

Abstract
Cognitive impairment is a quintessential feature of Alzheimer's disease (AD) and AD mouse models. The peroxisome proliferator-activated receptor-γ (PPARγ) agonist rosiglitazone improves hippocampus-dependent cognitive deficits in some AD patients and ameliorates deficits in the Tg2576 mouse model for AD amyloidosis. Tg2576 cognitive enhancement occurs through the induction of a gene and protein expression profile reflecting convergence of the PPARγ signaling axis and the extracellular signal-regulated protein kinase (ERK) cascade, a critical mediator of memory consolidation. We therefore tested whether PPARγ and ERK associated in protein complexes that subserve cognitive enhancement through PPARγ agonism. Coimmunoprecipitation of hippocampal extracts revealed that PPARγ and activated, phosphorylated ERK (pERK) associated in Tg2576 in vivo, and that PPARγ agonism facilitated recruitment of PPARγ to pERK during memory consolidation. Furthermore, the amount of PPARγ recruited to pERK correlated with the cognitive reserve in humans with AD and in Tg2576. Our findings implicate a previously unidentified PPARγ–pERK complex that provides a molecular mechanism for the convergence of these pathways during cognitive enhancement, thereby offering new targets for therapeutic development in AD.
Keywords: Alzheimer's, hippocampus, in vitro reconstitution, protein complex, transgenic
Introduction
Alzheimer's disease (AD) is a debilitating neurodegenerative disorder that manifests as cognitive impairment and brings with it a tremendous economic and social burden, as well as a tragic prognosis for increasing incidence in a burgeoning aging population (Thies and Bleiler, 2013). Many studies have suggested that a key causative factor in AD dementia is amyloid β (Aβ) derived from the amyloid precursor protein (Hsiao et al., 1996; Westerman et al., 2002; Sperling et al., 2011). Prompted by the realization that insulin resistance is another recognized risk factor in AD (van Himbergen et al., 2012) and that insulin resistance is a comorbidity factor in both diabetes and AD (Talbot et al., 2012), many studies have investigated insulin sensitizer therapies as therapeutics for AD (Craft, 2012). A popular target is the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ), a validated therapeutic target in type 2 diabetes, which regulates the expression of many genes critical to insulin sensitivity (Wu et al., 1999) as well as components of Aβ metabolism and toxicity (Mandrekar-Colucci and Landreth, 2011; Mandrekar-Colucci et al., 2012). While many large-scale clinical trials for dementia due to AD failed to show efficacy of PPARγ agonism, evolving consensus considers their ineffectiveness likely to be due to testing in late-stage disease, a fate similar to many other AD drug candidates (Becker and Greig, 2013). In contrast, clinical trials performed with patients having mild cognitive impairment obtained positive outcomes using insulin sensitizers (Stockhorst et al., 2004; Watson et al., 2005; Risner et al., 2006; Sato et al., 2011). Thus, before overt neurodegeneration, insulin sensitizers may impinge upon signaling axes to modulate memory in early AD (Watson and Craft, 2004; Craft et al., 2012).
It is established that PPARγ agonism enhances cognition in AD animal models (Pedersen et al., 2006; Jiang et al., 2008; Landreth et al., 2008; Escribano et al., 2009; Rodriguez-Rivera et al., 2011), and that extracellular signal-regulated protein kinase (ERK) is essential for several forms of hippocampus-dependent learning and memory that are impaired in AD (Dineley et al., 2002, 2007; Hamann et al., 2002; Westerman et al., 2002; Sweatt, 2004; Hort et al., 2007; Hoefer et al., 2008). Our work using the PPARγ agonist rosiglitazone (RSG) to enhance cognition in the Tg2576 mouse model of AD demonstrated convergence between the hippocampal PPARγ and ERK signaling pathways (Denner et al., 2012). Since proper ERK2 activity is a requisite for hippocampus-dependent learning and memory in rodents (Atkins et al., 1998; Selcher et al., 2001), we speculated that PPARγ may serve to rein in dysregulated ERK2 to enhance hippocampal cognition. Here we show that RSG cognitive enhancement leads to increased recruitment of PPARγ to activated, phosphorylated ERK (pERK) in a multiprotein complex during memory consolidation for a hippocampus-dependent cognitive task. Acute inhibition of hippocampal PPARγ, which blocks this type of memory consolidation, also prevented the increased recruitment of PPARγ to pERK, suggesting that formation of this protein complex is requisite for memory formation. We also show that these complexes correlate with cognitive reserve in human AD and AD model animals. Further, we demonstrate the ability to reconstitute the PPARγ–pERK association using in vitro recombinant protein pull-down assays, revealing that these two proteins have intrinsic properties for direct association.
Materials and Methods
Animals.
Tg2576 mice were bred in the University of Texas Medical Branch at Galveston (UTMB) animal care facility by mating hemizygous Tg2576 (Hsiao et al., 1996) males with B6SJL/F1J females (stock #100012, Jackson Laboratory). Mice were housed, n ≤ 5 per cage, with food and water ad libitum. UTMB operates in compliance with the US Department of Agriculture Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and Institutional Animal Care and Use Committee (IACUC)-approved protocols. Genotyping services were outsourced (Transnetyx), and genotypes were determined from tail clip biopsy specimens obtained at weaning; when mice were killed.
Rosiglitazone treatment.
Eight-month-old male and female Tg2576 and WT littermates were fed a control diet or a 30 mg/kg RSG diet (Bio-Serv) for 30 d, as previously described (Rodriguez-Rivera et al., 2011). Mouse food intake and body weights were monitored during the 30 d period, and no significant differences were observed by genotype or treatment group (data not shown). Additionally, RSG treatment did not confer any notable side effects, and age-related animal mortality rates were similar between groups. Animals were randomly assigned to receive control or RSG feed, and sample sizes were balanced by sex and genotype. Experimenters were blinded to treatment groups during key data acquisition and analysis steps.
Antibodies.
Antibodies to pERK (1:1000; catalog #9101), ERK (1:1000; catalog #9102), MEK1/2 (dual-specificity mitogen-activated protein kinase kinase 1/2; 1:1000; catalog #9122), RSK (1:1000; catalog #9333), anti-rabbit HRP secondary (1:20,000; catalog #7074), and anti-pERK-conjugated Sepharose bead slurry (catalog #3510) were from Cell Signaling Technology. Additional reagents included anti-PPARγ (1:500; catalog #07-466, Millipore) and anti-PPARγ-conjugated magnetic bead slurry (custom preparation, Affinity Life Sciences).
