

Abstract
Intracerebral hemorrhage (ICH) is a devastating type of stroke that lacks a specific treatment. An intense immune response develops after ICH, which contributes to neuronal injury, disability, and death. However, the specific mediators of inflammation-induced injury remain unclear. The objective of the present study was to determine whether blood-derived CCR2+Ly6Chi inflammatory monocytes contribute to disability. ICH was induced in mice and the resulting inflammatory response was quantified using flow cytometry, confocal microscopy, and neurobehavioral testing. Importantly, blood-derived monocytes were distinguished from resident microglia by differential CD45 staining and by using bone marrow chimeras with fluorescent leukocytes. After ICH, blood-derived CCR2+Ly6Chi inflammatory monocytes trafficked into the brain, outnumbered other leukocytes, and produced tumor necrosis factor. Ccr2−/− mice, which have few circulating inflammatory monocytes, exhibited better motor function following ICH than control mice. Chimeric mice with wild-type CNS cells and Ccr2−/− hematopoietic cells also exhibited early improvement in motor function, as did wild-type mice after inflammatory monocyte depletion. These findings suggest that blood-derived inflammatory monocytes contribute to acute neurological disability. To determine the translational relevance of our experimental findings, we examined CCL2, the principle ligand for the CCR2 receptor, in ICH patients. Serum samples from 85 patients were collected prospectively at two hospitals. In patients, higher CCL2 levels at 24 h were independently associated with poor functional outcome at day 7 after adjusting for potential confounding variables. Together, these findings suggest that inflammatory monocytes worsen early disability after murine ICH and may represent a therapeutic target for patients.
Keywords: bone marrow chimeras, CCL2, CCR2, intracerebral hemorrhage, monocytes, neuroinflammation
Introduction
Intracerebral hemorrhage (ICH) is an especially deadly type of stroke affecting 2 million people worldwide each year (Qureshi et al., 2009). ICH occurs when a blood vessel within the brain ruptures, causing blood to flow into the parenchyma. There is no specific treatment for ICH and the 7 d mortality rate remains >30% (Flaherty et al., 2006). Mediators of the coagulation cascade (Lee et al., 1996), combined with complement activation (Mack et al., 2007), the release of danger signals (Lei et al., 2013), toll-like receptor 4 activation (Sansing et al., 2011c), and other mechanisms are thought to initiate an inflammatory response. The resulting neuroinflammation is characterized by microglial activation, leukocyte infiltration, and edema formation. This response contributes to functional deficits in rodent models. Importantly, elevated immune responses are also associated with poor outcomes in patients (Castillo et al., 2002; Dziedzic et al., 2002; Silva et al., 2005; Zhou et al., 2010; Agnihotri et al., 2011).
In most ICH studies, microglia and monocytes are evaluated as a single population, despite potentially important functional differences. Murine monocytes are comprised of two populations, the CX3CR1+Ly6C− monocytes and the CCR2+Ly6Chi inflammatory monocytes. Ly6C− monocytes have high levels of the chemokine receptor CX3CR1, patrol the endothelium of blood vessels (Geissmann et al., 2003), and may aid in healing after myocardial infarction (Nahrendorf et al., 2007). Inflammatory monocytes require the chemokine receptor CCR2 to migrate out of bone marrow and toward CCL2 chemokine gradients (Shi and Pamer, 2011). Thus, Ccr2−/− mice have few circulating inflammatory monocytes (Boring et al., 1997; Kurihara et al., 1997). Microglia express similar cell surface markers as monocytes/macrophages and may also express CCR2 under certain conditions (Zhang et al., 2007). This leads to difficulty in distinguishing monocytes and microglia histologically and identifying their specific functions. The contributions of the monocyte and microglia populations to CNS injury are mostly unknown. This work aims to address this knowledge gap by specifically examining the roles of inflammatory monocytes after ICH.
CCR2+ inflammatory monocytes may be pro-inflammatory or regulatory depending on local cues (Shi and Pamer, 2011; Grainger et al., 2013; Riquelme et al., 2013). The present study discriminates between microglia and blood-derived inflammatory monocytes to determine how inflammatory monocytes influence neurological disability in the acute phase after ICH. We show that inflammatory monocytes are the most numerous blood-derived leukocytes in the brain at day 3 and that they produce tumor necrosis factor (TNF). Ccr2−/− mice exhibit only minor motor deficits following ICH compared with wild-type mice. Wild-type mice transplanted with Ccr2−/− bone marrow and wild-type mice depleted of circulating inflammatory monocytes also showed improved outcomes, localizing the protection to a reduction in blood-derived inflammatory monocytes. Finally, we assessed CCL2, the dominant chemokine for monocyte recruitment, in ICH patients and found that elevated serum CCL2 at 24 h was independently associated with poor functional outcome. We conclude that inflammatory monocytes worsen early motor deficits following ICH. Therapies that antagonize their recruitment may represent a new treatment strategy for this devastating type of stroke.
Materials and Methods
Mice.
C57BL/6J (WT CD45.2), B6.SJL-Ptprca Pepcb/BoyJ (WT CD45.1), C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ (GFP), B6.129S4-Ccr2tm1Ifc/J (Ccr2−/−), B6.129P-Cx3cr1tm1Litt/J (heterozygous, Cx3cr1GFP/+), and B6.129(Cg)-Ccr2tm2.1Ifc/J (heterozygous, Ccr2RFP/+) mice were purchased from The Jackson Laboratory and then bred in-house. All mice used in these experiments were males and were age matched with controls. Nonchimeric mice were 8–16 weeks old when subjected to ICH. Littermate controls (Ccr2+/+) were used in experiments with Ccr2−/− mice. Mice were randomized (ICH or sham, MC-21 or control) to treatment groups by coin flip when applicable and data were collected and processed blinded to genotype.
Bone marrow chimera generation.
WT CD45.1 mice (7–9 weeks old) were lethally irradiated (two doses of 5–6 Gy) in a Gammacell 40 research irradiator and 106 nucleated donor bone marrow (BM) cells were injected retro-orbitally. Chimeras were maintained on sulfamethoxazole/trimethoprim antibiotics in their drinking water 1 d before and 2 weeks following irradiation. Chimeras were used for experiments 8–16 weeks following transplantation.
Monocyte depletion.
