

Abstract
Objective
Although elevated plasma concentrations of serum amyloid A (SAA) are strongly associated with increased risk for atherosclerotic cardiovascular disease in humans, the role of SAA in the pathogenesis of lesion formation remains obscure. Our goal was to determine the impact of SAA deficiency on atherosclerosis in hypercholesterolemic mice.
Approach and Results
ApoE-/- mice, either wild type or deficient in both major acute phase SAA isoforms, SAA1.1 and SAA2.1 (SAAWT and SAAKO, respectively), were fed a normal rodent diet for 50 weeks. Female, but not male SAAKO mice had a modest increase (22%; p ≤ 0.05) in plasma cholesterol concentrations and a 53% increase in adipose mass compared to SAAWT mice that did not impact the plasma cytokine levels or the expression of adipose tissue inflammatory markers. SAA deficiency did not impact lipoprotein cholesterol distributions or plasma triglyceride concentrations in either male or female mice. Atherosclerotic lesion areas measured on the intimal surfaces of the arch, thoracic, and abdominal regions were not significantly different between SAAKO and SAAWT mice in either gender. To accelerate lesion formation, mice were fed a Western diet for 12 weeks. SAA deficiency had no effect on diet-induced alterations in plasma cholesterol, triglyceride or cytokine concentrationsn or on aortic atherosclerotic lesion areas in either male or female mice. In addition, SAA deficiency in male mice had no effect on lesion areas or macrophage accumulation in the aortic roots.
Conclusions
The absence of endogenous SAA1.1 and 2.1 does not impact atherosclerotic lipid deposition in apoE-/- mice fed either normal or Western diets.
Keywords: serum amyloid A, serum amyloid A-deficient mice, inflammation, acute phase, lipid deposition, atherosclerosis
Introduction
Serum amyloid A (SAA) comprises a family of proteins whose expression can be induced in the liver more than 1000-fold during an acute phase response. Plasma SAA concentrations are also modestly elevated in chronic inflammatory diseases including rheumatoid arthritis1, diabetes and insulin resistance2, 3, and obesity.3, 4 Two highly homologous acute phase SAA isoforms are expressed in humans (93% identical in amino acid sequence), designated SAA1 and SAA2, which correspond to mouse SAA1.1 and SAA2.1. A number of clinical studies have linked plasma SAA concentrations to atherosclerotic cardiovascular disease (CVD)5 with some studies reporting that SAA is more predictive than C-reactive protein (CRP) for cardiovascular outcomes.6, 7 Whether the link between elevated SAA concentrations and atherosclerosis is causal or merely associative has been the focus of extensive investigation. SAA has been detected by immunocytochemistry in mouse8, 9 and human10 atherosclerotic lesions. Based on in situ hybridization studies, SAA is expressed by macrophages, smooth muscle cells, and endothelial cells in the vessel wall.10 During systemic inflammation, the liver is the predominant source of plasma SAA, where it is almost exclusively associated with HDL. Thus, the presence of SAA in lesions could also reflect deposition of circulating SAA.
The physiological functions of SAA have yet to be firmly established. SAA reportedly promotes a number of potentially atherosclerotic effects including monocyte chemotaxis11, subendothelial lipoprotein retention8, 12, endothelial dysfunction13, 14 and pro-inflammatory cytokine15, 16 and matrix metalloproteinase17 induction. However, many of these effects were defined using commercially available SAA which is a hybrid molecule containing sequences corresponding to both human acute phase SAA isoforms. Recent evidence suggests that this recombinant SAA may exert pro-inflammatory activities not shared by native SAA.18 Simons et al recently described a transgenic mouse with inducible, liver-specific expression of SAA1.1.19 Notably, induction of SAA to levels corresponding to severe inflammation (>1000 mg/L) had no effect on plasma levels of murine serum amyloid P component, an acute phase reactant that is typically induced in response to inflammatory stimuli. Thus, evidence that SAA is intrinsically pro-inflammatory has come under question.