Fear conditioning.
Behavioral experiments were performed during the lights-on phase (6:00 A.M. to 6:00 P.M.) in the UTMB Rodent In Vivo Assessment Core (directed by K.T.D.) within the UTMB Center for Addiction Research (directed by Dr. Kathryn Cunningham). Based upon power analyses of previous data, 10 (WT) to 20 (Tg2576) mice per group (male and female) were subjected to our standard two-pairing fear conditioning (FC) training protocol as previously described (Dineley et al., 2002; Taglialatela et al., 2009; Rodriguez-Rivera et al., 2011; Denner et al., 2012). No statistical differences in performance or response to drug treatment were measured between male and female mice; therefore, the sex groups were collapsed for data reporting. All experimental groups were balanced to include approximately equivalent numbers of each gender. Twenty-four hours after training, hippocampus-dependent contextual learning was assessed by quantifying freezing behavior when the animals were placed back into the training chamber. Freezing behavior was analyzed using automated software (FreezeFrame/View, Actimetrics) from digitally recorded videos (Actimetrics).
Contextual FC is amenable to the testing of manipulations hypothesized to disrupt memory consolidation (e.g., GW9662) as FC training is achieved in a single training session as opposed to those cognitive tasks that require repeated training sessions (e.g., Morris water maze; Westerman et al., 2002). We previously established that Tg2576 mice, either untreated or treated with RSG with or without GW9662, have intact perception as they exhibit similar shock threshold to WT animals, and also freeze comparably to WT animals in response to shock during training (Rodriguez-Rivera et al., 2011; Denner et al., 2012). In the present study, mice were not subjected to the hippocampus-independent cued memory test since our previous work has determined that Tg2576 mice do not exhibit a deficit in this task compared with WT mice, and PPARγ agonism does not affect the performance of either genotype (Dineley et al., 2002; Rodriguez-Rivera et al., 2011; Denner et al., 2012).
Under deep anesthesia [1 ml of Avertin (Analytical 90710, Fluka) working solution (125 μl; 1.0 g of Avertin/ml tert-amyl-alcohol plus 9.88 ml of 0.9% NaCl)], animals were killed by transcardial perfusion with ice-cold PBS containing protease and phosphatase inhibitors [P8340 protease inhibitor cocktail (30 mm NaF, 10 mm Na3VO4, 1 mm PMSF, added fresh to perfusion buffer every 30 min); Sigma-Aldrich]. This is in contrast to previous work wherein animals were killed via decapitation without perfusion (Rodriguez-Rivera et al., 2011). Whole brains were extracted, and hippocampi were dissected in ice-cold saline (110 mm sucrose, 60 mm NaCl, 3 mm KCl, 1.25 mm sodium phosphate monobasic monohydrate, 28 mm sodium bicarbonate, 5 mm d-glucose, 1 mm l-ascorbic acid, 1 mm MgCl2, 1 mm CaCl2). All samples were frozen on dry ice and stored at −80°C until use.
Intracerebroventricular injection.
The PPARγ antagonist GW9662 (Sigma-Aldrich) and vehicle (1% dimethylsulfoxide) were directly infused into the lateral ventricles using a modified free-hand method (Clark et al., 1968; Taglialatela et al., 2009; Denner et al., 2012). Using an aseptic technique, mice were anesthetized (isoflurane, 1–4%) and the skull was exposed with a small incision along the midline to locate bregma (Paxinos et al., 1985). A 26 gauge needle was inserted 3 mm deep at 1 mm anterior and 1 mm lateral to bregma. GW9662 (32.5 pmol) or vehicle were delivered by an electronic programmable microinfuser (Harvard Apparatus) at 3 μl/min for 1 min. Doses and delivery rates were determined based on previous work using GW9662 to antagonize PPARγ function in the CNS (Bjorklund et al., 2012; Denner et al., 2012). Following infusion, the needle was stabilized for 1 min to ensure complete delivery and prevent reflux. No mice exhibited evidence of misplaced injections or brain hemorrhage. Following suturing, pain and local inflammation were attenuated via application of a topical NSAID (Neosporin) containing neomycin, bacitracin, and Polymyxin B antibiotics according to the IACUC-approved protocol. Intracerebroventricular injections were administered 4 h before FC training, and 8 h before killing of the animal and tissue harvest. This time point was chosen based on our previous work demonstrating that the GW9662 peak effect on PPARγ was 8 h postinjection (Denner et al., 2012), which also corresponds to the timeframe for hippocampal ERK-mediated memory consolidation (McGaugh, 2000; Trifilieff et al., 2007).
Protein extraction.
Nuclear extracts were isolated from hippocampi at 4°C using the Active Motif Nuclear Extract Kit (catalog #40010) then stored at −80°C. The resultant extracts were composed of nuclei (nuclear) and a separate fraction composed of the remaining cellular components (non-nuclear). Total protein concentrations in extracts were determined using a BCA protein assay kit (catalog #23225, Thermo Scientific).
Quantitative immunoprecipitation.
Hippocampal extracts were thawed on ice, and 200 μg (nuclear) or 500 μg (non-nuclear) of protein was suspended in 500 μl of extract buffer (25 mm HEPES, 0.1% Triton X-100, 10% glycerol), supplemented with 0.02 m Sigma protease inhibitor cocktail (P8340), 0.02 m NaF, and 0.02 m Na3VO4. Ten microliters of anti-pERK-conjugated Sepharose bead slurry (catalog #3510, Cell Signaling Technology) or 10 μl of anti-PPARγ-conjugated magnetic bead slurry (Affinity Life Sciences) was added, and this mixture was allowed to incubate in a rotating shaker at 4°C for ∼18 h. All remaining steps were performed at 4°C, unless otherwise noted. Following incubation, Sepharose bead samples were pelleted by centrifugation (14,000 × g for 1 min) or magnetic samples were isolated using a magnetic stand; in each case, the supernatant was then removed. The pelleted beads were washed by resuspension in extract buffer for 20 min then centrifuged (14,000 × g for 1 min) or placed in the magnetic stand to isolate washed beads. Bead wash was repeated four times. Protein was eluted in 30 μl of 2× Laemmli sample buffer (20% SDS, 20% glycerol, 1 m Tris, 5% β-mercaptoethanol, 8 m urea, double-distilled H2O, bromophenol blue) and incubated for 5 min at 95–100°C. One final bead pelleting step was performed to avoid loading beads onto SDS-PAGE gels.