CCR2+ monocytes were systemically depleted using the rat anti-mouse CCR2 antibody MC-21 (Mack et al., 2001; Mildner et al., 2007). Mice were injected intraperitoneally with 20 μg of MC-21 or rat IgG2b isotype control antibody dissolved in 100 μl PBS. Injections were performed 24 ± 2 h before ICH and again 24 h later. This protocol ensures effective monocyte depletion from the time of ICH until being killed.
ICH models.
Briefly, mice were anesthetized and maintained under 1–5% isoflurane as blood was injected 2.5 mm right and 3 mm deep to bregma at a 5° angle toward the midline, as previously described (Sansing et al., 2011b). For the blood injection ICH model, 20 μl of blood was injected, except for those in GFP chimeric mice, which received 25 μl of blood. Autologous blood was used to create the ICH in experiments shown in Figure 1. To standardize ICH composition, blood from naive C57BL/6J WT donor mice was used for ICH surgeries in all other blood injection ICH experiments. In additional experiments we used the collagenase injection ICH model, which results in breakdown of the vascular basement membrane (Rosenberg et al., 1990). Specifically, 0.05 U type VII collagenase (from Clostridium histolyticum; Sigma) in 1 μl PBS was injected at the same coordinates as for the blood injection model. Sham-treated mice received all experimental interventions (anesthesia and needle insertion) except for injection.
Figure 1.
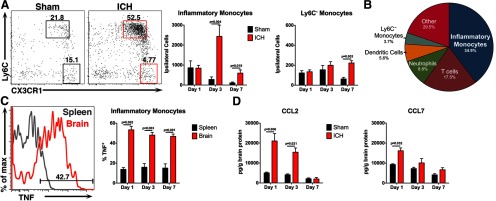
Inflammation following ICH is characterized by a monocyte-rich cellular infiltrate. Mice were subjected to sham or ICH and their brains were analyzed to characterize the cellular and molecular inflammatory composition. A, Representative flow cytometry plots from day 3 show that inflammatory monocytes (Ly6Chi CX3CR1low) outnumber Ly6C− monocytes (Ly6C− CX3CR1hi) in ICH-treated brains from Cx3cr1GFP/+ mice, whereas both populations are low in number in sham brains. Numbers in the plots indicate the percentage of cells in the plot located in each respective gate. Plots are gated on CD45hi, CD3−, Ly6G−. N = 5–10 mice per group. B, This pie chart shows the mean of each blood-derived cell population in Cx3cr1GFP/+ ICH brains at day 3. Each population was calculated as a percentage of total CD45hi cells in the ipsilateral brain hemisphere. N = 9. C, This representative histogram from WT brain and spleen 3 d after ICH shows inflammatory monocyte TNF staining. Roughly half of the inflammatory monocytes isolated from ICH brains were positive for TNF up to day 7, whereas significantly fewer inflammatory monocytes from spleens stained positive for TNF. N = 5–9 per group. D, By ELISA, the monocyte chemoattractant CCL2 is increased in WT ICH brains up to 3 d after ICH, whereas CCL7 is increased only at day 1. N = 4–6 per group. Error bars indicate SEM.
Flow cytometry.
Mice were killed by isoflurane overdose and intracardially perfused with 40 ml ice-cold PBS. The cerebellum and olfactory bulbs were removed from ipsilateral brain hemispheres, which were mechanically and enzymatically (collagenase, dispase, and DNase) digested to a single-cell suspension. The resulting suspension was suspended in a 30%/70% isotonic Percoll (GE Healthcare) gradient and spun at 500 × g. The interphase was collected, washed, and stained as previously detailed (Sansing et al., 2011c). Blood samples were lysed with a hypotonic solution and kept on ice until staining. Spleens were pulverized through 70 μm cell strainers and treated with a hypotonic solution to lyse red blood cells. For some experiments, intracellular cytokine staining was performed by incubating isolated cells with brefeldin A and monensin (BD Biosciences; each 1:1000) for 4 h at 37°C, 5% CO2 and then permeabilizing and staining cells as directed by the Cytofix/Cytoperm kit (BD Biosciences). Fluorophore-conjugated antibodies against CD45, CD45.1, CD11b, CD11c, Ly6G, Ly6C, and MHCII were obtained from eBioscience; CD45.2, CD3e, Ly6G (1A8), SIRPα, and TNF from BD Biosciences; CD19 from Invitrogen; CD36 from BioLegend; and CCR2 from R&D Systems. Alexa Fluor 350 carboxylic acid succinimidyl ester (Invitrogen) was used to exclude dead cells. CountBright counting beads (104 beads per sample; Invitrogen) were used to normalize each tube for the volume of cells run through an LSR II flow cytometer (BD Biosciences).
Flow cytometry data analysis.
Live singlets were analyzed with FlowJo software (Tree Star). Similar to the gating strategy described previously (Sansing et al., 2011c), CD45hi cells isolated from brain samples were classified as blood-derived leukocytes. Leukocyte populations were gated as follows: CD45hi, CD3+ (T-cells); CD45hi, CD19+ (B-cells); CD45hi, CD3−, Ly6C+, Ly6G+ (neutrophils); CD45hi, CD3−, Ly6G−, CD11b+, Ly6C−, CD11c+, MHCII+ (dendritic cells); CD45hi, CD3−, Ly6G−, CD11b+, Ly6Chi (inflammatory monocytes); and CD45hi, CD3−, Ly6G−, CD11b+, Ly6C−, CD11c− (Ly6C− monocytes). Microglia were gated as CD45low, CD11b+. Blood leukocyte populations were gated identically to CD45 hi brain samples.
ELISAs.
The perihematomal region (ipsilateral hemisphere ± 3 mm from needle insertion site) was isolated and homogenized for protein as described previously (Hammond et al., 2012). Briefly, samples were vigorously homogenized in radioimmunoprecipitation assay buffer (Cell Signaling Technology) with a complete protease inhibitor cocktail (Roche). Protein levels were quantified using a BCA Protein Assay Kit in triplicate (Thermo Scientific). CCL7 was quantified by mouse MCP-3 (CCL7) Instant ELISA (eBioscience) using 100 μg of total protein per well in duplicate. CCL2 was quantified by Luminex-based multiplex ELISA (Millipore), also with 100 μg of brain protein per well in duplicate.
Hemoglobin assay.