Many published in vitro studies have investigated lipid-free/lipid-poor SAA, and not HDL-associated SAA, which may be the more physiologically relevant form of SAA, since SAA in plasma is predominantly associated with HDL. In studies that investigated SAA complexed to HDL, the adverse effects observed for lipid-free/lipid-poor SAA appeared to be abrogated.20, 21Conversely, by associating with HDL, SAA may contribute to atherosclerotic processes by interfering with protective functions of HDL. For example, SAA has been suggested to modulate the ability of HDL to facilitate reverse cholesterol transport (RCT) to remove excess cholesterol from the periphery (including cholesterol-laden macrophages in an atherosclerotic plaque) to the liver for excretion from the body. Several groups have reported that macrophage to feces RCT is impeded in mice during inflammation.22-25 However, while SAA, in the absence of an acute phase response, modestly reduces RCT in mice23, impairment of RCT during an acute phase response does not require SAA.25
Direct evidence that SAA promotes atherogenic processes is supported by a recent study in which administration of a lentiviral vector expressing mouse SAA produced a modest, but significant increase in atherosclerotic lesion areas of apoE-/- mice.26 However, due to lack of suitable animal models, there has been no direct evidence to date that endogenous SAA is required for atherosclerotic lesion formation. We recently reported development of mice deficient in both acute phase SAA isoforms.27 Generation of these mice was complicated by the fact that SAA1.1 and SAA2.1 are located approximately 9 kb apart in opposite orientation on mouse chromosome 7, which hindered multiple attempts to target both genes simultaneously. For the current study, SAA-deficient mice were crossed with apoE-/- mice to determine whether lack of SAA alters the extent of either spontaneous or diet-induced atherosclerotic lesions.
Results
SAA deficiency does not impact atherosclerotic lipid deposition in apoE-/- mice fed a normal rodent diet
In a previous study, overexpression of SAA using a lentiviral vector enhanced atherosclerosis development in apoE-/- mice fed a normal rodentdiet.26 We detected SAA by immunohistochemical staining in aortic root lesions of 50 week old apoE-/- mice fed a normal diet, confirming an earlier report8 (Supplemental Figure I). To investigate whether endogenous SAA contributes to atherosclerotic lipid deposition in apoE-/- mice, we fed apoE-/- mice (SAAWT) and apoE-/- mice lacking both acute phase SAA isoforms (SAAKO mice) a normal rodent diet for 50 weeks. Neither strain exhibited any overt signs of adverse health during the course of the study. Body weights were determined at the onset of the experiment and monitored up to study termination at 50 weeks. As expected, male mice weighed more than female mice throughout the study (Table 1). Unexpectedly female SAAKO mice gained more weight than SAAWT mice during the course of the experiment. At study termination, female SAAKO mice exhibited a modest but significant 10.3% increase in weight compared to female SAAWT mice (Table 1, P<0.05). The difference in body weight was largely attributed to an increase in fat mass in female SAAKO mice (Supplemental Figure II), which was not accounted for by significant differences in FASN expression in adipose tissue (Supplemental Figure III A). The increased adiposity in female SAAKO mice was not associated with markedly increased markers of inflammation. Although plasma levels of IL-1β were higher in male SAAKO mice compared to SAAWT mice (p<0.05), the levels of both IL-1β and IL-6 in all groups of mice were barely above the level of detection (Table 1). Thus, the physiological relevance of differences in IL-1β levels in male mice is uncertain. There was a trend for decreased abundance of F4/80 mRNA, a macrophage marker, in gonadal fat from female SAAKO mice compared to SAAWT mice, however the difference did not reach statistical significance (p=0.06; Supplemental Figure III B). The abundance of arginase, IL-10, TNFα, and IL-6 mRNAs in adipose tissue of female SAAWT and SAAKO mice was also similar, suggesting that increased adiposity in SAAKO mice did not lead to changes in macrophage polarity (Supplemental Figure III C-F). Plasma total cholesterol concentrations were similar in male SAAKO and SAAWT mice and modestly but significantly higher in 50-week-old female SAAKO mice compared to SAAWT mice (22% increase, P<0.05; Table 1). Differences in plasma cholesterol concentrations was not reflected in altered lipoprotein cholesterol distributions of either male or female SAAWT and SAAKO mice (Supplemental Figure IV). While plasma triglyceride concentrations were higher in male mice compared to female mice, SAA deficiency had no effect on triglyceride concentrations in either gender (Table 1). Plasma SAA concentrations were detectable but low in 50 week old apoE-/- mice (SAAWT) fed a normal laboratory diet and did not differ between males and females (Table 1).
