARF1 and in yellow for those showing weak perturbations. FAPP1-PH is rotated approximately 180° relative to the depiction in Figure 2. Residue numbers are indicated for those discussed in the text.
ARF1 interaction surfaces from FAPP1 induced shifts
One of the advantages of using yARF1 is the availability of a complete set of resonance assignments for a GTP-loaded form (Liu et al., 2010). As in the previous sections dealing with FAPP1, perturbations of crosspeak positions in spectra of 15N-1H labeled myr-yARF1•GTPγS can be used to qualitatively map an interaction surface by adding FAPP1-PH to a bicelle preparation containing ARF1. Figure 4A shows chemical shift in TROSY spectra of myr-yARF1•GTPγS on adding FAPP1-PH in the presence of 10% DMPC/DHPC bicelles (q=0.25). The perturbations cluster in one region that encompasses switch I of ARF1 (G40-T55). This provides an immediate explanation for the observed preference for binding ARF1•GTP as opposed to ARF1•GDP. Switch I conformation changes dramatically in the process of communicating guanine nucleotide changes to the membrane interacting part of the ARF1 molecule. There is an additional, moderate perturbation of R19. R19 is near the connection between ARF1’s cytosolic domain and the N-terminal helix that anchors ARF1•GTP to the membrane surface. It is also near a hypothesized PIP binding site on ARF1. Alterations in membrane anchoring geometry or competition for PI4P on addition of FAPP1-PH may contribute to this remote perturbation. Interaction surfaces are depicted in Figure 4B with color coding as in previous figures: red strongest (E41, V42, E53, T55) and orange next strongest (R19, G40, I43, T45). It is worth noting that except for the conservative V to I and I to V changes at V42 and I43 respectively, all other perturbed residues are identical between yeast and human ARF proteins, indicating that this interface is likely conserved.
Figure 4.
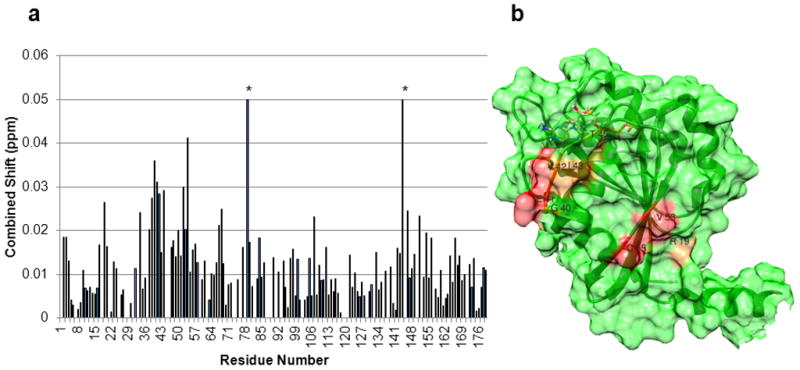
The FAPP1-PH binding interface of yARF1•GTPγS identified through chemical shift perturbations. A, Residue specific chemical shift perturbations in TROSY spectra of myr-yARF1•GTPγS in 10% DMPC/DHPC (q=0.25), 0.5% PI4P bicelles in the presence of 1.5mM FAPP1-PH. The bars marked with * are histidine residues whose shifts responded to small differences in pH (truncated to 0.05). B, The FAPP1-PH binding surface of ARF1 colored in red for residues of strong chemical shift perturbation and in orange for those of weak perturbations. Residue numbers are indicated for those discussed in the text.
FAPP1 interaction surfaces probed with spin-labeled ARF1
Chemical shift perturbations clearly provide useful qualitative information on potential interaction surfaces. However, chemical shift changes in heteronuclear HSQC and TROSY experiments result not just from proximity of an added binding partner, but from structural changes associated with binding. Some of these can be propagated to remote regions via allosteric effects. In an effort to support an argument for direct interaction we employed a spin-labeled version of myr-yARF1•GTPγS. We had previously designed spin-labeled versions of ARF1 to help locate the N-terminal helix in bicelle associated myr-yARF1•GTP (Liu et al., 2010). The native C159 residue was mutated to a serine and other strategically placed residues were mutated to cysteine (T55C, K59C, R83C, R117C or S176C). Reaction with a nitroxide carrying spin label reagent (MTSSL, 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate) adds a paramagnetic (unpaired electron) site capable of causing enhanced spin relaxation with an approximate 1/r6 dependence on the distance between the un-paired electron site and observed nuclear site. Intensities of HSQC crosspeaks from perturbed residues drop with an exponential dependence on this enhanced relaxation, and these reductions can be used to position residues in a structural model. To eliminate loss of intensity due to other processes such as broadening of peaks due to changes in dynamic processes and to facilitate comparison of properly paired crosspeaks intensity, comparisons are normally made between spectra taken with the label oxidized (carrying an unpaired electron) and with the label reduced (carrying no unpaired electron).
