

Abstract
Transforming growth factor β (TGFβ) plays critical roles in regulating a plethora of physiological processes in normal organs, including morphogenesis, embryonic development, stem cell differentiation, immune regulation, and wound healing. Though considered a tumor suppressor, TGFβ is a critical mediator of tumor microenvironment, in which it likewise mediates tumor and stromal cell phenotype, recruitment, inflammation, immune function and angiogenesis. The fact that activation of TGFβ is an early and persistent event in irradiated tissues and that TGFβ signaling controls effective DNA damage response provides a new means to understand both tissue and tumor responses to radiation. Here we discuss pre-clinical studies unraveling TGFβ effects in radiation responses and review TGFβ biology in lung cancer as an example of the opportunities for TGFβ pathway inhibition as a pharmaceutical approach to augment radiation therapy.
Keywords: TGFβ, lung cancer, tumor microenvironment
Genetic and cell biology studies indicate that cancer should be considered as an organ in which malignancy, like function, is not just determined by parenchymal cells themselves, but also by their stroma and microenvironment1. Tumor companion cells, such as myeloid cells, phagocytes and immune cells, intermingled in the tumor-associated fibroblastic stroma comprises most of the tumor mass in carcinomas and provides the soil in which malignant cells will grow, invade and metastasize2. In order to treat cancer, this growing body of evidence that solid tumors are organized in a manner that resembles an organ requires better understanding of the complex ways in which cancer cells interact with their surroundings, both locally and systemically.
Radiation therapy is prescribed to more than half of cancer patients3. Although mechanisms of radiation response and tumor cells are well understood on a molecular level, much remains uncertain concerning the interaction of cells within irradiated tumor microenvironment. In studies spanning two decades, we and others have identified transforming growth factor β (TGFβ) as a key extracellular signal in irradiated tissues and tumors. Among all radiation-induced cytokines in the tumor microenvironment, TGFβ arguably elicits the most complex and far-reaching effects in determining outcome. Evidence from pre-clinical models suggests that TGFβ inhibition may provide an effective means to improve the therapeutic response to radiation. Here we consider cellular and multicellular processes mediated by TGFβ in response to radiation, with particular reference to TGFβ biology and the therapeutic opportunity in treating non-small cell lung cancers.
TGFβ Signaling
The effects of TGFβ are mediated by three TGFβ ligands— TGFβ1, TGFβ2 and TGFβ3 — through TGFβ type 1 (TGFβR1) and type 2 receptors(TGFβR2)4. All three mammalian TGFß genes share a high degree of sequence homology in the ligand region and functional overlap. TGFß1 is the best studied protein of these three differentially expressed isoforms. During protein synthesis of the 390 amino acid polypeptide, intracellular proteolysis processes the precursor to yield a C-terminal peptide that dimerizes to form the mature 24 kDa cytokine (FIG 1). The remaining N-terminal peptide forms a 75 kDa glycosylated homodimer called the latency associated peptide (LAP) 5. LAP acts as a chaperone to ensure proper folding of TGFß and contains the signal peptide for secretion. Non-covalent association of LAP with its respective TGFß cytokine forms the latent TGFß (LTGFß) complex, referred to as the small latent complex. The so-called large latent complex in which LAP is covalently bound via a disulfide linkage to a protein called latent TGFß binding protein facilitates LTGFß sequestration within the extracellular matrix. Thus, biological activity of TGFß is restricted by the fact that it is secreted and sequestered in this latent state.
Figure 1. Schematic of TGFβ production as a latent complex.