Table 1. Body weights and plasma total cholesterol, triglyceride, SAA and cytokine concentrations in SAAWT and SAAKO mice fed a normal laboratory diet for 50 weeks.
| Females n = 8 - 11 |
Males n = 10 - 12 |
|||
|---|---|---|---|---|
| SAAWT | SAAKO | SAAWT | SAAKO | |
| Body weight g | 30.0 ± 1.2 | 33.1 ± 0.8* | 41.6 ± 2.0 | 38.7 ± 1.2 |
| TC mg/dl | 385.4 ± 26.8 | 469.9 ± 13.6* | 558.6 ± 37.1 | 487.4 ± 41.7 |
| TG mg/dl | 60.4 ± 5.9 | 73.6 ± 7.7 | 127.9 ± 13.7 | 140.8 ± 19.0 |
| SAA μg/ml | 17.5 ± 7.3 | - | 16.3 ± 4.0 | - |
| IL-6 pg/ml | 11.8 ± 4.0 | 6.2 ± 2.2 | 16.5 ± 5.6 | 5.5 ± 1.1 |
| IL-1beta pg/ml | 3.5 ± 0.7 | 2.2 ± 0.5 | 2.5 ± 0.3 | 3.7 ± 0.5* |
Values are mean ± SEM.
TC indicates total cholesterol; TG indicates triglyceride
denotes P< 0.05 vs SAAWT of the same gender.
Atherosclerotic lesion areas were measured on the intimal surfaces of the total aorta including the ascending aorta to bifurcation in all groups of mice by en face analyses (Figure 1). Lesion area as a percentage of the total aorta in male SAAWT (10.3 ± 1.7), male SAAKO (11.7 ± 1.1), female SAAWT (11.1 ± 1.2), and female SAAKO (11.5 ± 0.6) mice were not significantly different. Thus, endogenously expressed SAA in either male or female SAAWT mice fed a normal rodent diet does not impact the extent of aortic atherosclerotic lesions, despite modest increases in plasma cholesterol and body weight, at least in female mice.
Figure 1.

Absence of SAA does not affect en face lesion area in 50 week old apoE-/- mice fed a normal rodent diet. Circles correspond to lesion areas, expressed as the percent of the total intimal area from individual aortas of SAAWT and SAAKO mice (n = 10-12). Bars represent the mean ± SEM.
SAA deficiency does not impact atherosclerotic lesion development in apoE-/- mice fed a Western diet
The lack of an effect of SAA deficiency on atherosclerosis development in mice fed a normal rodent diet could be due to the relatively low concentrations of plasma SAA in this setting (∼17 μg/ml). Therefore, male and female SAAWT and SAAKO mice were challenged with a diet enriched in saturated fat for 12 weeks, which is known to increase plasma SAA concentrations.28 While there were gender differences in body weight, plasma lipid concentrations, and lean and fat mass in both strains after high fat feeding, SAA deficiency had no effect on any of these parameters in either male or female mice (Table 2 and Supplemental Figure V). Lipoprotein cholesterol distributions, as determined by size-exclusion chromatography, were similar in SAAWT and SAAKO mice (Supplemental Figure VI). Plasma cytokines concentrations (IL-6 and IL-1β) were determined as an index of systemic inflammation evoked by feeding a Western diet for 12 weeks (Table 2). Although somewhat elevated compared to mice fed a normal rodent diet for 50 weeks, plasma cytokine concentrations were low and not significantly different between the two strains. Based on quantitative immunoblotting, the Western diet increased plasma SAA concentrations ∼5.0-fold in male apoE-/- (SAAWT) mice and ∼10.6-fold in female SAAWT mice (Figure 2). As determined by ELISA, plasma SAA concentrations were highly variable in individual male and female SAAWT mice after high fat feeding, ranging from virtually undetectable values to > 1 mg/ml for both genders (Table 2). Plasma SAA concentrations in age-matched control male SAAWT mice were 18.8 ± 7.3 μg/ml. Mice with the largest induction of plasma SAA also exhibited the highest levels of IL-6 and IL-1β (data not shown).
Table 2. Body weights and plasma total cholesterol, triglyceride, SAA and cytokine concentrations in SAAWT and SAAKO mice fed a Western diet for 12 weeks.
| Females n = 9 - 10 |
Males n = 7 - 10 |
|||
|---|---|---|---|---|
| SAAWT | SAAKO | SAAWT | SAAKO | |
| Body weight g | 25.9 ± 0.7 | 24.6 ± 0.5 | 38.7 ±1.4 | 35.6 ± 1.4 |
| TC mg/dl | 1097 ± 49 | 1125 ± 80 | 1633 ± 116 | 1679 ± 91 |
| TG mg/dl | 87.1 ± 7.6 | 70.6 ± 4.9 | 174.7 ± 28.4 | 112.6 ± 13.4 |
| SAA μg/ml | 876 ± 690 | - | 440 ± 229 | - |
| IL-6 pg/ml | 19.7 ± 10.5 | 12.6 ± 6.2 | 19.9 ± 9.3 | 8.1 ± 1.8 |
| IL-1beta pg/ml | 5.0 ± 1.7 | 3.2 ± 0.8 | 4.0 ± 0.9 | 7.1 ± 1.2 |
Values are mean ± SEM.