One of the mutated sites, T55C, is near the site on ARF1 which shows major chemical shift perturbations on addition of FAPP1-PH. Reductions in FAPP1 HSQC peak intensities on adding this mutant of myr-yARF1•GTPγS in a bicelle were observed, as depicted in Figure 5 (most perturbed (Q33, K45, N58, G67, A78, A79) in red and the next most perturbed (V4, Y11, W15, D34, C49 and V53) in orange). The most tightly clustered set of residues showing a strong reduction in crosspeak intensity (N58, A78, A79) are to the right of the structure in Fig 5B. These are on the same face of the FAPP1-PH structure as residues showing perturbed chemical shifts but are displaced to the right and toward the membrane surface as compared to the region identified by chemical shift perturbation in Figure 3. Some displacement is not surprising as the spin label site (T55C) is at the lower edge of the site on ARF1 suggested to promote FAPP1 interactions. Some additional displacement toward the membrane may also result from the hydrophobic nature of the spin label and the rather long extension of the labeled side chain. There are a few more dispersed contact sites on the back side of the structure as depicted, possibly indicating the existence of other contact geometries. The decrease in specificity of interaction may have resulted from placing the spin label on ARF1 too close to the FAPP1-PH interaction site or from additional interactions promoted by its hydrophobic nature. Comparing FAPP1-PH chemical shifts induced by addition of reduced spin labeled ARF1 to those induced by non-labeled ARF1, does show some strong perturbations of residues 8, 63 and 65 that are in addition to shared perturbations in the 44–54 and 86–92 regions. These are difficult to interpret because of the absence of PI4P and possible changes of pH on addition of ascorbate in these particular samples, but they do occur in regions supporting an additional interaction site. Nevertheless, strong PRE effects on the face on which chemical shift perturbations occur in the non-labeled ARF1-FAPP1-PH system adds confidence to the chemical shift data. PRE perturbations on this face, along with the chemical shift perturbation data, will be used to model the FAPP1-PH-ARF1 interaction surface.
Figure 5.
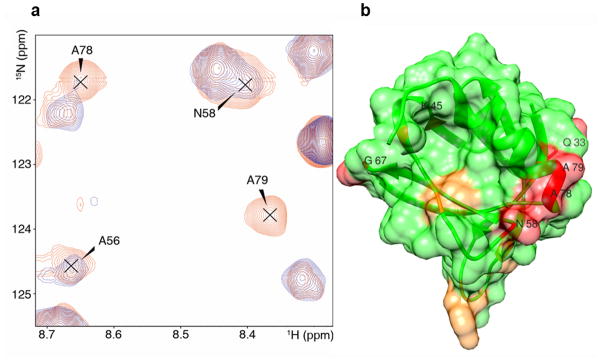
The ARF1 binding interface of FAPP1-PH identified through paramagnetic relaxation enhancements. A, Superimposed fast HSQC spectra of 0.3mM 2D-15N FAPP1-PH mixed with 0.9mM unlabeled oxidized yARF1.GTPγS -T55C-MTSSL (blue) and reduced yARF1.GDP-T55C-MTSSL (red). B, The binding surface of FAPP1-PH colored in red for residues undergoing strong PRE effects and in orange for those undergoing weaker PRE effects. PRE effects reflect spatial proximity to the spin-labeled site (T55) of ARF1. Residue numbers are indicated for those discussed in the text.
DISCUSSION
Interactions of ARF proteins with adaptor proteins and other effectors are key to ARF function in vesicle budding, cargo recruitment, and recycling to continue this process. Understanding the nature of these interactions can potentially lay a basis for intervention in a variety of diseases associated with vesicle traffic defects (Aridor and Hannan, 2002). Unlike the situation for several other families of GTPases, there is no consensus ARF-binding domain that is conserved or present in a substantial fraction of the >20 known effectors for ARFs. However, a large number of ARF-effector and ARF-ARF GEF interactions are influenced by or lead to changes in lipid signaling. Thus, the mechanisms involved in FAPP1/2 recruitment to membranes, involving coincidence detection of both activated ARF and PI4P may prove to be paradigmatic for ARF signaling (Godi et al., 2004; Menetrey et al., 2007). We have taken advantage of the high level of primary sequence (78% identity) and functional conservation (Kahn et al., 1991) between human and yeast ARFs to generate a protein amenable to production of a fully myristoylated version that can be examined in a native-like bicelle environment. Comparison of chemical shift perturbations using a soluble version of hARF1 and those from the bicelle anchored yARF1 further support the validity of using the yeast homolog and extrapolating results to the human protein. Residue-specific chemical shift perturbations of isotopically labeled yARF1 have identified a FAPP1 interaction site on yARF1 that includes residues within switch I. These residues change conformation dramatically on GTP exchange with GDP, and their presence in the interaction surface explains the preference of FAPP1 to interact with GTP vs GDP loaded ARFs. Residue specific chemical shift perturbations of isotopically labeled FAPP1-PH have likewise identified a site on FAPP1-PH that binds to ARF1 and the position of this site has been supported by independent PRE experiments using a spin-labeled version of yARF1. In addition, residue specific chemical shift perturbations of isotopically labeled FAPP1-PH have identified a site on FAPP1-PH that binds to PI4P. These two sites appear to be distinct and have minimal overlap on the FAPP1-PH surface. This explains the previously observed independence of these binding events and establishes a strong structural model for coincidence detection of PI4P and ARF•GTP to recruit FAPP1 to the TGN that is predicted to be relevant to other proteins homologous to FAPP1 (Godi et al., 2004).