Mice were intracardially perfused with 40 ml ice-cold PBS and ipsilateral brain hemispheres were isolated for homogenization. Each ipsilateral hemisphere, without the cerebellum and olfactory bulb, was homogenized with 1 mm glass beads and 325 μl ultrapure H2O in a Mini-Beadbeater (BioSpec Products) for 1 min. Samples were centrifuged for 30 min at 21,000 rcf and the supernatant, containing hemoglobin, was isolated for the assay. The assay was performed as previously described (Choudhri et al., 1997) with some modification. We added 20 μl of brain homogenate to 80 μl Drabkin's reagent (Sigma), incubated at room temperature for 15 min, and measured the absorbance at 540 nm. A standard curve was created by adding 20 μl of blood to a naive brain hemisphere before homogenization and serially diluting with untreated homogenate from a naive brain.
Neurobehavioral tests.
All tests were performed at the same time each day by a blinded observer. The cylinder test was performed by placing mice into a transparent glass cylinder and scoring 20 rears for paw placement on the wall of the cylinder as left, right, or both (Sansing et al., 2011a). A laterality index was calculated as (right − left) ÷ (right + left + both). Thus, a score of +1.0 indicates a severe left hemiparesis and exclusive use of the right forelimb, whereas a score of 0.0 indicates equal use of the forelimbs. The beam balance test (Zausinger et al., 2000) was performed by placing a mouse on a horizontal 1 inch diameter rod elevated 40 cm above the lab bench and scoring the longest distance traveled before falling in five walking trials. Mice were only tested once on the beam balance test due to training effects of repeated testing.
Immunohistochemistry.
Mice were intracardially perfused with 20 ml PBS followed by 20 ml 4% paraformaldehyde (PFA)/PBS. Brains were removed, postfixed in 4% PFA/PBS for 4 h, cryoprotected in 30% sucrose/PBS, embedded in Tissue-Tek OCT (Andwin Scientific), and cut into 7 μm sections on charged slides. Sections were blocked in 2% normal goat serum and 2% normal rat serum/PBS and then sequentially stained with rat anti-CD11b (1:100; eBioscience) and Cy3 anti-rat (1:100; Jackson ImmunoResearch) or Alexa Fluor 488 anti-rat (1:100; Invitrogen) antibodies. DAPI (1:10,000) was used as a nuclear co-stain. Sections were mounted in Cytoseal 60 (Thermo Scientific).
Microscopy.
An LSM 780 laser scanning microscope (Zeiss) with 405, 488, 561, and 633 nm laser lines was used for image acquisition at 20 ± 1°C. Tissue sections were imaged at 1 μm intervals in the Z-plane under a 20× objective lens or 0.55 μm under a 40× objective lens blinded to treatment. Images represent a collapsed Z-stack through each tissue section (maximum intensity isotropic orthogonal projection) taken at bregma. Zen and Zen Lite (Zeiss) software were used during acquisition and processing, respectively.
Human data collection.
Patients were prospectively enrolled from Hartford Hospital (2010–2013) and the Hospital of the University of Pennsylvania (2008–2011). Subjects' consent was obtained according to the Declaration of Helsinki. Patients with pre-existing severe disability (modified Rankin scale (mRS) score ≥4) were excluded. Blood was collected by peripheral venipuncture at 24 ± 6 h after ICH symptom onset (or last known well) and centrifuged at 2000 × g for 10 min. Serum supernatant was frozen at −80°C until analysis. Clinical data, including demographics, ICH size and location, pre-ICH functional status, and disability at 7 d by mRS score were prospectively collected by reviewers blinded to CCL2 levels. CCL2 levels were quantified by multiplex ELISA (Millipore) in a single batch.
Statistical methods in murine studies.
Differences were evaluated using ANOVA with individual t test post hoc analysis if data were normally distributed or by Kruskal–Wallis testing with individual Mann–Whitney U test post hoc analysis otherwise. Data in graphs are represented as mean ± SEM unless otherwise noted in the figure legend. Numerical data presented in the text indicate mean ± SD; p values <0.05 were considered statistically significant.
Statistical methods in human studies.
Univariate analyses were conducted to explore serum CCL2 levels and age, sex, ICH volume, time to sample collection, intraventricular extension, and other potential confounding variables using linear regression or Mann–Whitney U test, as appropriate. A multivariable model was created to determine the independent association of CCL2 levels with functional disability at 7 d by ordered logistical regression while adjusting for known predictors of poor outcome after ICH. All statistical analyses were performed using Stata v 11.
Study approval.
All mouse procedures were approved by the University of Connecticut Health Center Animal Care and Use Committee and were performed in strict compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Institutional review boards at the University of Connecticut Health Center, Hartford Hospital, and the Hospital of the University of Pennsylvania approved the clinical study.
Results
Inflammatory monocytes dominate the cellular infiltrate in the days after ICH
To better understand the innate immune response after ICH, we subjected Cx3cr1GFP/+ reporter mice to either ICH surgery using autologous blood or to sham surgery (Sansing et al., 2011b). These mice facilitate classification of the various monocyte populations by CX3CR1 expression (Jung et al., 2000). The cellular infiltrate in the brain was analyzed using flow cytometry. Ly6Chi Inflammatory monocytes constituted the major blood-derived cell population in the brain, peaking at 3 d (Fig. 1A). Notably, the CX3CR1+ Ly6C− monocytes constituted a much smaller population. Inflammatory monocytes, however, comprised 34.9% of leukocytes that trafficked into the brain at day 3, making them the most numerous blood-derived cell type (Fig. 1B). Approximately half of the inflammatory monocytes found in WT brains up to day 7 after ICH were found to be producing the pro-inflammatory cytokine TNF. In comparison, fewer inflammatory monocytes isolated from spleens, a monocyte reservoir (Swirski et al., 2009), were TNF+, indicating that these cells do not produce TNF until they traffic to the site of injury (Fig. 1C). In concordance with our finding of intense inflammatory monocyte infiltration, the two chemokines mainly responsible for recruitment of these monocytes, CCL2 and CCL7 (Tsou et al., 2007), were both increased in perihematomal brain tissue at day 1 and CCL2 remained elevated at day 3 (Fig. 1D).