TC indicates total cholesterol; TG indicates triglyceride
Figure 2.
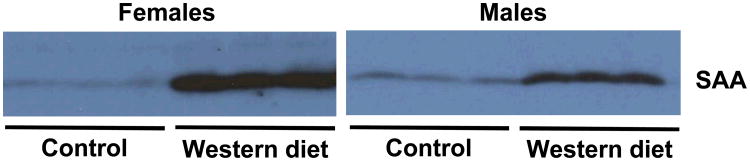
Plasma SAA is increased in female and male apoE-/- mice fed a Western diet. Plasma was collected from male and female apoE-/- mice before (control) and after feeding a Western diet for 12 weeks. Due to large variations in plasma SAA concentrations in Western diet-fed mice (see text and Table 2), individual plasmas were pooled for each group, and triplicate aliquots (1.5 μl) from each pool were subjected to SDS-PAGE and immunoblotted for SAA using rabbit anti-mouse antisera (De Beer laboratory).
Atherosclerotic lesion area, expressed as the percent of total aortic surface of male SAAWT (14.1 ± 1.3), male SAAKO (14.5 ± 1.1), female SAAWT (13.3 ± 0.8), and female SAAKO (14.7 ± 1.0) mice (Figure 3), was not significantly influenced by gender or genotype. Atherosclerotic lesion areas within the arch region, where lesions were relatively more abundant, were also similar for the four groups (not shown). Additional analyses of male mice indicated that lesion areas in the aortic root, quantified after ORO staining of neutral lipids, as well as the macrophage content of the aortic root lesions, quantified by CD68 immunostaining, was also not significantly different in SAAWT compared to SAAKO mice (Figure 4 and Supplemental fig VII). In SAAWT mice, plasma SAA concentrations did not correlate with atherosclerotic lesion area measured en face (Figure 5) or within aortic roots (not shown). Taken together, these data indicate that endogenous SAA does not significantly impact body weights, plasma lipid concentrations and lipoprotein profiles, or atherosclerotic lesion development in either male or female apoE-/- (SAAWT) mice fed an atherosclerotic diet for 12 weeks.
Figure 3.

Absence of SAA does not affect en face lesion area in apoE-/- mice fed a Western diet for 12 weeks. Circles correspond to lesion areas, expressed as the percent of the total intimal area, from individual aortas of SAAWT and SAAKO mice (n = 8 - 10). Bars represent the mean ± SEM.
Figure 4.

Absence of SAA does not alter atherosclerotic lesion formation in the aortic sinuses of male apoE-/- mice fed a Western diet for 12 weeks. Atherosclerotic lesion area in aortic root sections of male mice fed a diet enriched in saturated fats for 12 weeks were determined after ORO staining. A: Circles represent the means and bars the SEM of sections located ∼ 80 μm apart starting 400 μm before the transition and ending 400μm after the transition. The transition zone is defined by the disappearance of the valve cusps and is labeled “0” on the x-axis, n=5. B: Representative Oil Red O stained sections at the transition zone from 2 SAAWT mice and 2 SAAKO mice.
Figure 5.

Plasma SAA concentration does not correlate with en face lesion area in apoE-/- mice fed a Western diet for 12 weeks. Plasma SAA concentrations were measured by ELISA and en face lesions expressed as a percentage of the total aortic intimal surface in male and female apoE-/- mice, n=19. There is no significant relationship between SAA and atherosclerosis (r=0.35, P=NS).
Discussion
It is widely recognized that inflammation contributes significantly to the initiation, progression and rupture of atherosclerotic plaques. SAA is an inflammatory marker that has been strongly associated with increased risk for clinical cardiovascular events. However, whether SAA is actively involved in atherogenesis or merely serves as a marker of atherogenic processes has not been clearly established. In this study, we determined whether apoE-/- mice lacking SAA are protected from atherosclerosis. Our data clearly show that deficiency of both acute phase forms of SAA does not impact the extent of atherosclerotic lipid deposition that spontaneously occurs in apoE-/- mice fed a normal rodent diet, or that is induced by a Western diet. Our results also indicate that plasma SAA levels do not correlate with atherosclerotic lesion area in aortas of apoE-/- mice, in contrast to what was reported for LDLR-/- mice fed a high fat diet with or without addition of cholesterol.28 Despite the observation by Dong et al that increased plasma SAA concentration contributes to accelerated atherosclerosis,26 we conclude that endogenous SAA is not required for atherosclerotic lipid deposition in this murine model. The lack of reduction of atherosclerosis in SAAKO mice does not negate a role for SAA in atherogenesis, but demonstrates that it is not required.