Identification of ARF1 and PI4P binding sites on FAPP1-PH has been achieved before, but only in solution and thus not in a membrane-like environment in which these molecules normally function. yARF1 interacts with a membrane surface though a N-terminal myristoylated amphipathic α-helix that is absent in the previously used constructs. Similarly, FAPP1 is believed to interact with a membrane through an N-terminal loop that is rich in hydrophobic and positively charged residues. PI4P normally is esterified with a distribution of long chain fatty acids that associate strongly with a membrane, as opposed to the short chain fatty acids used in previous studies. It might be expected that the preferred positioning of ARF1, FAPP1, and PI4P in a membrane-like environment would alter sites available for their interaction. While the sites identified in the membrane-like environment of a bicelle are found to overlap extensively with those identified in solution, the ARF1 binding site on FAPP1-PH is shifted distinctly away from the membrane surface while the PI4P binding site is shifted distinctly toward the membrane surface. These altered interaction sites, along with the newly observed site on myr-yARF1•GTP allows the construction of a more specific model for ARF1-FAPP1-PH interactions (Figure 6).
Figure 6.
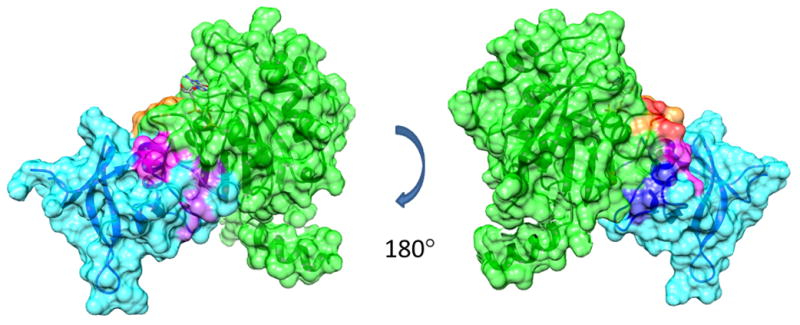
The FAPP1-PH/yARF1 complex modeled by HADDOCK using chemical shift perturbation and PRE data as the experimental constraints. FAPP1-PH is colored in cyan with residues of strong and weak chemical shift perturbations upon adding yARF1•GTPγS and used in the calculations shown in purple and magenta respectively; residues strongly affected by PREs and used in the calculations are shown in blue. yARF1•GTPγS is colored in green with residues of strong and weak chemical shift perturbations upon adding FAPP1-PH shown in red and orange respectively.
The use of qualitative information on interaction surfaces and loose distance constraints from PREs involving pairs of specifically identified residues to construct models for protein-protein interactions is becoming more feasible with the advent of better docking software and molecular dynamics refinement options. We have employed the HADDOCK docking program (De Vries et al., 2010) along with chemical shift perturbation and residue-specific PRE distance constraints to generate the model shown in Figure 6. Strong and medium perturbations on ARF1 (green ribbon) are shown in red and orange, respectively, and strong and medium perturbations on FAPP1-PH (blue ribbon) are shown in magenta and purple, respectively. The model shows a rather well formed interface of approximately 1880 Å2. While the energies calculated by the program are intended primarily for scoring purposes and not thermodynamic interpretation, they indicate substantial electrostatic contributions (−250 kcal/mol) as well as van der Waals contributions (−63 kcal/mol) to the interaction energy. A predominantly positive surface on FAPP1-PH and a predominantly negative surface on ARF1 are also evident.