Both microglia and blood-derived macrophages express common myeloid cell-surface markers, including CD11b and F4/80 (Saijo and Glass, 2011), making histologic differentiation of these populations difficult. To further characterize monocyte recruitment in the brain after ICH, we generated BM chimeric mice. Nucleated BM cells from a mouse ubiquitously expressing GFP under control of the β-actin promoter (Okabe et al., 1997) were transplanted into WT hosts. After 8 weeks, 97.5 ± 0.62% of circulating leukocytes originated from the GFP+ donor marrow (Fig. 2A). Most blood-derived leukocytes isolated from the brains of these mice 3 d after ICH were GFP+ (81.2 ± 13.7%; Fig. 2B, population I). Radio-resistant microglia were found to be CD45low GFP− (population II), consistent with our and others' previous classification of microglia (Renno et al., 1995; Sansing et al., 2011c) suggesting that microglia do not appreciably upregulate CD45 when activated after ICH. Using immunohistochemistry for the myeloid marker CD11b, GFP+ blood-derived myeloid cells were found within and around the hematoma (Fig. 2C). While these cells also appear in the needle track in the brains of sham mice, consistent with focal injury from needle insertion, they were far more abundant and widespread after ICH. These findings confirm that ICH causes robust blood-derived myeloid cell recruitment to the region surrounding the hematoma.
Figure 2.

Blood-derived monocytes are abundant in the brain after ICH. A, WT mice transplanted with BM from a mouse constitutively expressing GFP were used to distinguish between resident microglia and blood-derived leukocytes. At least 8 weeks after transplantation, the vast majority of leukocytes (CD45+ cells) were found to originate from GFP donor cells. B, Three days following ICH, the majority of CD45hi peripheral leukocytes in the brain were GFP+ (population I), rather than GFP− (population III). Microglia were GFP−, CD45low (population II). C, Staining for CD11b (red), GFP (green), and DAPI (blue) shows that many of the globoid CD11b+ cells within the hematoma are also GFP+ (arrows and upper insets), indicating their BM origin. Some GFP− microglia can also be seen (arrowheads and lower insets). Dotted lines in sham indicate localized inflammation due to injury from the needle insertion. D, Ccr2RFP/+ mice 3 d following injury have CD11b+ (green) and CCR2+ (red) inflammatory monocytes present around the hematoma. DAPI is shown in blue. Scale bars: 50 μm.
We next wanted to confirm histologically that the blood-derived myeloid cells in the perihematomal region were indeed inflammatory monocytes. We used Ccr2RFP/+ mice, which express red fluorescence protein under control of the Ccr2 promoter (Saederup et al., 2010). In the brains of these mice 3 d after ICH, we found numerous CCR2+CD11b+ inflammatory monocytes (Fig. 2D). Rarely was a CCR2+ cell observed that was not CD11b+. Some inflammatory monocytes were also found in sham brains, but again they were localized to the needle insertion site. Together these data show that ICH results in robust inflammatory monocyte recruitment to the site of hemorrhage.
Mice lacking inflammatory monocytes are protected from early motor deficits
Because inflammatory monocytes represent the largest leukocyte population recruited to the brain after ICH, we next determined the contribution of these cells to secondary injury. Ccr2−/− mice and littermate controls were subjected to ICH and the inflammatory responses and functional outcomes were quantified using flow cytometry and neurobehavioral testing. Brains from Ccr2−/− mice contained significantly fewer inflammatory monocytes following either blood injection ICH or collagenase ICH (Fig. 3A). Consistent with our histologic data, the majority of CCR2+ leukocytes from control brains were inflammatory monocytes, whereas few CCR2+ T-cells or B-cells were identified (Fig. 3B). While approximately one-third of Ly6C− monocytes isolated from control brains at day 1 stained positive for CCR2, this population expressed low levels of the receptor (mean fluorescence intensity 820 ± 313 A.U. inflammatory monocytes vs 369 ± 207 A.U. Ly6C− monocytes, n = 4, p = 0.043). Functionally, Ccr2−/− mice displayed less severe left forelimb weakness at day 1 (Fig. 3C,D) and could walk farther on the beam balance test at day 3 (Fig. 3E). To account for other potential leukocyte abnormalities in Ccr2−/− mice, we quantified leukocyte levels in the blood. Ccr2−/− mice had similar numbers of B-cells, T-cells, and neutrophils as littermate controls (Fig. 3F). However, Ccr2−/− mice had many fewer inflammatory monocytes in blood and a slight decrease in Ly6C− monocytes (Fig. 3G).
Figure 3.
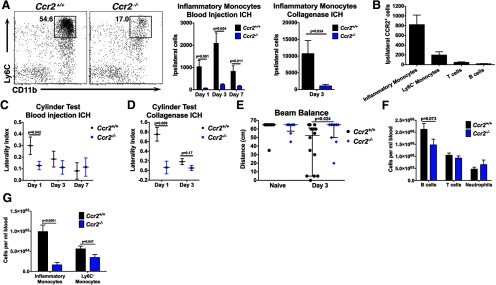
Ccr2−/− mice exhibit decreased inflammatory monocyte recruitment and are protected from early motor deficits. Because Ccr2−/− mice have few circulating inflammatory monocytes, they were used to evaluate the functional and inflammatory effects of monocytes following ICH. A, Inflammatory monocytes are significantly less abundant in Ccr2−/− brains following either blood injection ICH or collagenase ICH. Plots are representative from day 3 blood injection ICH and are gated on CD45hi, CD3−, Ly6G−. N = 7–9 blood injection ICH; n = 3–4 collagenase ICH. B, The majority of CCR2+ cells in ipsilateral control brains are inflammatory monocytes. N = 3–9. C, One day following blood injection ICH, Ccr2−/− mice display a decreased left forelimb deficit. N = 9–15 per group. D, In the collagenase ICH model, Ccr2−/− mice also display a decreased forelimb deficit at day 1. N = 3–5. E, Three days following blood injection ICH Ccr2−/− mice perform better on the beam balance test, indicating that mice with fewer inflammatory monocytes have abrogated secondary injury. Horizontal lines indicate median and bars show interquartile range. N = 8–14. F, Ccr2−/− mice have similar lymphocyte and neutrophil numbers in blood compared with littermate controls. N = 11–17. G, Ccr2−/− mice have many fewer inflammatory monocytes and slightly fewer Ly6C− monocytes in blood. N = 13–17. Error bars in A–D, F, and G indicate SEM.