We noted that female SAAKO mice had a modest but significant increase in weight gain and percent body fat during the 50-week study compared to female SAAWT mice. In contrast, 50-week old male SAAKO mice did not show a significant difference in weight compared to SAAWT mice. It should be noted, however, that the modest increase in adiposity in 50-week old female SAAKO mice was not associated with increased plasma levels or adipose tissue expression of inflammatory cytokines. Thus, the extent of atherosclerosis in female SAAWT and SAAKO mice fed the normal rodent diet was not influenced in the two strains by major differences in systemic inflammation. In the course of studying the impact of SAA deficiency in normolipidemic (C57BL/6) mice, we have observed that both male and female SAAKO mice gained less weight compared to their wild-type counterparts. The difference in weight gain can be attributed to differences in fat mass (unpublished data). Thus, SAA may regulate adiposity through mechanisms that are modulated by hyperlipidemia and gender. Abundant data from both human and mouse studies establish that increased adiposity is associated with increased SAA concentrations. Inflammatory cytokines produced by adipose tissue can act locally to induce SAA expression, or enter the circulation and stimulate hepatic secretion of SAA. A strong correlation between BMI and plasma SAA concentrations is seen in children and adults29, 30 and weight loss is accompanied by a reduction in plasma SAA.4, 31 Obese mice also have elevated circulating SAA concentrations32 and we previously reported an association between elevated SAA in obesity and increased atherosclerosis in apoE-/- mice.9 Our data in SAAKO mice highlight the need for further studies addressing the complex relationship between SAA and adiposity.
The wide variations in plasma SAA concentrations in apoE-/- mice after 12 weeks on the Western diet provided the opportunity to determine whether circulating SAA concentrations predicted the extent of atherosclerotic lesion area in our study. The finding that plasma SAA concentrations did not correlate with atherosclerotic lesion area in apoE-/- mice fed a high-fat diet supports our conclusion that SAA does not play a major role in atherosclerotic lipid deposition in this mouse strain. However, in an earlier study, Lewis et al. determined that plasma concentrations of SAA, but not cholesterol, correlated with the extent of atherosclerosis in LDLR-/- mice fed a normal, high fat, or high-fat and high-cholesterol diet for 10 weeks.28 This finding led the authors to conclude that SAA may have a causal role in atherosclerosis. In that study, atherosclerotic lipid deposition was expressed as total lesion area in the aortic arch region, which complicates our ability to make direct comparisons with our data. The possibility that SAA may contribute to atherosclerotic mechanisms in LDLR-/- mice, but not apoE-/- mice, merits further investigation.
In a recently published study, Dong et al. used a gain-of-function approach to investigate whether SAA promotes atherosclerosis in mice26. At 8 weeks of age, male apoE-/- mice were administered a lentiviral vector expressing mouse SAA1.1 or a control lentiviral vector. After 14 weeks on a normal rodent diet, plasma SAA levels in mice administered the SAA-expressing lentivirus were significantly increased compared to control mice (35.5 ± 9.6 μg/ml and 8.5 ± 2.8 μg/ml, respectively). Increased SAA was associated with significantly increased lesion areas on both the luminal surface of the aorta as well as in the aortic root. These findings contrast to our results, which show no effect of SAA deficiency on atherosclerotic lesion area in apoE-/- mice fed a normal rodent diet for 50 weeks, or in mice fed a Western diet for 12 weeks. The reason for the discrepant results from the two studies is unclear. Although no data supports this suggestion, we cannot rule out the possibility that SAA1.1 and SAA2.1 have opposing effects on atherosclerosis in vivo such that overexpression of SAA1.1 increases atherosclerosis whereas deficiency of both SAA isoforms has no effect. More likely, however, is the possibility that plasma levels of SAA in 50 week-old SAAWT mice (∼17 μg/ml) were too low to have a major effect on atherosclerosis in our study. On the other hand, the extreme hypercholesterolemia evoked by high fat diet feeding may have overwhelmed any modest atherogenic effects exerted by endogenous SAA. Notably, Dong et al. reported no effect of exogenous SAA expression on accelerated atherosclerosis produced by high fat diet feeding, supporting the conclusion that SAA plays a minor role in this setting. As another approach to investigate the role of endogenous SAA in atherogenesis, we recently carried out studies to determine whether SAAKO mice are protected from angiotensin II (AngII)-induced atherosclerosis (manuscript in preparation). AngII infusion resulted in plasma SAA levels (192.1 ± 126.9 μg/ml) in male SAAWT mice that were in the range of values induced by 12-week Western diet feeding (Table 2) without altering plasma cholesterol levels. However, despite the increase in circulating SAA, atherosclerotic lesion areas were similar in SAAWT and SAAKO mice after AngII infusion, indicating that endogenous SAA does not play a role in accelerated atherosclerosis induced by AngII. Taken together, comprehensive studies under three experimental conditions strongly support the conclusion that endogenous SAA, expressed at levels achieved in our studies, does not impact atherosclerotic lipid deposition in apoE-deficient mice.