The model generated can be compared to at least one structure involving a complex between an ARF protein and a PH domain from an effector protein. There is a crystal structure of a complex between the ARF binding domain of ARHGAP21, a Rho family GTPase-activating protein involved in the control of F-actin dynamics at the Golgi, and ARF1(2J59) (Menetrey et al., 2007). The Arf-binding domain of ARHGAP21 contains a PH domain in addition to an α-helix that is important to interaction. The interaction in this structure also involves binding to switch I in ARF. Superimposing the PH domain of FAPP1 in our model, with the PH domain in the ARHGAP21 complex, results in the ARF component of our model occupying essentially the same space as the ARF component of the ARHGAP21 complex, but rotated by approximately 90 degrees. The regions of major chemical shift perturbation on FAPP1-PH make good contact with ARF1 in the ARHGAP21-PH complex, but in a region on ARF1 displaced more toward the top of the switch I region, and a less than optimal contact is made by FAPP1-PH with regions on ARF1 showing chemical shift perturbations. Paramagnetic perturbations are poorly satisfied and it would be difficult to keep both FAPP1 and ARF1 in contact with a membrane in this structure. Thus, we believe that the binding of ARF1•GTP with these two different PH domains involve quite distinct interaction surfaces, and reveal two different ways that ARFs may bind to PH domains. Diversity in interaction surfaces may be essential to numerous and complex protein-protein interactions with which ARF proteins are involved.
A PI4P molecule can be docked with FAPP1-PH using HADDOCK in a similar way. Regions of major chemical shift perturbations on FAPP1-PH are shown in Figure 7 in magenta. The PI4P molecule (truncated to octyl chains for display here) is shown as a ball and stick model. Major contacts are satisfied although a few seem to be involved in a secondary site on the far side of the model shown in the figure. The location of the potentially anomalous perturbation of E50, because of its contact with the C-terminal extension, is shown in the upper left. Surface area contacts in the model are substantial (392 Å2) with predominantly electrostatic contributions to interaction energies (−361 kcal/mol scoring energy).
Figure 7.
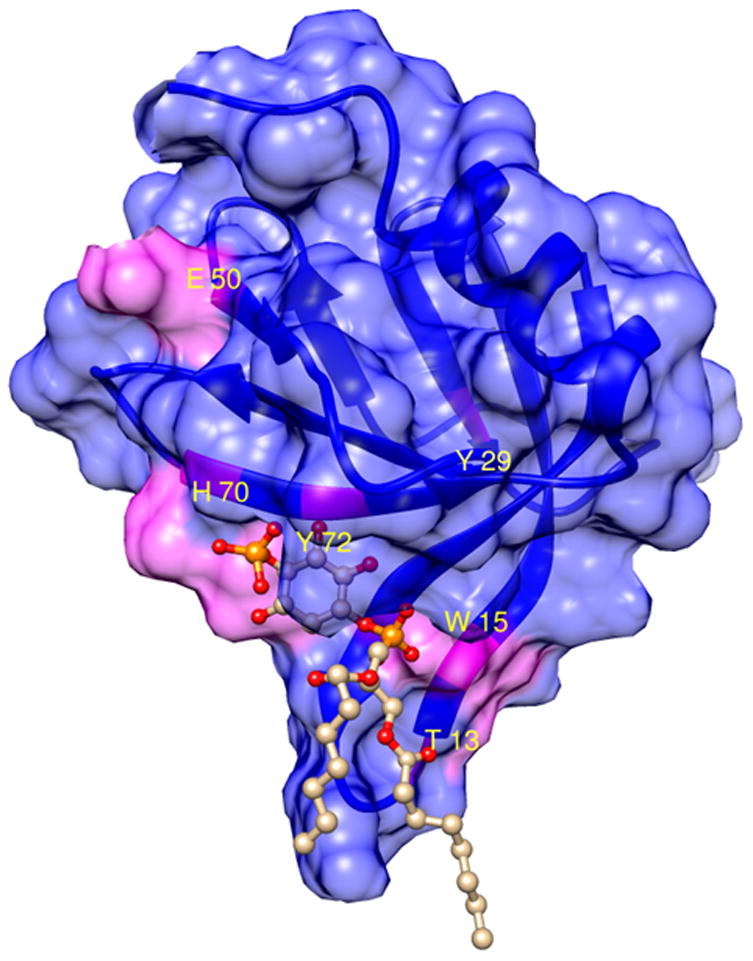
The FAPP1-PH/PI4P complex modeled by HADDOCK using chemical shift perturbation data as the experimental constraints. Residues of significant chemical shift perturbations upon adding PI4P and used in the calculation are shown in magenta. Residue numbers are indicated for those discussed in the text.