Quantitative immunoblot following immunoprecipitation.
Extracted proteins were resolved by SDS-PAGE (Bio-Rad, 7.5% Mini-PROTEAN TGX) and electroblotted onto nitrocellulose. To quantitatively compare between immunoblot film bands, a crude whole-brain lysate (20 μg/well) prepared from a homogenate of ∼40 C57BL/6J brains was included in triplicate on each gel as an internal standard (further described below).
Following electrophoresis and transfer, each membrane was blocked (2% Advanced ECL blocking solution, GE Healthcare), and incubated with primary and secondary antibodies. Samples were visualized via chemiluminescence using the GE Healthcare ECL Western blotting reagent system, according to the manufacturer's instructions. Exposure to GE Healthcare Hyperfilm ECL was performed to obtain band intensities within the linear range of the antibody combinations used.
Immunoblot membranes were scanned at 300 dpi, and numeric band density and background values were acquired using ImageJ software [National Institutes of Health (NIH)]. The numeric values for the loading control (LC) protein from each of the three identical C57BL/6J internal standards was averaged (LC), and all other samples (e.g., PPARγ or pERK2 from the immunoprecipitation) were normalized to the LC average. The PPARγ value in our control sample was chosen as a normalization value to remain in the linear range of the samples we were investigating. Because immunoprecipitates (IPs) were loaded with 200 or 500 μg of protein, a standard loading control such as actin would have generated a signal too intense to accurately quantify the PPARγ and pERK that is present. Thus, this method allows for more precise quantitative comparison between different gels and across different experiments. After normalizing PPARγ and pERK2 protein density values for each sample, the amount of PPARγ that co-immunoprecipitated with pERK was determined by taking the ratio of normalized PPARγ and normalized pERK2. This step corrected for any variation in immunoprecipitation efficiency. Thus, the final value represents the relative amount of PPARγ that is associated with pERK2 in a given sample normalized to a low-abundance protein whose expression level is not subject to the effects of the pharmacological manipulations used. Our quantification and normalization procedure was calculated as follows:
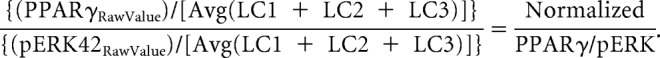 |
An example of this procedure can be found in Figure 2A. Quadruplicate runs on six individual animals using this approach yielded a coefficient of variation between 1% and 4.8% for the four PPARγ/pERK ratios calculated, thus demonstrating the reproducibility and accuracy of immunoprecipitation (see Fig. 2B).
Figure 2.

Quantification method to determine PPARγ/pERK2 ratios. A, Shown is an example Western blot for PPARγ and pERK in pERK IPs from four individual mice. For quantification across multiple immunoblots of IP material, a homogenate prepared from pooled brains from C57BL/6J mice was used as a LC and was resolved in triplicate (lanes 1, 4, and 7) on each SDS-PAGE gel. pERK IPs from four individual mice (lanes 2, 3, 5, and 6) are depicted. For the data described herein, immunoblots for PPARγ or pERK2 from the IPs were normalized relative to the LC. PPARγ in the LC lanes was chosen as the normalization protein because it tracked in the linear range with immunoprecipitated PPARγ and pERK2 for their respective exposures. After acquiring normalized values for immunoprecipitated PPARγ and pERK2 proteins for each individual animal's hippocampal extract, the amount of PPARγ that coimmunoprecipitates with pERK was calculated by taking the ratio of normalized PPARγ to normalized pERK2. In the example above, Mouse 2 has a hippocampal PPARγ/pERK2 ratio of 0.658. B, PPARγ/pERK2 ratios are highly reproducible. Western blots of PPARγ and pERK in four independent pERK IPs from six individual animals (lanes 2, 3, 4, 6, 7, and 8) and the triplicate LC (lanes 1, 5, 9) resolved by four separate gels. The PPARγ/pERK2 ratios were calculated as in Figure 2A, and the coefficient of variation for each individual animal was determined. All replicate IPs yielded a coefficient of variation of ≤4.8%.
Recombinant protein and in vitro GST pull-down assay.
In vitro recombinant protein association studies were performed using Pierce glutathione agarose beads (catalog #16100, Thermo Scientific), recombinant human PPARγ (catalog #RCP9207, Randox Life Sciences), GST-tagged (N-terminal) recombinant human active ERK2 (catalog #1230-KS, R&D Systems), and GST-tagged (N-terminal) recombinant human ERK2 (catalog #10030-H09B, Sino Biological). All steps were performed at 4°C, unless otherwise specified. Glutathione beads were suspended in 250 μl 1× TBS (0.02 m Tris, 0.14 m NaCl) and incubated with 100 ng of recombinant PPARγ and 100 ng of either recombinant GST-pERK or GST-ERK protein on a rocker overnight. Controls were prepared to include all possible combinations of glutathione beads and recombinant proteins (see Fig. 5). Beads were pelleted by centrifugation (700 × g for 2 min), and the supernatant was removed. Samples were washed (4 × 1 min) in 1× TBS, followed by centrifugation (700 × g for 2 min) to pellet beads. Bound proteins were eluted with 2× sample buffer (30% glycerol, 2% SDS, 62.5 mm Tris, pH 6.8, bromophenol blue) and heated for 5 min at 95–100°C.
Figure 5.

PPARγ and pERK recombinant proteins associate in vitro. A, Western blot for pERK (top) and PPARγ (bottom) following incubation of recombinant human GST-pERK2 with increasing amounts of human PPARγ followed by glutathione bead affinity isolation. B, Input–output relationship for PPARγ pulldown from GST-pERK2 IP. Dotted lines represent 95% confidence intervals. C, Western blot for nonphosphorylated ERK (top) and PPARγ (bottom) following incubation of recombinant GST-nonphosphorylated ERK2 with increasing amounts of human PPARγ followed by glutathione bead affinity isolation.
Human brain tissue.