Although our data clearly establish that SAA is not essential for atherosclerotic lipid deposition in a widely utilized mouse model, we cannot ignore abundant evidence that this acute phase reactant is strongly linked to human CVD. Patients with elevated plasma SAA concentrations have higher rates of adverse events (death, myocardial infarction, or urgent target-vessel revascularization) at 30 days irrespective of whether another acute phase reactant, CRP, is elevated.7 In patients with acute myocardial infarction, concentrations of SAA, but not CRP, are increased at the site of plaque rupture.33 Acute phase SAA has been conserved through ∼ 500 million years of evolution, and during severe inflammation it can account for as much as 2.5% of hepatic protein production.34 Thus, from an evolutionary perspective, acute phase SAA should perform an important survival role in the systemic response to acute injury and infection, with HDL likely serving as the vehicle for transporting SAA from the liver to sites of tissue injury. Consistent with this concept, SAA has been implicated in a number of processes important for tissue remodeling and repair, including leukocyte chemotaxis35, 36, inflammatory cytokine induction15, 16, 26, and up-regulation of genes involved in extracellular matrix remodeling, including transforming growth factor-β (TGF-β)12 and matrix metalloproteinases (MMPs).17 However, many of these potentially atherogenic effects were based on studies using a commercially available hybrid SAA that is not identical to any known SAA isoform. A recent study suggests that this recombinant molecule may exert some effects not elicited by human or mouse SAAs.18 In our studies the absence of endogenous acute phase SAA did not impact the macrophage content of atherosclerotic lesions in the aortic root of male mice fed a Western diet for 12 weeks, nor the expression of macrophage and inflammatory markers in the adipose tissue of female mice fed a normal rodent diet for 50 weeks. In a recent study, high-level expression of SAA1.1 in a transgenic mouse did not evoke an inflammatory response, providing additional evidence that SAA may not be intrinsically pro-inflammatory.19 Nevertheless, in the setting of chronic inflammation, such as rheumatoid arthritis1, obesity3, 4, and diabetes and insulin resistance2, 3, modest and sustained increases in SAA may contribute to maladaptive remodeling of atherosclerotic lesions, leading to plaque instability and rupture. Thus, further studies are needed to determine whether SAA promotes CVD events through pathological actions that occur during later stages of atherosclerosis
Supplementary Material
Significance.
Cardiovascular disease is a major cause of death worldwide. Atherosclerosis, characterized by inflammation and accumulation of lipids and fibrous material in the arterial wall, is the leading underlying contributor to cardiovascular disease. Plasma concentrations of serum amyloid A (SAA), an inflammatory acute phase reactant, are strongly associated with increased risk for cardiovascular disease. However, whether elevated SAA concentration is a consequence of inflammation or contributes to a therogenesis has not been clearly established, in part due to a lack of a suitable animal model. We generated mice that do not express acute phase SAA (SAAKO mice) and crossed them with hyperlipidemic apoE-/- mice. Our studies show that endogenous acute phase SAA does not impact atherosclerotic lesion formation in apoE-/- mice fed either normal or fat-enriched diets
Acknowledgments
Sources of funding: This work was supported by National Institutes of Health Grant P01HL086670. The studies were supported with resources and facilities provided by the Lexington, Kentucky Veterans Affairs Medical Center and by a grant from the National Institute of General Medical Sciences (8 P20 GM103527-05) of the National Institutes of Health.