The models in Figures 6 and 7 can be combined with a model of a bicelle to depict possible modes of membrane surface interaction (Figure 8). The bicelle was constructed manually from 88 DMPC molecules and 134 DHPC molecules and then subjected to a cycle of energy minimization. The ratio of DMPC to DHPC is higher than the 1:3 preparation ratio, to account for the substantial critical micelle concentration of DHPC. The total size of the assembly (135 kDa) is consistent with the apparent correlation time for the ARF1-bicelle complex if one assumes a reduction of approximately a factor of 2 due to internal motion at the micelle surface. The N-terminal helix of ARF1 is depicted in a position represented by one member of the previously determined distribution for N-terminal conformations needed to model the membrane associated structure (Liu et al., 2010). The selected position allows FAPP1-PH and PI4P contacts depicted in Figures 6 and 7 while maintaining hydrophobic membrane contacts of the N-terminal helix and exposing the hydrophilic surface to solvent. The model produced is clearly not an ideal representation of the ARF1-FAPP1-PH complex on a planar bilayer as the small radius of curvature of the bicelle departs from that of a more extended lipid bilayer, but it does maintain a bilayer-like distribution of lipid head groups and does allow membrane insertion of the putative FAPP1-PH loop and the bound PI4P molecule in geometries as determined by the currently presented data.
Figure 8.
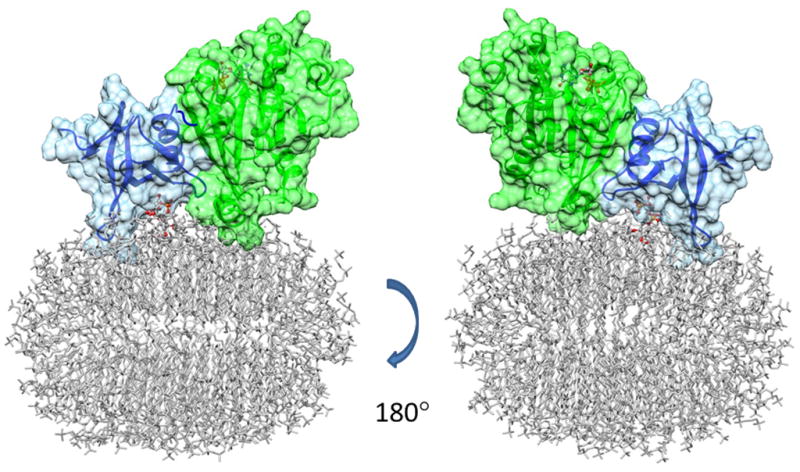
A model of FAPP1-PH/PI4P/ARF1 ternary complex on a small bicelle surface. The HADDOCK models for the FAPP1-PH/yARF1 complex and the FAPP1-PH/PI4P complex are superimposed through the common partner FAPP1-PH, to obtain a model for the ternary complex. This complex is further superimposed through ARF1 structures with our previously published ARF1/bicelle model, to obtain the FAPP1-PH/PI4P/ARF1 ternary complex on the small bicelle surface.
The interactions depicted provide a model that may be further tested and refined through designed mutations and further structural studies. Expansion of the modeling to include larger proteins, e.g., full length FAPP1/2 or other effectors, GAPs, or GEFs, will clearly increase our understanding of these essential molecular interactions and the means of their regulation and importantly, on a biological membrane. The methods used can of course be applied to other ARF interacting molecules to explore a wider range of functions for ARF dependent vesicle transport, lipid metabolism, and other outputs of ARF signaling in all eukaryotic cells.
EXPERIMENTAL PROCEDURES
Materials
Isotopes for NMR labeling were obtained from Cambridge Isotope Laboratories or Sigma Aldrich isotopes. PI4P isolated from brain was obtained from Avanti Polar Lipids, Inc. All other materials were from Sigma Aldrich.
Protein Expression and Purification
The PH domain of FAPP1 (GenBank: AAG15199.1) was cloned into the pET20(b) vector (Novagen, Inc) using NdeI and XhoI sites incorporating the sequence of a C-terminal Hisx6 tag before the stop codon. The resulting protein expressed, MEGVLYKWTNYLTGWQPRWFVLDNGILSYYDSQDDVCKGSKGSIKMAVCEIKVHSAD NTRMELIIPGEQHFYMKAVNAAERQRWLVALGSSKACLTDTLEHHHHHH, begins with the first 98 residues at the N-terminus of the 300 residue human protein and has eight extra amino acids at the C-terminus including two residues from the XhoI cloning site and the Hisx6 tag. It differs from the sequence used in the published X-ray structure of FAPP1 by having those 8 residues instead of a 5 residue sub-cloning extension and from the sequence used in the previous NMR study by having those 8 residues and not having an 8 amino acid N-terminal extension introduced by sub-cloning.