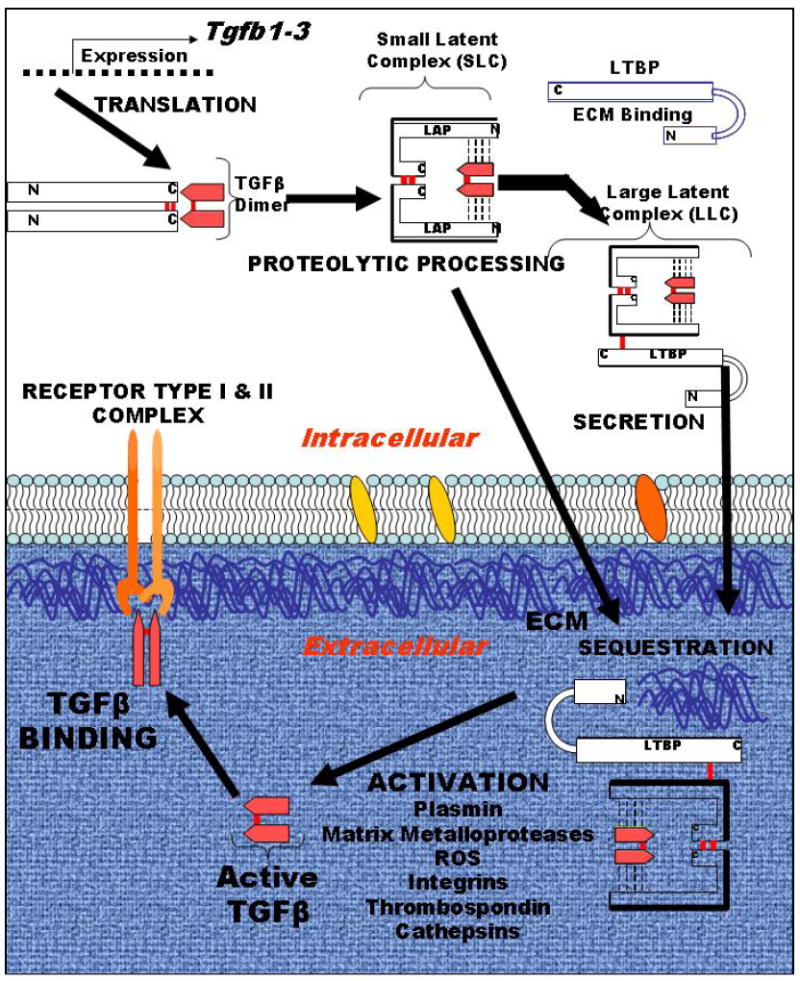
TGFβ 1-3 isoform genes encode a single polypeptide that is processed, cleaved to make a homodimer of the pre-pro peptide, which is designated as latency associated protein (LAP), that is non-covalently bound to TGFβ proper, which is a 24 kD homodimer, which associate in a non-covalent bound complex. LAP acts as a chaperone for proper folding, contains the signal sequence for secretion and sequesters TGFβ until activation. This so-called small latent complex can also form the large latent complex upon covalently binding the latent TGFβ binding proteins (LTBP), which sequester latent TGFβ in the extracellular matrix (ECM). Activation occurs by multiple relatively understudied mechanisms, as listed, to release TGFβ for binding to its receptors, type I and II, which then initiates the signaling cascade.
Latent TGFβ1 is abundant and stored in extracellular reservoirs until activation by extracellular stimuli, such as proteases, reactive oxygen or radiation, that release TGFΒ to bind to cell surface receptors. TGFΒ signals via ubiquitously expressed cell surface type I and type II receptors that in turn phosphorylate Smad proteins that translocate to the nucleus to mediate transcription directing differentiation, function, proliferation and apoptosis depending on cell type and context (reviewed in6). Despite 25 years of study, TGFΒ actions are still being defined and new activities are being discovered.
Release of TGFß from its LAP, referred to as activation, is required before the cytokine can bind to cell surface receptors, which places the modes of activation as critical determinants of its biological activity5, 7. Although all three isoforms bind to the same cell surface receptors, gene knockout of the three genes results in distinct phenotypes8-10, suggesting that specificity of action may reside at the level of susceptibility to different modes of activation. LTGFß can be activated in solution using highly acidic or basic conditions or by heat treatment11 but these activation mechanisms are not physiologically relevant. Most activation mechanisms require the participation of one or more additional proteins generally localized to the cell surface and these activation modes are relatively inefficient 5.
Rapid activation of LTGFß, as demonstrated by an increase in reactivity of TGFß epitopes that are masked by the latent complex, is observed in vivo after exposure to IR12, 13. Because reactive oxygen species (ROS) are a product of the interaction of IR with water or with cell membranes, we postulated that the rapid activation of TGFß in vivo could be due to ROS generated by IR if the protein itself contained redox sensitive amino acids. Redox switches that alter conformation and activity are found in a variety of proteins. Examples include the clotting cascade mediator, thrombomodulin and transcription factors like Sox and p53. We showed that solution sources of ROS generated by Fenton chemistry efficiently release biologically active TGFß from recombinant LTGFΒ in the absence of cells or other proteins14. Indeed, solution sources of ROS generate more biologically active TGFΒ as measured by bioassay than does heat or acid, possibly because the latter can also denature the ligand. This mode of activation has been confirmed in recent studies using asbestos generated ROS in solution15. Since ROS induces very efficient LTGFΒ activation that does not require participation of additional cellular machinery and since ROS are widely generated as by products of cellular metabolism, inflammation, ischemia/reperfusion injury, as well as exposure to exogenous agents such as IR, chemotherapeutic agents and asbestos, LTGFΒ could function as an extracellular sensor of oxidative stress14.