BM-derived inflammatory monocytes promote early disability following ICH
Microglia (Eltayeb et al., 2007; Zhang et al., 2007) and endothelial cells (Weber et al., 1999) express CCR2 in certain circumstances and Ccr2−/− mice lack CCR2 on these cells as well as monocytes. We sought to determine what source of CCR2 signaling promoted disability after ICH. Ccr2−/− BM chimeras were created by lethally irradiating WT CD45.1 mice and engrafting BM from either WT or Ccr2−/− (both CD45.2+) donors. In the resulting Ccr2−/− BM chimeras, microglia were of CD45.1 (WT) origin and the majority of peripheral CD45+ leukocytes were derived from donor BM (93.5 ± 3.5% WT vs 94.9 ± 0.95% Ccr2−/−, n = 6–8, p = 0.61; Fig. 4A). At 12 h after ICH, Ccr2−/− BM chimeras showed significantly fewer inflammatory monocytes in the brain compared with control chimeras (1949 ± 1779 cells WT vs 74 ± 28 Ccr2−/−, n = 4, p = 0.021; Fig. 4B) but no difference in the numbers of neutrophils (408 ± 334 cells WT vs 358 ± 133 Ccr2−/−, n = 4, p = 0.77), indicating that the neutrophilic response to ICH is unaltered in mice with Ccr2−/− leukocytes. Ccr2−/− BM chimera brains contained similar amounts of hemoglobin relative to controls at day 1 (2.73 ± 1.52 μl hemoglobin WT vs 3.90 ± 2.63 μl Ccr2−/−, n = 8, p = 0.53), confirming equivalent initial hemorrhage volumes between strains. More than half of the monocytes isolated from the brains of WT control chimeras produced TNF at day 3, whereas few found in blood or spleen were TNF+, again confirming the production of TNF by inflammatory monocytes is localized to the site of injury (Fig. 4C). Behaviorally, Ccr2−/− BM chimeras displayed a less severe left hemiparesis compared with controls for the first 3 d after ICH (Fig. 4D). These data demonstrate that blood-derived inflammatory monocytes traffic to the brain and promote acute neurological disability following ICH.
Figure 4.

Blood-derived inflammatory monocytes contribute to secondary disability after ICH. WT CD45.1 mice were lethally irradiated, transplanted with Ccr2−/− BM, and subjected to ICH at least 8 weeks later. A, The majority of leukocytes in the blood of chimeric mice in both genotypes were derived from donor BM. B, Mice given Ccr2−/− BM were found to have fewer inflammatory monocytes in their brains as early as 12 h following ICH. Plots are gated on CD45hi, CD3−, Ly6G−. C, This representative histogram from a chimeric mouse with WT BM shows that inflammatory monocytes isolated from brain 3 d after ICH make TNF, but those found in blood and spleen do not make appreciable levels. N = 5. D, Chimeric mice given Ccr2−/− BM display a decreased left forelimb deficit 1 and 3 d following ICH, suggesting that blood-derived inflammatory monocytes are responsible for inflicting secondary injury following ICH. Both groups improve from day 1 to 3. N = 8–18. Error bars indicate SEM.
To confirm our findings in a WT mouse with normal hematopoietic development, we used the anti-CCR2 antibody MC-21 to deplete circulating CCR2+ cells (Mack et al., 2001; Mildner et al., 2007). Mice were treated with MC-21 or isotype control antibody 1 d before ICH and again immediately following ICH to ensure that CCR2+ monocytes were absent for the duration of the experiment. Significantly fewer inflammatory monocytes were found in blood samples from MC-21 mice (13,179 ± 6934 cells/ml blood control vs 2566 ± 1778 MC-21, n = 5–7, p = 0.0074), but no differences were seen in T-cells (5.33 ± 2.58 × 105 cells/ml blood control vs 6.15 ± 2.84 × 105 cells/ml blood MC-21, n = 5–7, p = 0.68) or neutrophils (4.13 ± 1.02 × 105 cells/ml blood control vs 3.78 ± 1.09 × 105 cells/ml blood MC-21, n = 5–7, p = 0.29). Brains from MC-21-treated mice contained significantly fewer inflammatory monocytes 1 d after ICH, but no change was seen in the numbers of neutrophils (Fig. 5A). Importantly, MC-21-treated mice displayed a significantly reduced left forelimb deficit as assessed by the cylinder test (Fig. 5B). This experiment in a WT mouse strongly suggests that inflammatory monocytes facilitate acute neurological disability.
Figure 5.

MC-21 treatment diminishes the inflammatory monocyte response to ICH. A, WT mice were treated with the anti-CCR2 depletion antibody MC-21 before and after ICH. Fewer CD11b+, Ly6Chi inflammatory monocytes were detected in the brains of MC-21-treated mice, but no differences were seen in neutrophils. The plots are gated on CD45hi, CD3−, Ly6G− cells. N = 5–7. B, Mice treated with MC-21 display a significantly reduced left forelimb deficit 1 d after ICH. N = 7–8. Error bars indicate SEM.
Inflammatory monocytes in the brain change phenotype over time
The most robust cellular recruitment to the brain occurs during the first 3 d after ICH (Fig. 1). Ccr2−/− mice showed a decreased motor deficit compared with WT mice during this time period, but all mice improved over time (Fig. 3). Thus, we were initially puzzled by the increase in brain monocytes between days 1 and 3 and concomitant improvement in functional deficits in the WT mice. To determine the activities of inflammatory monocytes in the brain following ICH, we isolated brains from WT mice up to day 7 and prepared them for flow cytometry. When gating specifically on the Ly6Chi inflammatory monocytes, we found that the proportion of cells staining positive for CCR2 decreased with time after ICH (Fig. 6A). In contrast, the percentage of inflammatory monocytes staining positive for the class II scavenger receptor CD36 increased with time (Fig. 6A,B). CD36 expression is induced by peroxisome proliferator-activated receptor-γ after ICH and is involved in the phagocytosis of red blood cells (Zhao et al., 2007), but has also been implicated in activation of the inflammasome in various sterile injury models (Sheedy et al., 2013). The staining intensity of SIRPα, an initiator of phagocytosis (Han et al., 2012), also increased on inflammatory monocytes in the brain from day 1 to 7 (Fig. 6C). Similarly, CD11c staining intensity increased with time, consistent with the phenotype of regulatory macrophages (Riquelme et al., 2013). Thus we propose that the initial recruitment of the pro-inflammatory monocytes contributes to early disability after ICH. However, over several days the population increases expression of molecules that potentially aid in long-term repair.
Figure 6.