FAPP1-PH was expressed using the Rosetta DE3 cell line (EMD Biosciences, Inc) grown in a minimal medium supplemented with 1gr/L 15N-ammonium chloride and 2gr/L uniformly 13C labeled glucose. Induction was initiated by 0.5mM IPTG as OD600nm reached 0.8, and expression continued overnight at 28°C. As the current investigation was conducted in a lipid bicelle system, resulting in broader lines, perdeuteration was also employed in most preparations by substituting 2H2O for 1H2O. Perdeuteration follows a previously documented protocol (Liu et al., 2010). After cell lysis by French Pressing at 1000psi, the Hisx6 tagged protein was enriched on a Ni-column. A second cleanup was then performed on an ion exchange (Q-Sepharose) column. The yield of perdeuterated, 15N, 13C labeled material was typically 5mg/L The procedures for the expression and purification of myristoylated yARF1, in vitro GDP/GTPγs exchange, and MTSSL spin-labeling, have also been described previously (Liu et al., 2010).
NMR Spectroscopy
Backbone resonance assignment was obtained for FAPP1 in the presence of 10% DMPC/DHPC (q=0.25), with and without 8mM PI4P respectively. In addition to the lipids, the sample also contained 0.8mM FAPP1 PH domain dissolved in a NMR buffer containing 10mM K2HPO4-KH2PO4 (pH 7.0), 50 mM NaCl, 10mM K2SO4, 2mM MgCl2, 5mM DTT, and 5% D2O. Initially a 13C, 15N labeled sample was used to establish the majority of assignments based on HNCACB and CBCA(CO)NH assignments. Due to reduced signal/noise ratio in the presence of lipids, the missing assignments were completed from a 2D, 13C, 15N labeled sample through TROSY-based HNCACB experiments. All experiments were conducted at 25°C on Varian 800 and 900 MHz spectrometers unless otherwise noted. Assignments have been deposited in the BioMagResBank (BMRB) with accession number 19576.
31P observe experiments for FAPP1-PH/PI4P titration were conducted on a Varian 300MHz spectrometer. The buffer contained 20mM MES (pH6.5), 50mM NaCl, 5mM DTT, 10% DMPC/DHPC(q=0.25), and 2mM PI4P. The spectra were externally referenced with 10mM K2HPO4-KH2PO4 buffer (pH 7.0).
For the titration experiments to map the ARF1-binding interface on FAPP1, the two proteins were first dialyzed in the same beaker containing the NMR buffer (no D2O) in order to minimize any mismatch in buffer conditions. FAPP1 and ARF1 were then mixed with ARF1 in excess. Lipids and D2O were added and GDP-for-GTPγS exchange was performed for ARF1 in the mixture. A dummy “exchange” step was also performed for FAPP1 with lipids and D2O added, after which FAPP1 was properly diluted with the dialysis buffer so that its concentration would match that in the mixture. This dummy operation ensures that samples being compared have nearly identical chemical composition, which is important as the chemical shift changes being observed in these studies are quite small. For serial titration experiments, the sample with excess ARF1 was measured first; then a fixed volume of the sample was taken out and replaced with free FAPP1 prepared in the manner mentioned above. The process was repeated to give spectra of serially diluted ARF1 with a constant FAPP1 concentration in an identical buffer condition. The FAPP1 binding site on myr-yARF1•GTP was derived through a comparison of TROSY spectra collected without pH equilibration on a sample of 0.5mM myr-yARF1•GTP and another sample of 0.5mM myr-yARF1•GTP plus 0.75mM FAPP1-PH, both in NMR buffer with 10% DMPC/DHPC (q=0.25). Combined chemical shift perturbations are reported as the root of the sum of the squares of 1H and 15N perturbations with the 15N shifts weighted by 0.17 (Farmer, 1996). In the PRE based interface mapping experiment, a mixture was first made with 0.3mM FAPP1 and 0.9mM yARF1.GDP-T55C-MTSSL. Lipids were added and guanine nucleotide exchange was conducted prior to HSQC spectrum collection. To blank out the PRE effect, 3.6mM ascorbic acid was added to the same sample for the collection of the control HSQC spectrum. As the peak intensities rather than chemical shifts were of interest here, small chemical shift perturbations that might arise from the addition of ascorbic acids were of no major concern.
Modeling of Complexes
Modeling of the complexes was accomplished using the web server version of HADDOCK (De Vries et al., 2010). One thousand docking poses were generated by rigid body energy minimization using a combination of ambiguous (chemical shift perturbation derived) and explicit distance constraints (PRE derived). The best scoring 200 structures were subjected to refinement based on the CNS package and force constants taken from the PRODRG server (Schuttelkopf and van Aalten, 2004). These were subsequently subjected to refinement in the presence of explicit water. Active residues (for ambiguous constraints) were explicitly defined and semi-flexible (passive) residues were defined automatically using a 7.5Å radius from active residues. Fully flexible residues were designated for the C-terminus of FAPP1-PH (88–96) and the switch region (40–55) plus N-terminus (2–20) for yARF1. The resulting structures were clustered based on ligand positions and scored based on average energies. The lowest energy poses in the best or second best clusters were used for models depicted in the figures.