Frozen human cortex was acquired from the Oregon Brain Bank at Oregon Health and Science University (OHSU) in Portland, OR, as previously described (Bjorklund et al., 2012). Briefly, all donor subjects were enrolled and evaluated in studies at the NIH-sponsored C. Rex and Ruth H. Layton Aging Alzheimer's Disease Center at OHSU. Subjects were evaluated for neurological and neuropsychological competency annually and subsequently assigned a clinical dementia rating (CDR) by an experienced clinician. AD subjects were diagnosed by a clinical team consensus conference, met National Institute for Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorder Association diagnostic criteria for clinical AD, had a CDR > 1.0, and had AD status confirmed at autopsy following informed consent. All tissue was examined by a neuropathologist to confirm neurodegenerative pathology including neurofibrillary tangles and neuritic plaques. Amyloid score was assessed using standardized Consortium to Establish a Registry for Alzheimer's Disease criteria (0, no plaques; 1, sparse plaques; 2, moderate plaques; 3, dense plaques), and a Braak stage (0–6 with 6 being the most severe) indicative of the level and location of hyperphosphorylated tau tangles. In addition to the pathological information detailed above, demographic data including age, sex, and Mini Mental State Examination (MMSE) score were received along with the frozen tissue, and details can be found in Table 1. Power analysis on these data, which were subjected to correlation analyses, found that seven Alzheimer's disease samples provided >95% confidence that a type I error did not occur.
Table 1.
Demographic and cognitive data for control and AD subject cortical samples
| Case no. | Diagnosis | Age at onset (years) | Age (years) | Sex | PMI | Braak stage | Plaque stage | MMSE |
|---|---|---|---|---|---|---|---|---|
| 1008 | Control | 77.4 | F | 12 | 0 | 4 | >25 | |
| 1525 | Control | 88.7 | F | 3 | 1 | 4 | 29 | |
| 1029 | Control | 73 | F | 4 | 0 | 4 | >25 | |
| 767 | Control | 86 | F | 8 | 2 | 4 | >25 | |
| 1775 | Control | 85 | M | 38.5 | 3 | 3 | 28 | |
| 1013 | Control | >89 | M | 6 | 1 | 0 | 29 | |
| 1052 | Control | 87.7 | M | 8 | 2 | 1 | 29 | |
| 1766 | AD | 57.3 | 63 | F | 3.5 | 6 | 1 | 18 |
| 1770 | AD | 70.2 | 82 | F | 6.5 | 6 | 1 | 15 |
| 1811 | AD | 87.3 | >89 | M | 18 | 6 | 2 | 21 |
| 1774 | AD | >89 | M | 3.25 | 6 | 1 | 2 | |
| 1742 | AD | 48.6 | 64 | M | 9.25 | 6 | 1 | 1 |
| 1777 | AD | 67 | F | 20.5 | 6 | 3 | 9 | |
| 1827 | AD | >89 | F | 5 | 6 | 2 | 16 |
F, Female; M, male; PMI, post-mortem interval.
Statistics.
Data are reported as the mean ± SEM. Statistical analyses were conducted using GraphPad Prism 6. Where indicated, a one-way or two-way ANOVA was performed for group analyses followed by either Tukey or Bonferroni post hoc comparison. Correlations were determined by Pearson correlation test for linearity, and coefficient of variation was assessed by calculating the average percentage deviation from the respective group mean.
Results
While previous reports have described many binding partners for PPARγ (Miyamoto-Sato et al., 2010) and pERK (Yoon and Seger, 2006; von Kriegsheim et al., 2009), our observations regarding convergence of these signaling axes during cognitive enhancement with RSG (Denner et al., 2012) led us to test whether PPARγ and pERK (pERK2) were associated with each other in multiprotein complexes. We found that pERK multiprotein complexes immunoprecipitated from Tg2576 hippocampal extracts (Fig. 1A) also contained PPARγ (Fig. 1B). We established that the pERK immunoprecipitation exhibited a linear input–output relationship up to 750 μg of input protein (r2 = 0.991; Fig. 1C). In reciprocal studies, PPARγ IPs contained pERK and the PPARγ IPs exhibited linearity up to 500 μg of input (r2 = 0.849, data not shown). Given the narrower confidence intervals with the pERK IPs, we developed a quantitative method to assess the PPARγ/pERK ratio in pERK IPs using 200 μg of hippocampal protein (Fig. 2A), thereby ensuring that our IPs were within the linear range and exhibited high reproducibility (individual animal coefficients of variation <4.8%; Fig. 2B).
Figure 1.

PPARγ associates with pERK in vivo in Tg2576 hippocampal multiprotein complexes. A, B, Western immunoblots (IBs) for pERK and PPARγ in pERK IPs from Tg2576 using anti-pERK-conjugated Sepharose beads with increasing input of hippocampal nuclear extract. C, Input–output IP linear relationship for pERK IPs (r2 = 0.991 up to 750 μg of input). Densitized Western blot values were normalized to the loading control described in Materials and Methods and Fig. 2A. Dotted lines represent the 95% confidence intervals. IgHC, Ig heavy chain.
We next examined the PPARγ/pERK ratio in postmortem human brain samples from AD and age-matched control subjects and found a significant correlation between nuclear PPARγ/pERK ratio in AD brain and MMSE, a measure of cognitive reserve (Fig. 3A,B). No correlation was found between MMSE score and the PPARγ/pERK ratio in the aged-matched control group. In agreement, we observed similar relationships in mouse hippocampus where the PPARγ/pERK ratio correlated with cognitive performance in Tg2576 mice but not in WT mice (Fig. 3C). Since RSG treatment alleviated Tg2576 cognitive deficits and the amount of hippocampal nuclear PPARγ in complex with pERK correlated with better hippocampus-dependent cognitive performance, we tested whether RSG treatment simply led to an increase in the steady-state PPARγ/pERK ratio and found that it was not affected by RSG treatment in either WT or Tg2576 mice (Fig. 3D), leading to the conclusion that the cognitive enhancing effects of PPARγ agonism were not due to increased constitutive formation of PPARγ–pERK complexes. These observations provide the first evidence for a physical interaction between PPARγ and pERK, and provide a molecular mechanism for the convergence of these two pathways in RSG-mediated cognitive enhancement in the Tg2576 mouse model for AD.
Figure 3.