The specific mediators and requirements for the ROS mechanism of activation showed that redox sensitivity is restricted to LTGFβ116. Although the three latent TGFΒ proteins share 75% sequence identity, their respective LAP exhibit only 34-38% identity, which supports the idea that the isoforms differ in structural susceptibilities to modes of activation. Specific radical scavengers pinpointed hydroxyl radical as the critical ROS for activation in the Fenton chemistry cell-free model for generating ROS. It was postulated that oxidation of specific amino acids within LTGFβ1 causes a conformational change in the latent complex, allowing release of active TGFβ. The target of ROS modification is LAP.
Comparison of the primary amino acid sequences of the three LAP regions of the TGFß isoforms. Each monomer of LAPβ1 contains five methionine residues, which are susceptible to oxidation. Of these five, two methionines (132 and 253) are unique to LAPβ1. Single point amino acid changes to alanine were made to methionines at positions 132 or 253 unique to LAPβ1. Like wild type, the expressed proteins containing site-directed mutations formed latent complexes that could be activated by heat treatment, however mutant M253A LAPβ1 was resistant to ROS mediated activation. The specificity and efficiency of the oxidative activation mechanism indicates that LTGFβ1 operates as a tissue sensor of oxidative damage and that TGFß1 release is a signal transducer17. The redox switch provides a highly efficient mechanism by which radiation increases TGFΒ activity.
Once released from the latent complex, TGFβ ligands bind to TGFβ receptor II (TBRII) with high affinity. Both type I and type II receptors contain serine/threonine kinase domains in their intracellular portions4. Binding of the ligand causes the formation of heterotetrameric active receptor complexes that result in the phosphorylation of the type I receptor by the type II receptor. TGF-βs elicit their effects by binding to cell surface receptors. The tumor suppressor activity of TGFβ is predominantly mediated through its effect on cell cycle progression and apoptosis. Inhibition of c-Myc, which is known to promote cell cycle entry into S phase by regulating various cell cycle related genes, by TGFβ signaling is very well established18. Repression complex of SMAD3, E2F4/5 and p107 which are induced upon TGFβ signaling repress the expression of c-Myc19. Stimulation of p15ink4b and p21cip1 expression by TGFβ leads to inhibition of cyclin D-CDK4/6 and Cyclin A/E-CDK2 activity20.
Although TGFβ has an anti-proliferative effect on most epithelial cells and haematopoietic cells, it promotes proliferation of certain mesenchymal cells, including smooth muscle cells, through the induction of platelet-derived growth factor (PDGF)21. Similarly, TGFβ induces the proliferation of certain types of cancer cells, including glioma and osteosarcoma cells, through the induction of PDGFA or PDGFB22, 23. Abnormal proliferation observed in cancer cells is caused by mutations that either increase positive growth signals or decrease negative growth control signals or a combination of both events. Tumors may use various mechanisms anywhere along alterations in TGFβ receptors, its primary cytoplasmic signal transducers, the Smad proteins, to circumvent the growth inhibitory effects of TGFβ24. TGFβR2 mutations have been reported in several epithelial type human malignancies, such as pancreatic, and breast cancer, which lose their growth inhibitory response to TGFβ25-27.
Almost all cancer cells lose the proliferative arm of TGFβ signaling by one means or another, but maintain some degree of response, and in general produce and activate significantly more TGFβ than surrounding normal tissue; these events characterize the so-called TGFβ switch from a tumor suppressor to a tumor promoter (reviewed in28-30)
TGFβ is strongly implicated in the acquisition of invasive and metastatic disease, in large part via activation of the epithelial to mesenchymal transition (EMT) phenotypic program, usurped from development and wound healing where it provides a means for transient motility of epithelial cells.
TGFβ’s Role in the Composition of the Tumor Microenvironment
The normal tissue stroma is essential for maintenance and integrity of epithelial tissues and contains a multitude of cells that collaborate to sustain normal tissue homeostasis. There is a continuous and bilateral molecular crosstalk between normal epithelial cells and cells of the stromal compartment, mediated through direct cell-cell contacts or by secreted molecules. There is a similarity between stroma from normal organs and tumors, because both entities have active angiogenesis and numerous proliferating fibroblasts secreting a complex extracellular matrix (ECM)31. Fibroblasts are mesenchymally derived cells present in the stroma of most tissues. During lung development they or their progenitors are involved in epithelial-mesenchymal crosstalk, helping to shape the organ.
TGFβ also plays a central role in physiological fibrogenesis and pathological pulmonary fibrosis by promoting the activation, proliferation, and differentiation of epithelial cells and collagen-producing myofibroblasts32. After lung injury, TGFβ from the recruited leukocytes trigger fibroblasts to proliferate and differentiate into myofibroblasts, which release ECM components. However, if this process becomes dysregulated it can lead to the development of a permanent lung pathogenesis such as pulmonary fibrosis and chronic obstructive pulmonary disease33.