Inflammatory monocytes isolated from ICH brains change phenotype over time. WT mice were subjected to ICH and brains were analyzed by flow cytometry. A, These plots are gated on CD45hi, CD3−, Ly6G−, Ly6Chi inflammatory monocytes isolated at the indicated time points. CCR2 decreases as CD36 increases between day 1 and 7. Numbers indicate the percentage of total cells on each plot represented in each respective quadrant. B, Quantification from the data shown in A indicates that the percentage of CCR2+ inflammatory monocytes decreases as the percentage of CD36+ inflammatory monocytes increases. Graphs depict mean ± SD. N = 3–6. C, The mean fluorescence intensity (MFI) of SIRPα and CD11c on inflammatory monocytes increases from day 1 to 7. Graphs depict mean ± SD. N = 3–6.
High serum CCL2 levels are associated with worse early outcomes in patients with ICH
To determine whether our findings are relevant in human ICH patients, we measured serum CCL2 levels in a prospective cohort of ICH patients enrolled from two centers. Functional outcome was quantified using the mRS, which has been extensively validated in stroke outcome studies (Rankin, 1957; Banks and Marotta, 2007). The mRS is a seven point ordinal scale in which 0 indicates full recovery with no residual symptoms and 6 indicates death. The median age of patients was 67.1 [interquartile range 57.8–76.1] years, 53.5% were male, and median ICH volume was 14.1 [5–38.5] ml. The median CCL2 level 24 h after ICH was 320 [226–478] pg/ml serum. CCL2 levels did not correlate with already established predictors of outcome after ICH, supporting its value as an independent biomarker of prognosis. Indeed, we found no correlation between CCL2 levels and patient age (r2 = 0.001, p = 0.78) or volume of ICH (r2 = 0.01, p = 0.38). There was also no association between CCL2 levels and premorbid functional status (p = 0.19) or extension of the hemorrhage into the ventricular system (p = 0.45) in univariate analysis. Using a multivariable model to adjust for known predictors of outcome after ICH, high CCL2 levels were independently associated with higher mRS scores at day 7 (Table 1). Thus, patients with higher levels of the principal chemokine for CCR2+ monocyte recruitment were more likely to die or be severely disabled, suggesting that activation of the CCL2–CCR2 axis worsens acute injury in patients with ICH.
Table 1.
Serum CCL2 levels are independently associated with functional outcome 7 d after ICH in patients
| Odds ratio | 95% CI | p | |
|---|---|---|---|
| Age, per year | 1.0 | 0.97–1.04 | 0.903 |
| ICH volume, per 10 ml | 1.6 | 1.3–2.0 | ***<0.001 |
| Intraventricular extension | 2.4 | 1.0–5.9 | *0.047 |
| CCL2 at 24 h, per 100 pg/ml serum | 1.2 | 1.02–1.4 | *0.032 |
An ordered logistic regression was performed factoring in age, ICH volume, intraventricular extension, and CCL2 level versus day 7 mRS scores. These data show that higher day 1 CCL2 levels are independently associated with worse mRS scores one week later. N = 85 patients. CI, confidence interval.
Discussion
The specific cellular mediators of brain injury after ICH are largely unknown due to traditional grouping of microglia and blood-derived monocytes as a single cell type. Here, we show for the first time that blood-derived inflammatory monocytes are present in highest numbers throughout the initial inflammatory period, underscoring the importance of understanding their contribution to acute disability. We also show that these monocytes produce TNF once they have trafficked into the perihematomal brain. Further, using both genetic knock-outs and BM chimeras to reduce the circulating inflammatory monocyte pool while maintaining WT microglia and endothelial cells, we determined that these monocytes promote neurological disability in the first days after ICH. Notably, we showed in two distinct models of ICH that Ccr2−/− mice have decreased functional deficits and fewer recruited monocytes. These findings were supported by studies that used immunodepletion of CCR2+ cells, which confirmed the early behavioral benefit of loss of CCR2+ circulating cells in WT mice. Finally, we confirmed the potential therapeutic relevance of these findings in a human cohort. Higher levels of CCL2, the principle chemokine for monocyte chemotaxis, at 24 h after ICH were independently associated with worse functional disability at day 7.
It was recently shown that inflammatory monocytes are essential for recovery from bacterial infections in mice (Dunay et al., 2010; Shi et al., 2011). However, others have shown that these cells initiate secondary injury in models of ischemic stroke, myocardial infarction, and traumatic brain injury (Dimitrijevic et al., 2007; Semple et al., 2010; Leuschner et al., 2011). Results from these studies suggest inflammatory monocytes, while necessary during infection, may cause additional damage following sterile injury. However, this paradigm has not been consistent in all models. Depletion of inflammatory monocytes during spinal cord injury results in larger lesion sizes and worsened functional outcomes (Shechter et al., 2009). Similarly, inflammatory monocytes prevent hemorrhagic transformation and neurological deterioration in a model of ischemic stroke (Gliem et al., 2012). These findings highlight the many possible contributions inflammatory monocytes offer in diverse injury models and at various times following injury. In the present study, inflammatory monocytes consistently exacerbated early disability after ICH in two distinct models. The long-term function of these cells requires further study.
Our finding of early protection in Ccr2−/− mice is attributed to a reduction in CCR2+ inflammatory monocytes. However, some endothelial cells (Dzenko et al., 2001) and a small population of T-cells also express CCR2 (Brühl et al., 2004). Our interpretation that the inflammatory monocytes are responsible for acute disability is supported by our findings in Ccr2−/− BM chimeras, which have WT microglia and endothelial cells but demonstrated similar inflammatory and neurobehavioral improvements as Ccr2−/− mice. Many studies have shown that endothelial cells are relatively radio-resistant to the irradiation doses used to generate BM chimeras (Kapessidou et al., 2006; Perry et al., 2009; Azcutia et al., 2012; Fukuhara et al., 2012; Kamei et al., 2012; Flynn et al., 2013; Wood et al., 2013), supporting Ccr2−/− BM chimeras as a useful tool to localize the CCR2 effect to leukocytes. Similarly, acutely depleting circulating CCR2+ cells in WT mice also confers significant protection from neurological disability. CCR2+ T-cells make up ∼5% of T-cells in the blood, making it possible that differential T-cell responses contribute to our findings in Ccr2−/− mice and Ccr2−/− BM chimeras. However, we think this is unlikely since there are on average <50 CCR2+ T-cells in the brain at day 1 and T-cells typically take much longer to form an adaptive response. Thus, while it is possible that a small population of other CCR2+ cells contributed to our findings, it seems most likely that the CCR2+ inflammatory monocytes are responsible for instigating neurological disability following ICH.