PPARγ/pERK2 ratios in human AD brains and Tg2576 mouse hippocampi correlate with cognitive performance. A, Correlation between AD human brain PPARγ/pERK2 ratios and MMSE, a measure of cognitive reserve in humans (n = 7, r2 = 0.87, p = 0.003, power > 80%; Cohen, 1992). No correlation was found between complex ratios and cognitive reserve in control human brains (n = 7). B, Western blots for PPARγ and pERK as a function of MMSE score. C, Correlation between Tg2576 mouse hippocampal PPARγ/pERK2 ratios and contextual freezing, a measure of cognitive reserve in mice (n = 7, r2 = 0.59, p = 0.043). No correlation was found between complex ratios and cognitive reserve in control, WT hippocampi (n = 9). D, Hippocampal PPARγ/pERK2 ratios in WT mice (Tg2576) and Tg2576 mice treated with (+) or without (−) RSG. No significant interaction between genotype or treatment on PPARγ/pERK2 ratios. Two-way ANOVA, n = 7–12/group, p = 0.565, F(1,34) = 0.3375. Densitometric analysis of the Western blots are presented as the mean ± SEM. ns, Nonsignificant.
Since (1) RSG treatment enhances hippocampus-dependent cognition in both AD animal models and some humans with AD (Watson and Craft, 2004; Pedersen et al., 2006; Jiang et al., 2008; Landreth et al., 2008; Escribano et al., 2009; Rodriguez-Rivera et al., 2011), (2) ERK phosphorylation-dependent activation is necessary for hippocampal memory consolidation (Atkins et al., 1998; Sweatt, 2004), and (3) PPARγ associates with pERK in protein complexes, we analyzed the dynamics of these complexes during memory consolidation. RSG-treated and RSG untreated 9-month-old Tg2576 and WT littermates were subjected to two-pair FC training, wherein acquisition of the task was unaffected (Rodriguez-Rivera et al., 2011), and then were killed 4 h later (Fig. 4A) at a time point that correlated with the peak effect of PPARγ agonism on FC consolidation (Denner et al., 2012). Animals that were not exposed to the training chamber context served as controls. RSG-treated Tg2576 mice subjected to FC training exhibited significantly increased PPARγ/pERK ratios in both the nuclear (Fig. 4B) and non-nuclear fractions (Fig. 4C) compared with untreated Tg2576 mice. Two-way ANOVA revealed a significant interaction between RSG treatment and training in regard to the Tg2576 PPARγ/pERK ratio, demonstrating that PPARγ agonism facilitated PPARγ recruitment to pERK during Tg2576 memory consolidation.
Figure 4.

PPARγ agonism increases the recruitment of PPARγ to pERK during memory consolidation in Tg2576 mice. A, Experimental paradigm: Tg2576 mice fed control (−) or RSG (+) diet were either naive (FC−) or trained in the FC task (FC+), then were killed 4 h post-training during consolidation to determine hippocampal PPARγ/pERK2 ratios. For PPARγ antagonism studies, 4 h before training vehicle (GW−) or GW9662 (GW+) were intracerebroventricularly administered, and ratios were determined 4 h after training. B, C, Effects of RSG and fear conditioning on nuclear ratios (two-way ANOVA, n = 7–8/group, F(1,26) = 11.28, p = 0.002, 0.025, 0.002 for interaction, treatment, and training, respectively; B) and non-nuclear ratios (two-way ANOVA, n = 7/group, F(1,24) = 8.155, p = 0.009, 0.064, 0.015 for interaction, treatment, and training, respectively; C). D, E, Effects of PPARγ antagonism on nuclear ratios (two-way ANOVA, F(1,40) = 5.705, p = 0.022, 0.121, 0.559 for interaction, treatment, and intracerebroventricular injection, respectively; D) and non-nuclear ratios (two-way ANOVA, ns, F(1,31) = 1.016; E). F, G, Neither RSG treatment nor GW9662 antagonism had any effect on WT PPAR/pERK2 ratios in nuclear (two-way ANOVA, p = 0.41, ns, F(1,37) = 0.694; F) or non-nuclear (two-way ANOVA, p = 0.78, ns, F(1,24) = 0.074; G) fractions. H, I, pERK association MEK (n = 5/group, representative blot; H) or p90RSK (n = 2/group; I). *p < 0.05; **p ≤ 0.01.
To establish the specificity of RSG induction of PPARγ recruitment to pERK during memory consolidation, we performed intracerebroventricular injection of the PPARγ antagonist GW9662 4 h before FC training, an intervention that does not affect acquisition but blocks RSG-mediated cognitive enhancement (Denner et al., 2012) and is much more rapid than genetic intervention (Ryan et al., 2011). In a complementary manner, PPARγ antagonism blocked training-induced increased recruitment of PPARγ to pERK in the nuclear fraction (Fig. 4D), with a similar trend in the non-nuclear fraction (Fig. 4E). In agreement with previous reports that PPARγ agonism does not affect WT cognitive performance (Denner et al., 2012), the nuclear (Fig. 4F) and non-nuclear (Fig. 4G) hippocampal PPARγ/pERK2 ratios were unaffected by RSG treatment or GW9662 during WT memory consolidation. Two-way ANOVA and post hoc analysis revealed an interaction between RSG treatment and GW9662 intracerebroventricular injection on Tg2576 PPARγ/pERK ratios, indicating that RSG and GW9662 had significant effects on the complexes. Together, these results suggest that PPARγ agonism with RSG facilitates the association of hippocampal PPARγ with pERK to restore proper memory consolidation in the Tg2576 mouse model of AD.
We next investigated whether RSG treatment had an effect on other members of the ERK cascade. An essential mediator of ERK activation is phosphorylation by the upstream binding partner and kinase MEK1/2 (Canagarajah et al., 1997), which is essential for FC (Shalin et al., 2004). Indeed, we found MEK associated with pERK, an effect enhanced by RSG (Fig. 4H). We next tested for pERK binding to ribosomal S6 kinase protein 1α, MAPK-activated protein kinase-1a (p90RSK), a downstream pERK binding partner and effector kinase (Gavin and Nebreda, 1999; Smith et al., 1999) that is also required for memory consolidation (Morice et al., 2013). RSK was associated with pERK, again increased in response to RSG agonism of PPARγ (Fig. 4I).
To further understand the interaction between PPARγ and pERK, we next tested whether these proteins had the intrinsic ability to directly associate in the absence of other proteins. We used recombinant GST-tagged pERK (GST-pERK2) and PPARγ proteins in an in vitro glutathione bead pull-down assay in an attempt to reconstitute the in vivo interaction detected by hippocampal co-immunoprecipitation. We found that increasing amounts of input PPARγ resulted in increased GST-pERK pulldown of PPARγ (Fig. 5A) in a linear response (Fig. 5B). Control reactions demonstrated that PPARγ only associated with the beads in the presence of pERK, suggesting that the observed PPARγ signal was due to a direct association between the two proteins. When similar binding studies were performed with PPARγ and GST-tagged non-phosphorylated ERK2, no association with PPARγ was detected (Fig. 5C). Thus, ERK activation/phosphorylation is necessary for PPARγ binding, providing an intriguing level of specificity to these complexes.