Abbreviations
- SAA
Serum amyloid A
- SAAKO mice
apoE-/- mice deficient in SAA1.1 and SAA2.1
- SAAWT mice
apoE-/- mice expressing SAA1.1 and SAA2.1
- FASN
fatty acid synthase
- ORO
Oil Red O
- RCT
Reverse cholesterol transport
Footnotes
Disclosures: none
References
- 1.Wong M, Toh L, Wilson A, Rowley K, Karschimkus C, Prior D, Romas E, Clemens L, Dragicevic G, Harianto H, Wicks I, McColl G, Best J, Jenkins A. Reduced arterial elasticity in rheumatoid arthritis and the relationship to vascular disease risk factors and inflammation. Arthritis Rheum. 2003;48:81–89. doi: 10.1002/art.10748. [DOI] [PubMed] [Google Scholar]
- 2.Ebeling P, Teppo AM, Koistinen HA, Viikari J, Ronnemaa T, Nissen M, Bergkulla S, Salmela P, Saltevo J, Koivisto VA. Troglitazone reduces hyperglycaemia and selectively acute-phase serum proteins in patients with Type II diabetes. Diabetologia. 1999;42:1433–1438. doi: 10.1007/s001250051315. [DOI] [PubMed] [Google Scholar]
- 3.Leinonen E, Hurt-Camejo E, Wiklund O, Hulten LM, Hiukka A, Taskinen MR. Insulin resistance and adiposity correlate with acute-phase reaction and soluble cell adhesion molecules in type 2 diabetes. Atherosclerosis. 2003;166:387–394. doi: 10.1016/s0021-9150(02)00371-4. [DOI] [PubMed] [Google Scholar]
- 4.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, Snitker S, Horenstein RB, Hull K, Goldberg NH, Goldberg AP, Shuldiner AR, Fried SK, Gong DW. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006;3:e287. doi: 10.1371/journal.pmed.0030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 6.Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, Pepine CJ, Sharaf B, Bairey Merz CN, Sopko G, Olson MB, Reis SE. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women's Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109:726–732. doi: 10.1161/01.CIR.0000115516.54550.B1. [DOI] [PubMed] [Google Scholar]
- 7.Kosuge M, Ebina T, Ishikawa T, Hibi K, Tsukahara K, Okuda J, Iwahashi N, Ozaki H, Yano H, Kusama I, Nakati T, Umemura S, Kimura K. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ J. 2007;71:186–190. doi: 10.1253/circj.71.186. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien KD, McDonald TO, Kunjathoor V, Eng K, Knopp EA, Lewis K, Lopez R, Kirk EA, Chait A, Wight TN, deBeer FC, LeBoeuf RC. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:785–790. doi: 10.1161/01.ATV.0000158383.65277.2b. [DOI] [PubMed] [Google Scholar]
- 9.King VL, Hatch NW, Chan HW, de Beer MC, de Beer FC, Tannock LR. A murine model of obesity with accelerated atherosclerosis. Obesity (Silver Spring) 2010;18:35–41. doi: 10.1038/oby.2009.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, Kelvin DJ, Oppenheim JJ. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–209. doi: 10.1084/jem.180.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson PG, Thompson JC, Webb NR, de Beer FC, King VL, Tannock LR. Serum amyloid A, but not C-reactive protein, stimulates vascular proteoglycan synthesis in a pro-atherogenic manner. Am J Pathol. 2008;173:1902–1910. doi: 10.2353/ajpath.2008.080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Chai H, Wang Z, Lin PH, Yao Q, Chen C. Serum amyloid A induces endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H2399–2408. doi: 10.1152/ajpheart.00238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witting PK, Song C, Hsu K, Hua S, Parry SN, Aran R, Geczy C, Freedman SB. The acute-phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-density lipoprotein. Free Radic Biol Med. 2011;51:1390–1398. doi: 10.1016/j.freeradbiomed.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 15.Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun. 2000;268:405–408. doi: 10.1006/bbrc.2000.2143. [DOI] [PubMed] [Google Scholar]
- 16.Song C, Hsu K, Yamen E, Yan W, Fock J, Witting PK, Geczy CL, Freedman SB. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis. 2009;207:374–383. doi: 10.1016/j.atherosclerosis.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JI, Kwak JY, Bae YS. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem Biophys Res Commun. 2005;330:989–998. doi: 10.1016/j.bbrc.2005.03.069. [DOI] [PubMed] [Google Scholar]