The inflammatory monocytes are found in highest numbers at day 3 in control mice, a time point at which they seem to be recovering in the cylinder test. This suggests that CCR2+ inflammatory monocytes are not consistently injurious over time. Indeed, we provide evidence of a changing inflammatory monocyte phenotype from day 1 to 7. In addition to decreasing CCR2 levels, inflammatory monocytes upregulate CD36 and SIRPα, two molecules involved in phagocytosis. Thus, it is possible that these monocytes shift toward an alternative phenotype involved in clearing hemorrhagic debris, rather than continuing to worsen injury over time. If inflammatory monocytes are important for recovery, then perhaps the best therapeutic strategy would be to inhibit the early response or acutely modulate these cells such that they promote recovery. Further studies are needed to fully understand the long-term influences of inflammatory monocytes on chronic ICH recovery.
Our work builds on a recent study by another group examining the CCR2-CCL2 system in a model of collagenase-induced ICH (Yao and Tsirka, 2012). Their work identified plasmin-mediated truncation of CCL2 as critical to the chemokine function. Interestingly, Ccr2−/− mice exhibited delayed hemorrhage expansion and delayed hematoma clearance in this model, suggesting a role for CCR2 in vascular integrity and recovery. The delay in ICH clearance was associated with enhanced inflammation and neuronal injury 7 d after ICH. Although Ccr2−/− mice displayed a behavioral benefit at early time points in that study, the day 1 hemorrhages were smaller in this cohort hindering the interpretation of neurobehavioral differences between the genotypes. The present study primarily used the blood injection model to standardize hemorrhage sizes. Similar to our findings, microglia/macrophage numbers were reduced at early time points after ICH. Our work builds on this study by discriminating between microglia and monocytes, allowing us to identify the peripheral monocyte population as trafficking into the brain and facilitating early neurological disability in a model with consistent hematoma size. Identifying which CCR2+ cell populations are involved in long-term hematoma clearance and recovery warrants further study.
The brain has limited recovery capacity. Preventing additional injury during the acute phase may provide long-term benefit. However, many studies on neuroprotection after stroke have failed to translate into improved patient care. One potential reason is pathophysiologic differences between rodent models of injury and human disease. To determine whether the CCL2-CCR2 pathway is relevant in patients, we not only used two distinct ICH models experimentally, but we also investigated activation of this pathway in the serum of patients with ICH. Importantly, we found elevations in CCL2 levels in ICH patients who had poor functional outcomes at 7 d. CCL2 levels did not merely correlate with initial measures of ICH severity. Rather, early elevation in CCL2 was independently associated with poor outcome 1 week later, after adjusting for relevant potential confounding patient variables.
In summary, our work demonstrates that inflammatory monocytes contribute to acute neurological disability after ICH in two murine models and that this pathway has importance in human ICH patients. Our findings suggest that antagonizing or modulating this early target has the potential to reduce neurological disability after ICH.
Footnotes
This work was supported by National Institutes of Health grants 1F31NS083231 (M.D.H.), 5T32AI7080 (M.D.H.), and 1K08NS078110 (L.H.S.); an American Heart Association predoctoral fellowship (M.D.H.); a University of Connecticut Gala Funds Junior Pilot Award (L.H.S.); a Hartford Hospital Medical Staff Award; and the Marlene L. Cohen and Jerome H. Fleisch Scholar Grant (L.H.S.). We thank all those who contributed to the clinical study, including Lori Capozzi, Brett Cucchiara, Alexandra Czap, Sharon DiMauro, Inam Kureshi, Joshua Levine, Jeanie Luciano, Steven Messe, Larami MacKenzie, James McKinney, Lena O'Keefe, Swaroop Pawar, Jonathan Raser, David Z. Rose, Igor Rybinnik, Neelofer Shafi, Benjamin Scott, Sami Tarabishy, Linda Wendell, Eugene Yu, and Neer Zeevi. We would also like to thank patients and their families who participated in the study.
The authors declare no competing financial interests.
References
- Agnihotri S, Czap A, Staff I, Fortunato G, McCullough LD. Peripheral leukocyte counts and outcomes after intracerebral hemorrhage. J Neuroinflammation. 2011;8:160. doi: 10.1186/1742-2094-8-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcutia V, Stefanidakis M, Tsuboi N, Mayadas T, Croce KJ, Fukuda D, Aikawa M, Newton G, Luscinskas FW. Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J Immunol. 2012;189:2553–2562. doi: 10.4049/jimmunol.1103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke. 2007;38:1091–1096. doi: 10.1161/01.STR.0000258355.23810.c6. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brühl H, Cihak J, Schneider MA, Plachý J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlöndorff D, Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- Castillo J, Dávalos A, Alvarez-Sabin J, Pumar JM, Leira R, Silva Y, Montaner J, Kase CS. Molecular signatures of brain injury after intracerebral hemorrhage. Neurology. 2002;58:624–629. doi: 10.1212/WNL.58.4.624. [DOI] [PubMed] [Google Scholar]
- Choudhri TF, Hoh BL, Solomon RA, Connolly ES, Jr, Pinsky DJ. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997;28:2296–2302. doi: 10.1161/01.STR.28.11.2296. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- Dunay IR, Fuchs A, Sibley LD. Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect Immun. 2010;78:1564–1570. doi: 10.1128/IAI.00472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzenko KA, Andjelkovic AV, Kuziel WA, Pachter JS. The chemokine receptor CCR2 mediates the binding and internalization of monocyte chemoattractant protein-1 along brain microvessels. J Neurosci. 2001;21:9214–9223. doi: 10.1523/JNEUROSCI.21-23-09214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke. 2002;33:2334–2335. doi: 10.1161/01.STR.0000027211.73567.FA. [DOI] [PubMed] [Google Scholar]
- Eltayeb S, Berg AL, Lassmann H, Wallström E, Nilsson M, Olsson T, Ericsson-Dahlstrand A, Sunnemark D. Temporal expression and cellular origin of CC chemokine receptors CCR1, CCR2 and CCR5 in the central nervous system: insight into mechanisms of MOG-induced EAE. J Neuroinflammation. 2007;4:14. doi: 10.1186/1742-2094-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty ML, Haverbusch M, Sekar P, Kissela B, Kleindorfer D, Moomaw CJ, Sauerbeck L, Schneider A, Broderick JP, Woo D. Long-term mortality after intracerebral hemorrhage. Neurology. 2006;66:1182–1186. doi: 10.1212/01.wnl.0000208400.08722.7c. [DOI] [PubMed] [Google Scholar]