Discussion
Identification of the molecular mechanisms that contribute to memory impairment in AD elucidate therapeutic strategies for the ever-expanding population of humans in whom the disease was diagnosed (Thies and Bleiler, 2013). In the past several years, many studies have shown that agonists of PPARγ enhance memory in some patients (Watson et al., 2005; Risner et al., 2006; Sato et al., 2011) and in genetic AD mouse models in tasks that require intact ERK MAPK signaling (e.g., associative learning in the contextual FC paradigm and spatial navigation in the Morris water maze; Pedersen et al., 2006; Jiang et al., 2008; Landreth et al., 2008; Escribano et al., 2009; Rodriguez-Rivera et al., 2011). Still, the therapeutic mechanism by which PPARγ agonism led to improved cognition remains poorly understood.
It is well established that consolidation after a learning event is an essential phase in the formation of new memories, a process that requires dynamic phosphorylation-dependent activation of hippocampal ERK (Atkins et al., 1998; Sweatt, 2004; Trifilieff et al., 2007). Prior studies in our laboratory demonstrated that PPARγ-mediated cognitive enhancement linked the hippocampal PPARγ and ERK MAPK signaling pathways by promoting the transcription of peroxisome proliferator response element-containing PPARγ target genes and Cre-containing ERK-regulated target genes (Denner et al., 2012). Given that ERK/CREB/CBP/Cre-dependent signaling is requisite for hippocampal memory consolidation (Atkins et al., 1998; McGaugh, 2000; Vecsey et al., 2007), the current study investigated whether PPARγ agonism can directly modulate ERK to enhance this process.
Here we found that PPARγ agonism induced recruitment of PPARγ to pERK during memory consolidation, and that these complexes correlated with cognitive reserve in humans with AD and in a genetic AD mouse model. The fact that hippocampal PPARγ association with pERK during memory consolidation increased only in RSG-treated Tg2576 mice implies that PPARγ-mediated effects on ERK happen selectively during AD-related hippocampal dysfunction. In this regard, we observed that acute pharmacological antagonism of PPARγ with GW9662 only blocked hippocampal memory consolidation in RSG-treated Tg2576 (Denner et al., 2012) via prevention of hippocampal PPARγ association with pERK during this process. Thus, we conclude that hippocampal PPARγ activity is necessary to enhance the formation of complexes during memory consolidation. Direct binding of recombinant PPARγ and pERK in vitro suggests an intrinsic affinity that may underlie the cognitive enhancing activity of RSG.
Cognitive reserve in AD is a measure of the ability of the brain to resist damage inflicted by AD pathology (Sperling et al., 2011). The observation that 9-month-old Tg2576 mice on average perform poorly in the hippocampus-dependent contextual FC task, while individual animals vary considerably, led us to hypothesize that if the PPARγ/pERK ratio was relevant to cognitive performance, individual human or animal ratios would correlate with their respective performance. Indeed, we found in humans with AD that cognitive performance, assessed by the MMSE score, positively correlated with the PPARγ/pERK ratio. In further support of this hypothesis, we found that Tg2576 mice contextual freezing behavior, a reflection of cognitive performance, also positively correlated with the PPARγ/pERK ratio. Notably, neither age-matched human control subjects nor WT littermates of Tg2576 mice exhibited such a correlation. Coupled with our previous observation that a subset of hippocampal PPARγ target genes are also CREB/CBP target genes (Denner et al., 2012), which are known to be regulated by ERK MAPK during memory consolidation (Guzowski and McGaugh, 1997; Ahi et al., 2004), these data suggest that PPARγ participation in a pERK complex may serve a compensatory role to re-establish proper ERK signaling that is disrupted by AD pathology. Previous studies found reduced levels of ERK2 protein and mRNA in AD hippocampus compared with controls (Trojanowski et al., 1993; Hyman et al., 1994), suggesting that the observed reduced interaction between PPARγ and pERK as AD progresses may result from an overall reduction in hippocampal ERK. Our previous bioinformatics analysis of transcriptomic and proteomic data from Tg2576 hippocampus following RSG treatment identified ERK signaling components and placed ERK as a central node in the signaling networks identified with cognitive enhancement (Denner et al., 2012). Thus, PPARγ agonism may serve to re-establish not only ERK2 levels, but also the dynamic range for ERK activation (e.g., recruitment of MEK) during memory consolidation.
Nuclear receptor interaction with ERK is not unprecedented as exemplified by the estrogen (Hashimoto et al., 2012), glucocorticoid (Strawhecker et al., 1989; Revest et al., 2005), and progesterone (Vicent et al., 2009) receptors. It is also becoming evident that ERK can be regulated through protein–protein interactions via well defined protein motifs. ERK has been shown to interact with numerous proteins, including Elk-1 and p90rsk (Sheridan et al., 2008), through both ERK-exclusive docking sites, known as DEF sites and defined by an FX(F/Y)P amino acid motif and generally located the C terminus to an ERK phosphorylation site (Sheridan et al., 2008), and the more general MAPK recognition D sites, defined by the amino acid sequence K/R K/R K/R X(1–5) L/I X L/I (Reményi et al., 2005; Garai et al., 2012).
We identified putative DEF and D sites within the N terminus of PPARγ at amino acids FHYG119–122 and RRTIRLKL136–143, respectively. It is noteworthy that proteins that contain both D and DEF sites generally exhibit more specific and higher-affinity interaction with ERK than those that contain only one of the sites (Jacobs et al., 1999). That PPARγ contains consensus sequences, which are predicted to mediate direct interaction with ERK, provides a potential mechanism for their interaction during cognitive enhancement. Further, binding of proteins to the N terminus of nuclear receptors stabilizes the intrinsic disorder of this domain (Khan et al., 2012), suggesting an additional mechanism whereby pERK may facilitate complex stability by conferring order to the PPARγ N-terminal domain.