ARF1-FAPP1 docking was initiated using model 20 of the bicelle associated myristoylated and GTPγS loaded form of yARF1 as determined by NMR methods (2KSQ) and model 1 of the FAPP1 PH domain as determined by X-ray crystallography (3RCP). The NMR models of yARF1 show a number of possible positions of the N-terminal membrane associated helix. Model 20 was initially selected based on a position of the N-terminal helix that allowed maximum access to the residues perturbed on FAPP1-PH addition. In the end, docking was done with the terminal helix removed (last 17 residues plus myristic acid), then reattached after docking. The C-terminal 5 residues in the FAPP1-PH crystal structure were also removed and the 91–96 segment moved away from residues perturbed on yARF1 addition to facilitated docking. During docking runs the following were designated active residues on ARF1 (G40, E41, V42, I43, T45, E53, T55). The following were designated active residues on FAPP1 (A47, V48, E50, K52, E80, R83, V86, L88, G89, S90, S91 92, 94, 95). Paramagnetic distance constraints were set at 7 +/− 2 Å between residue T55 on ARF1 and the following residues on FAPP1 (N58, A78, A79).
For FAPP1-PI4P docking the PI4P headgroup from pdb structure 3W68 was used during docking and the diacylglycerol structure from pdb entry 4J7Q, truncating the acyl chains to 8 carbons was added subsequent to docking. The entire PI4P headgroup was designated active and the following residues from FAPP1 were designated active (T13, W15, Y29, H70, Y72). Semi-flexible residues were selected automatically based on a 7.5Å cutoff.
HIGHLIGHTS.
Chemical shift perturbation and PRE data identify interaction sites on ARF1 and FAPP1
Sites on FAPP1 for ARF1 and PI4P have minimal overlap and support coincidence detection
A model of FAPP1, ARF1, and PI4P interacting on a bicelle surface has been constructed
Acknowledgments
This work was supported by a grant from the National Institutes of General Medical Sciences, GM061268.
Footnotes
Abbreviations used in the text include: ARF, ADP-ribosylation factor; FAPP1 four-phosphate-adaptor protein 1; HSQC, heteronuclear single quantum coherence; PH, pleckstrin homology (domain); PIPs, phosphatidylinositol phosphates; PI4P, phosphatidylinositol 4-phosphate; PRE, paramagnetic spin relaxation enhancement; TROSY, transverse relaxation optimized spectroscopy
The authors declare that they have no competing financial interests.
ACCESSION CODES
Assignments of NMR resonances for FAPP1 in bicelle environments have been deposited in the BioMagResBank (BMRB) with accession number 19576.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambroggio EE, Sillibourne J, Antonny B, Manneville JB, Goud B. Arf1 and Membrane Curvature Cooperate to Recruit Arfaptin2 to Liposomes. Plos One. 2013:8. doi: 10.1371/journal.pone.0062963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor JC, Harrison DH, Kahn RA, Ringe D. Structure of the human ADP-ribosylation factor 1 complexed with GDP. Nature. 1994;372:704–708. doi: 10.1038/372704a0. [DOI] [PubMed] [Google Scholar]
- Aridor M, Hannan LA. Traffic jams II: An update of diseases of intracellular transport. Traffic. 2002;3:781–790. doi: 10.1034/j.1600-0854.2002.31103.x. [DOI] [PubMed] [Google Scholar]
- Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Campelo F, Malhotra V. Membrane Fission: The Biogenesis of Transport Carriers. In: Kornberg RD, editor. In Annual Review of Biochemistry. Vol. 81. 2012. pp. 407–427. [DOI] [PubMed] [Google Scholar]
- Cherfils J, Zeghouf M. Regulation of smallGTPases BY GEFs, GAPs, AND GDIs. Physiological Reviews. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- Clore GM, Iwahara J. Theory, Practice, and Applications of Paramagnetic Relaxation Enhancement for the Characterization of Transient Low-Population States of Biological Macromolecules and Their Complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Tang C, Iwahara J. Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr Opin Struc Biol. 2007;17:603–616. doi: 10.1016/j.sbi.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries SJ, van Dijk M, Bonvin A. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nature Reviews Molecular Cell Biology. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East MP, Kahn RA. Models for the functions of Arf GAPs. Seminars in Cell & Developmental Biology. 2011;22:3–9. doi: 10.1016/j.semcdb.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer BT. Localizing the NADP(+) binding site on the MurB enzyme by NMR. Nature Structural Biology. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol. 2004;6:393–404. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- Goldberg J. Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell. 1998;95:237–248. doi: 10.1016/s0092-8674(00)81754-7. [DOI] [PubMed] [Google Scholar]