- Flynn KM, Michaud M, Madri JA. CD44 deficiency contributes to enhanced experimental autoimmune encephalomyelitis: a role in immune cells and vascular cells of the blood-brain barrier. Am J Pathol. 2013;182:1322–1336. doi: 10.1016/j.ajpath.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, Uemura A, Kiyonari H, Abe T, Fukamizu A, Hirashima M, Sawa H, Aoki J, Ishii M, Mochizuki N. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122:1416–1426. doi: 10.1172/JCI60746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71:743–752. doi: 10.1002/ana.23529. [DOI] [PubMed] [Google Scholar]
- Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, Koo LY, Brenchley JM, Fraser ID, Belkaid Y. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med. 2013;19:713–721. doi: 10.1038/nm.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond MD, Ai Y, Sansing LH. Gr1+ macrophages and dendritic cells dominate the inflammatory infiltrate 12 hours after experimental intracerebral hemorrhage. Transl Stroke Res. 2012;3:s125–s131. doi: 10.1007/s12975-012-0174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Lundgren DH, Jaiswal S, Chao M, Graham KL, Garris CS, Axtell RC, Ho PP, Lock CB, Woodard JI, Brownell SE, Zoudilova M, Hunt JF, Baranzini SE, Butcher EC, Raine CS, Sobel RA, Han DK, Weissman I, Steinman L. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J Exp Med. 2012;209:1325–1334. doi: 10.1084/jem.20101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei N, Kwon SM, Kawamoto A, Ii M, Ishikawa M, Ochi M, Asahara T. Contribution of bone marrow-derived endothelial progenitor cells to neovascularization and astrogliosis following spinal cord injury. J Neurosci Res. 2012;90:2281–2292. doi: 10.1002/jnr.23113. [DOI] [PubMed] [Google Scholar]
- Kapessidou Y, Habran C, Buonocore S, Flamand V, Barvais L, Goldman M, Braun MY. The replacement of graft endothelium by recipient-type cells conditions allograft rejection mediated by indirect pathway CD4(+) T cells. Transplantation. 2006;82:582–591. doi: 10.1097/01.tp.0000184444.93108.d1. [DOI] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KR, Betz AL, Kim S, Keep RF, Hoff JT. The role of the coagulation cascade in brain edema formation after intracerebral hemorrhage. Acta Neurochir. 1996;138:396–401. doi: 10.1007/BF01420301. [DOI] [PubMed] [Google Scholar]
- Lei C, Lin S, Zhang C, Tao W, Dong W, Hao Z, Liu M, Wu B. High-mobility group box1 protein promotes neuroinflammation after intracerebral hemorrhage in rats. Neuroscience. 2013;228:190–199. doi: 10.1016/j.neuroscience.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachý J, Brühl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlöndorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Mack WJ, Ducruet AF, Hickman ZL, Garrett MC, Albert EJ, Kellner CP, Mocco J, Connolly ES., Jr Early plasma complement C3a levels correlate with functional outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;61:255–261. doi: 10.1227/01.NEU.0000255518.96837.8E. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/S0014-5793(97)00313-X. [DOI] [PubMed] [Google Scholar]
- Perry TE, Song M, Despres DJ, Kim SM, San H, Yu ZX, Raghavachari N, Schnermann J, Cannon RO, 3rd, Orlic D. Bone marrow-derived cells do not repair endothelium in a mouse model of chronic endothelial cell dysfunction. Cardiovasc Res. 2009;84:317–325. doi: 10.1093/cvr/cvp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin J. Cerebral vascular accidents in patients over the age of 60. II. Prognosis. Scott Med J. 1957;2:200–215. doi: 10.1177/003693305700200504. [DOI] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-a expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Riquelme P, Tomiuk S, Kammler A, Fändrich F, Schlitt HJ, Geissler EK, Hutchinson JA. IFN-gamma-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol Ther. 2013;21:409–422. doi: 10.1038/mt.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–807. doi: 10.1161/01.STR.21.5.801. [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Sansing LH, Harris TH, Kasner SE, Hunter CA, Kariko K. Neutrophil depletion diminishes monocyte infiltration and improves functional outcome after experimental intracerebral hemorrhage. Acta Neurochir. 2011a;(Suppl 111):173–178. doi: 10.1007/978-3-7091-0693-8_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansing LH, Kasner SE, McCullough L, Agarwal P, Welsh FA, Kariko K. Autologous blood injection to model spontaneous intracerebral hemorrhage in mice. J Vis Exp. 2011b;pii:2618. doi: 10.3791/2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. 2011c;70:646–656. doi: 10.1002/ana.22528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J Cereb Blood Flow Metab. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Hohl TM, Leiner I, Equinda MJ, Fan X, Pamer EG. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J Immunol. 2011;187:5293–5298. doi: 10.4049/jimmunol.1101721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Dávalos A. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke. 2005;36:86–91. doi: 10.1161/01.STR.0000149615.51204.0b. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KS, Nelson PJ, Gröne HJ, Weber C. Expression of CCR2 by endothelial cells: implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arteriosclerosis, thrombosis, and vascular biology. 1999;19:2085–2093. doi: 10.1161/01.ATV.19.9.2085. [DOI] [PubMed] [Google Scholar]
- Wood KC, Cortese-Krott MM, Kovacic JC, Noguchi A, Liu VB, Wang X, Raghavachari N, Boehm M, Kato GJ, Kelm M, Gladwin MT. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb Vasc Biol. 2013;33:1861–1871. doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. The CCL2-CCR2 system affects the progression and clearance of intracerebral hemorrhage. Glia. 2012;60:908–918. doi: 10.1002/glia.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zausinger S, Hungerhuber E, Baethmann A, Reulen H, Schmid-Elsaesser R. Neurological impairment in rats after transient middle cerebral artery occlusion: a comparative study under various treatment paradigms. Brain Res. 2000;863:94–105. doi: 10.1016/S0006-8993(00)02100-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Xiong KL, Lin S, Zhong Q, Lu FL, Liang H, Li JC, Wang JZ, Yang QW. Elevation of high-mobility group protein box-1 in serum correlates with severity of acute intracerebral hemorrhage. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/142458. [DOI] [PMC free article] [PubMed] [Google Scholar]