Our finding that PPARγ association with pERK in vivo was increased in RSG-treated Tg2576 mice only during memory consolidation suggests a dynamic ligand-dependent (RSG) mechanism for recruitment of pERK and other signaling partners (e.g., MEK and p90RSK). The conformational change conferred upon PPARγ through ligand binding (Choi et al., 2011) may make the ERK docking domains within PPARγ more accessible and could therefore increase the binding. Furthermore, ERK interacts with substrate DEF sites via a hydrophobic pocket adjacent to the kinase active site cleft (Lee et al., 2004) that is exposed following MEK phosphorylation (Canagarajah et al., 1997). This phosphorylation-induced conformational change in ERK may account for our observation of a direct interaction in in vitro reconstitution studies between recombinant PPARγ and pERK2 that was not recapitulated with nonphosphorylated ERK2. Together, our data suggest that PPARγ and pERK directly interact in vitro and in vivo, and that this interaction contributes to cognitive enhancement with RSG treatment.
Since PPARγ agonism improved performance in an ERK-dependent learning and memory task, and since PPARγ has a higher affinity for phosphorylated ERK compared with nonphosphorylated ERK, MEK is likely an important mediator of PPARγ–pERK recruitment. While our studies did not directly address the involvement of MEK, our findings do indicate that MEK is likely a dynamic component of the PPARγ–pERK complex, since we detected increased steady-state MEK in pERK immunoprecipitation material from RSG-treated Tg2576 hippocampus yet decreased MEK when we probed pERK immunoprecipitation material at the 4 h memory consolidation time point (data not shown). In addition, MEK has been demonstrated to shuttle both ERK and PPARγ between nuclear and cytosolic compartments (Burgermeister and Seger, 2007), suggesting that MEK may regulate PPARγ–pERK complex localization. Alternatively, PPARγ–pERK association may facilitate the downstream activity of pERK through improved pERK-dependent phosphorylation activation of Elk-1 and p90RSK (Frödin and Gammeltoft, 1999; Ahi et al., 2004).
Memory formation begins with an acquisition phase followed by a consolidation phase in the ensuing hours to form a memory trace that can be retrieved at a later time. ERK phosphorylation is required for memory through transcriptional regulation of target genes essential for coding a new memory trace. In the Tg2576 model of AD, ERK is dysregulated and unable to properly function in new memory formation. Thus, we propose a model in which ligand-activated PPARγ restores dysfunctional ERK-dependent signaling to facilitate memory consolidation through the recruitment of binding partners to a pERK multiprotein complex (Fig. 6). One potential binding partner in this process is the histone acetyltransferase CBP, which serves as a transcriptional cofactor for both CREB and PPARγ (Vecsey et al., 2007; Bugge et al., 2009), and may be a convergent central node between the PPARγ and ERK pathways (Denner et al., 2012). The identification of these novel PPARγ–pERK complexes provides unique opportunities for newly targeted therapeutics to improve memory in AD and warrants further investigation.
Figure 6.

Working model for PPARγ-mediated enhancement of memory consolidation in AD. In the cognitively impaired Tg2576 AD model mice, ligand-bound PPARγ is recruited to activated ERK following a learning event. The complex recruits a number of other transcriptional regulatory proteins, ultimately increasing ERK downstream efficiency, including Cre-mediated gene transcription, as well as activation of p90RSK and Elk-1.
Footnotes
This work was supported by the National Institutes of Health under Grant R01-AG031859 (to K.T.D. and L.D.). Additional funding was provided by the American Health Assistance Foundation, The Sealy Foundation for Biomedical Research, and a gift from J.W. Mohn (to K.T.D. and L.D.); by the Emmett and Miriam McCoy Foundation (to L.D.); and by the Cullen Trust for Health Care to the Mitchell Center. Behavioral testing was performed in the University of Texas Medical Branch at Galveston Rodent In Vivo Assessment Core directed by K.T.D. We thank Drs. Nicole Bjorklund and Giulio Taglialatela for generously providing the human tissue samples investigated herein. We also thank Wei Song for expert technical assistance and Ibdanelo Cortez for a number of insightful discussions.
The authors declare no competing financial interests.
References
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behav Brain Res. 2004;149:17–31. doi: 10.1016/S0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Becker RE, Greig NH. Fire in the ashes: can failed Alzheimer's disease drugs succeed with second chances? Alzheimers Dement. 2013;9:50–57. doi: 10.1016/j.jalz.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund NL, Sadagoparamanujam VM, Taglialatela G. Selective, quantitative measurement of releasable synaptic zinc in human autopsy hippocampal brain tissue from Alzheimer's disease patients. J Neurosci Methods. 2012;203:146–151. doi: 10.1016/j.jneumeth.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge A, Grøntved L, Aagaard MM, Borup R, Mandrup S. The PPAR-gamma2 a/B-domain plays a gene-specific role in transactivation and cofactor recruitment. Mol Endocrinol. 2009;23:794–808. doi: 10.1210/me.2008-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgermeister E, Seger R. Mapk kinases as nucleo-cytoplasmic shuttles for Ppargamma. Cell Cycle. 2007;6:1539–1548. doi: 10.4161/cc.6.13.4453. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the map kinase Erk2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/S0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidović D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist Ppargamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WG, Vivonia CA, Baxter CF. Accurate freehand injection into the lateral brain ventricle of the conscious mouse. J Appl Physiol. 1968;25:319–321. doi: 10.1152/jappl.1968.25.3.319. [DOI] [PubMed] [Google Scholar]
- Cohen J. A power primer. Psychol Bull. 1992;112:155–159. doi: 10.1037/0033-2909.112.1.155. [DOI] [PubMed] [Google Scholar]
- Craft S. Alzheimer disease: insulin resistance and AD—extending the translational path. Nat Rev Neurol. 2012;8:360–362. doi: 10.1038/nrneurol.2012.112. [DOI] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon BA, Wood TG, Dineley KT. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J Neurosci. 2012;32:16725–16735a. doi: 10.1523/JNEUROSCI.2153-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of Alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano L, Simón AM, Pérez-Mediavilla A, Salazar-Colocho P, Del Río J, Frechilla D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochem Biophys Res Commun. 2009;379:406–410. doi: 10.1016/j.bbrc.2008.12.071. [DOI] [PubMed] [Google Scholar]