- Haucke V. Phosphoinositide regulation of clathrin-mediated endocytosis. Biochemical Society Transactions. 2005;33:1285–1289. doi: 10.1042/BST0331285. [DOI] [PubMed] [Google Scholar]
- He J, Scott JL, Heroux A, Roy S, Lenoir M, Overduin M, Stahelin RV, Kutateladze TG. Molecular Basis of Phosphatidylinositol 4-Phosphate and ARF1 GTPase Recognition by the FAPP1 Pleckstrin Homology (PH) Domain. Journal of Biological Chemistry. 2011;286:18650–18657. doi: 10.1074/jbc.M111.233015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA. Toward a model for Arf GTPases as regulators of traffic at the Golgi. Febs Letters. 2009;583:3872–3879. doi: 10.1016/j.febslet.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Kern FG, Clark J, Gelmann EP, Rulka C. Human ADP-ribosylation factors. A functionally conserved family of GTP-binding proteins. J Biol Chem. 1991;266:2606–2614. [PubMed] [Google Scholar]
- Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. In: Wakelam MJO, editor. Cell Biology of Inositol Lipids and Phosphates. 2007. pp. 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M, Coskun U, Grzybek M, Cao XW, Buschhorn SB, James J, Simons K, Overduin M. Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. Embo Reports. 2010;11:279–284. doi: 10.1038/embor.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kahn RA, Prestegard JH. Structure and membrane interaction of myristoylated ARF1. Structure. 2009;17:79–87. doi: 10.1016/j.str.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kahn RA, Prestegard JH. Dynamic structure of membrane-anchored Arf*GTP. Nature structural & molecular biology. 2010;17:876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaby AW, van den Berg B, Lambright DG. Structural basis for membrane recruitment and allosteric activation of cytohesin family Arf GTPase exchange factors. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14213–14218. doi: 10.1073/pnas.1301883110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetrey J, Perderiset M, Cicolari J, Dubois T, Elkhatib N, El Khadali F, Franco M, Chavrier P, Houdusse A. Structural basis for ARF1-mediated recruitment of ARHGAP21 to Golgi membranes. The EMBO journal. 2007;26:1953–1962. doi: 10.1038/sj.emboj.7601634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno-Yamasaki E, Rivera-Molina F, Novick P. GTPase Networks in Membrane Traffic. In: Kornberg RD, editor. In Annual Review of Biochemistry. Vol. 81. 2012. pp. 637–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo PA. Functional interaction of ADP-ribosylation factor 1 with phosphatidylinositol 4,5-bisphosphate. Journal of Biological Chemistry. 1997;272:7688–7692. [PubMed] [Google Scholar]
- Richardson BC, McDonold CM, Fromme JC. The Sec7 Arf-GEF Is Recruited to the trans-Golgi Network by Positive Feedback. Developmental Cell. 2012;22:799–810. doi: 10.1016/j.devcel.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D-Biological Crystallography. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Snyder JT, Singer AU, Wing MR, Harden TK, Sondek J. The pleckstrin homology domain of phospholipase C-beta(2) as an effector site for Rac. Journal of Biological Chemistry. 2003;278:21099–21104. doi: 10.1074/jbc.M301418200. [DOI] [PubMed] [Google Scholar]
- Terui T, Kahn RA, Randazzo PA. Effects of acid phospholipids on nucleotide exchange properties of ADP-ribosylation factor-1 - evidence for specific interaction with phosphatidylinositol 4,5-bisphosphate. Journal of Biological Chemistry. 1994;269:28130–28135. [PubMed] [Google Scholar]
- Touhara K, Inglese J, Pitcher JA, Shaw G, Lefkowitz RJ. Binding of G-protein beta-gamma-subunits to plextrin homology domains. Journal of Biological Chemistry. 1994;269:10217–10220. [PubMed] [Google Scholar]
- Wang DS, Shaw R, Winkelmann JC, Shaw G. Binding of PH domains of beta-adrenergic-receptor kinase and bets-spectrin to WD40/beta-transducin repeat containing regions of the beta-subunit of trimeric G-proteins. Biochemical and Biophysical Research Communications. 1994;203:29–35. doi: 10.1006/bbrc.1994.2144. [DOI] [PubMed] [Google Scholar]
- Wieffer M, Haucke V, Krauss M. Regulation of Phosphoinositide-Metabolizing Enzymes by Clathrin Coat Proteins. In: DiPaolo G, Wenk MR, editors. In Lipids. Vol. 108. 2012. pp. 209–225. [DOI] [PubMed] [Google Scholar]
- Yao L, Kawakami Y, Kawakami T. The pleckstrin homology domain of bruton tyrosine kinase interacts with protein-kinase-C. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9175–9179. doi: 10.1073/pnas.91.19.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]