- 18.Christenson K, Bjorkman L, Ahlin S, Olsson M, Sjoholm K, Karlsson A, Bylund J. Endogenous Acute Phase Serum Amyloid A Lacks Pro-Inflammatory Activity, Contrasting the Two Recombinant Variants That Activate Human Neutrophils through Different Receptors. Front Immunol. 2013;4:92. doi: 10.3389/fimmu.2013.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simons JP, Al-Shawi R, Ellmerich S, et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc Natl Acad Sci U S A. 2013;110:16115–16120. doi: 10.1073/pnas.1306621110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashby D, Gamble J, Vadas M, Fidge N, Siggins S, Rye K, Barter PJ. Lack of effect of serum amyloid A (SAA) on the ability of high-density lipoproteins to inhibit endothelial cell adhesion molecule expression. Atherosclerosis. 2001;154:113–121. doi: 10.1016/s0021-9150(00)00437-8. [DOI] [PubMed] [Google Scholar]
- 21.Kim MH, de Beer MC, Wroblewski JM, Webb NR, de Beer FC. SAA does not induce cytokine production in physiological conditions. Cytokine. 2013;61:506–512. doi: 10.1016/j.cyto.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, Rothblat GH, Reilly MP. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119:1135–1145. doi: 10.1161/CIRCULATIONAHA.108.810721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, van der Giet M, Tietge UJ. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2) J Lipid Res. 2010;51:743–754. doi: 10.1194/jlr.M000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik P, Berisha SZ, Santore J, Agatisa-Boyle C, Brubaker G, Smith JD. Zymosan-mediated inflammation impairs in vivo reverse cholesterol transport. J Lipid Res. 2011;52:951–957. doi: 10.1194/jlr.M011122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Beer MC, Wroblewski JM, Noffsinger VP, Ji A, Meyer JM, van der Westhuyzen DR, de Beer FC, Webb NR. The Impairment of Macrophage-to-Feces Reverse Cholesterol Transport during Inflammation Does Not Depend on Serum Amyloid A. J Lipids. 2013;2013:283486. doi: 10.1155/2013/283486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, Zhang Y, Zhang C, An F. Serum amyloid a directly accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Mol Med. 2011;17:1357–1364. doi: 10.2119/molmed.2011.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Beer MC, Webb NR, Wroblewski JM, Noffsinger VP, Rateri DL, Ji A, van der Westhuyzen DR, de Beer FC. Impact of serum amyloid A on high density lipoprotein composition and levels. J Lipid Res. 2010;51:3117–3125. doi: 10.1194/jlr.M005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O'Brien KD, Chait A. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. 2004;110:540–545. doi: 10.1161/01.CIR.0000136819.93989.E1. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Ambrosi J, Azcona C, Patino-Garcia A, Fruhbeck G. Serum Amyloid A concentration is increased in obese children and adolescents. J Pediatr. 2008;153:71–75. doi: 10.1016/j.jpeds.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y, He X, Shi X, Huang C, Liu J, Zhou S, Heng CK. Association between serum amyloid A and obesity: a meta-analysis and systematic review. Inflamm Res. 2010;59:323–334. doi: 10.1007/s00011-010-0163-y. [DOI] [PubMed] [Google Scholar]
- 31.O'Brien KD, Brehm BJ, Seeley RJ, Bean J, Wener MH, Daniels S, D'Alessio DA. Diet-induced weight loss is associated with decreases in plasma serum amyloid a and C-reactive protein independent of dietary macronutrient composition in obese subjects. J Clin Endocrinol Metab. 2005;90:2244–2249. doi: 10.1210/jc.2004-1011. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A, 3rd, Kirk EA, O'Brien KD, Chait A. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:685–691. doi: 10.1161/ATVBAHA.107.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, Sutsch G, Roffi M, Neidhart M, Eberli FR, Tanner FC, Gobbi S, von Eckardstein A, Luscher TF. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. 2005;111:1355–1361. doi: 10.1161/01.CIR.0000158479.58589.0A. [DOI] [PubMed] [Google Scholar]
- 34.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee HY, Kim SD, Shim JW, Lee SY, Lee H, Cho KH, Yun J, Bae YS. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J Immunol. 2008;181:4332–4339. doi: 10.4049/jimmunol.181.6.4332. [DOI] [PubMed] [Google Scholar]
- 36.Badolato R, Johnston JA, Wang JM, McVicar D, Xu LL, Oppenheim JJ, Kelvin DJ. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J Immunol. 1995;155:4004–4010. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.