- Frödin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (Rsk) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/S0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Garai Á, Zeke A, Gógl G, Törő I, Fördős F, Blankenburg H, Bárkai T, Varga J, Alexa A, Emig D, Albrecht M, Reményi A. Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci Signal. 2012;5:ra74. doi: 10.1126/scisignal.2003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Nebreda AR. A map kinase docking site is required for phosphorylation and activation of P90(Rsk)/MAPKAP kinase-1. Curr Biol. 1999;9:281–284. doi: 10.1016/S0960-9822(99)80120-1. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal camp response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci U S A. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S, Monarch ES, Goldstein FC. Impaired fear conditioning in Alzheimer's disease. Neuropsychologia. 2002;40:1187–1195. doi: 10.1016/S0028-3932(01)00223-8. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Yu J, Koizumi H, Ouchi Y, Okabe T. Ginsenoside Rb1 prevents Mpp(+)-induced apoptosis in Pc12 cells by stimulating estrogen receptors with consequent activation of ERK1/2, Akt and inhibition of SAPK/Jnk, P38 MAPK. Evid Based Complement Alternat Med. 2012;2012:693717. doi: 10.1155/2012/693717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefer M, Allison SC, Schauer GF, Neuhaus JM, Hall J, Dang JN, Weiner MW, Miller BL, Rosen HJ. Fear conditioning in frontotemporal lobar degeneration and Alzheimer's disease. Brain. 2008;131:1646–1657. doi: 10.1093/brain/awn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hort J, Laczó J, Vyhnálek M, Bojar M, Bures J, Vlcek K. Spatial navigation deficit in amnestic mild cognitive impairment. Proc Natl Acad Sci U S A. 2007;104:4042–4047. doi: 10.1073/pnas.0611314104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Elvhage TE, Reiter J. Extracellular signal regulated kinases. Localization of protein and MRNA in the human hippocampal formation in Alzheimer's disease. Am J Pathol. 1994;144:565–572. [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by Erk Map kinase. Genes Dev. 1999;13:163–175. doi: 10.1101/gad.13.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-Gamma (Ppargamma) in Alzheimer's disease: therapeutic implications. CNS Drugs. 2008;22:1–14. doi: 10.2165/00023210-200822010-00001. [DOI] [PubMed] [Google Scholar]
- Khan SH, Awasthi S, Guo C, Goswami D, Ling J, Griffin PR, Simons SS, Jr, Kumar R. Binding of the N-terminal region of coactivator Tif2 to the intrinsically disordered Af1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J Biol Chem. 2012;287:44546–44560. doi: 10.1074/jbc.M112.411330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth G, Jiang Q, Mandrekar S, Heneka M. Ppargamma agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics. 2008;5:481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in Map kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/S1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Nuclear receptors as therapeutic targets for Alzheimer's disease. Expert Opin Ther Targets. 2011;15:1085–1097. doi: 10.1517/14728222.2011.594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Miyamoto-Sato E, Fujimori S, Ishizaka M, Hirai N, Masuoka K, Saito R, Ozawa Y, Hino K, Washio T, Tomita M, Yamashita T, Oshikubo T, Akasaka H, Sugiyama J, Matsumoto Y, Yanagawa H. A comprehensive resource of interacting protein regions for refining human transcription factor networks. PLoS One. 2010;5:e9289. doi: 10.1371/journal.pone.0009289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice E, Farley S, Poirier R, Dallerac G, Chagneau C, Pannetier S, Hanauer A, Davis S, Vaillend C, Laroche S. Defective synaptic transmission and structure in the dentate gyrus and selective fear memory impairment in the Rsk2 mutant mouse model of Coffin-Lowry syndrome. Neurobiol Dis. 2013;58:156–168. doi: 10.1016/j.nbd.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Reményi A, Good MC, Bhattacharyya RP, Lim WA. The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol Cell. 2005;20:951–962. doi: 10.1016/j.molcel.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8:664–672. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rivera J, Denner L, Dineley KT. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav Brain Res. 2011;216:255–261. doi: 10.1016/j.bbr.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nat Med. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Zirrgiebel U, Honsa KJ, Julien JP, Miller FD, Kaplan DR, Sweatt JD. Neuronal MEK is important for normal fear conditioning in mice. J Neurosci Res. 2004;75:760–770. doi: 10.1002/jnr.20052. [DOI] [PubMed] [Google Scholar]
- Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J Biol Chem. 2008;283:19511–19520. doi: 10.1074/jbc.M801074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. doi: 10.1074/jbc.274.5.2893. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association Workgroups on Diagnostic Guidelines for Alzheimer's Disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockhorst U, de Fries D, Steingrueber HJ, Scherbaum WA. Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol Behav. 2004;83:47–54. doi: 10.1016/j.physbeh.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Strawhecker JM, Betz NA, Neades RY, Houser W, Pelling JC. Binding of the 97 kDa glucocorticoid receptor to the 5′ upstream flanking region of the mouse C-Ha-Ras oncogene. Oncogene. 1989;4:1317–1322. [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res. 2009;200:95–99. doi: 10.1016/j.bbr.2008.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, Irs-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies W, Bleiler L. 2013 Alzheimer's disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Trifilieff P, Calandreau L, Herry C, Mons N, Micheau J. Biphasic Erk1/2 activation in both the hippocampus and amygdala may reveal a system consolidation of contextual fear memory. Neurobiol Learn Mem. 2007;88:424–434. doi: 10.1016/j.nlm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Mawal-Dewan M, Schmidt ML, Martin J, Lee VM. Localization of the mitogen activated protein kinase ERK2 in Alzheimer's disease neurofibrillary tangles and senile plaque neurites. Brain Res. 1993;618:333–337. doi: 10.1016/0006-8993(93)91286-2. [DOI] [PubMed] [Google Scholar]
- van Himbergen TM, Beiser AS, Ai M, Seshadri S, Otokozawa S, Au R, Thongtang N, Wolf PA, Schaefer EJ. Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: results from the Framingham Heart Study. Arch Neurol. 2012;69:594–600. doi: 10.1001/archneurol.2011.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent GP, Zaurin R, Ballaré C, Nacht AS, Beato M. ERK signaling and chromatin remodeling in MMTV promoter activation by progestins. Nucl Recept Signal. 2009;7:e008. doi: 10.1621/nrs.07008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, Gilbert D, Kolch W. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11:1458–1464. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GS, Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. Eur J Pharmacol. 2004;490:97–113. doi: 10.1016/j.ejphar.2004.02.048. [DOI] [PubMed] [Google Scholar]
- Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–958. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Cross-regulation of C/Ebp alpha and Ppar gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. doi: 10.1016/S1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]