Most epithelial-derived tumors are characterized by the generation of mesenchymal-derived stromal cells, including intratumoral and peritumoral carcinoma-associated fibroblasts (CAF) associated with tumor growth, angiogenesis, invasion, and metastasis34, 35. CAF are phenotypically distinguishable from normal fibroblasts. Although their heterogeneity is yet to be fully explored, at least a subset of CAF have been characterized as myofibroblasts based on expression of α-smooth muscle actin (α-SMA). The mechanism of fibroblast activation in cancer is not completely understood. Indeed, even the origins of activated fibroblasts in tumors are incompletely described. Currently it is believed that the majority of CAF arise from the activation of resident fibroblasts. However, activated fibroblasts can also originate from pericytes, vascular smooth muscle cells, bone marrow-derived mesenchymal cells, and from epithelial to mesenchymal cell transition36. In adult tissues, differentiation from resident stromal fibroblasts into activated myofibroblasts occurs through paracrine signaling with TGFβ generated by damaged or inflamed tissues37-39. Such TGFβ-mediated activation of CAFs may occur in tumors. Both TGFβ and interleukin-1β induce differentiation of quiescent fibroblasts into activated myofibroblasts, but TGFβ is considered the predominate inducer40.
Importantly, the FAP promoter has an EGR-1 binding site, and EGR-1 binding is important for FAP expression41. It is rational to assume that FAP may be induced in resident tissue fibroblasts that are activated by TGFβ released from stromal cells or epithelial cancer cells. The autocrine-signaling TGFβ-SDF-1 loops initiate and maintain the differentiation of fibroblasts into myofibroblasts which gives rise to tumor-promoting CAF myofibroblasts during tumor progression42. CAFs also promote tumor progression through communications with pericytes and endothelial cells that promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion43. Communication between fibroblasts and cancer cells often involves other cell types: cancer cell-secreted platelet-derived growth factor (PDGF) can recruit macrophages, which then produce TGFβ that, in turn, induces development of reactive fibroblasts44. These examples underscore how TGFβ activity is often amplified in the tumor microenvironment. TGFβ produced by resistant cancer cells induces an activated phenotype, e.g. CAF, that then contributes more TGFβ to the microenvironment, which promotes more recruitment, e.g. BMDC, and remodeling, the dysfunctional cancer dynamic leading to ”the wound that does not heal” 31.
TGFβ pathway as an emerging target for anti-angiogenic therapy
Angiogenesis is the formation of new capillaries from pre-existing vessels which is of fundamental importance in development, normal organ maintenance and survival. This is a complex process that requires interaction between different cell types, the extracellular matrix and several cytokines and growth factors. Tumor angiogenesis relies on many of the same processes as those involved in physiological angiogenesis. Ischemia and hypoxic conditions, which initiate a cascade of highly coordinated cellular functions resulting in the establishment of new blood vessels and oxygen and nutrient supply, are major drivers of both physiological and tumor angiogenesis. Under physiological conditions, the chain of events and change in cellular function and composition recede following vascular perfusion.
By contrast, during tumor angiogenesis, the angiogenic cascade is persistent and unresolved, fuelled in part by tumor-secreted factors and tumor hypoxia. Angiogenesis contributes to the progression of cancer from a dormant in situ lesion to a life-threatening metastatic disease. The key role of vascular endothelial growth factor A (VEGFA) and its receptor VEGF receptor 2 (VEGFR2) in tumor angiogenesis is firmly established. VEGFA expression has been correlated with both vessel count and poor prognosis in patients with NSCLC, and anti-VEGF therapy has been approved by the FDA in this tumor type.
The ability of tumor cells to induce new blood vessel formation is essential for progressive tumor growth and blood-bone metastasis. Genetic studies in mouse and human have provided much evidence of the strong link between the TGFβ signaling pathway and vascular morphogenesis and dysfunction. Deletion of the Tgfb1 gene in the mouse results in embryonic lethality because of defective yolk sac vasculogenesis. Targeted deletions of Acvrl1 (ALK 1), Tgfbr1 (ALK 5), Tgfbr2 and Eng endoglin in mice result in vascular abnormalities45. Studies in animal models and in humans suggest that the pro-angiogenic effects of TGFβ1 are dependent on the activation of its downstream signaling molecules: ALK1, which functions as a positive regulator of endothelial cell migration and proliferation, and ALK5, which promotes vessel maturation and is anti-angiogenic46, 47. Therefore, the ratio of TGFβ signaling through ALK1 and ALK5 probably determines its effect on angiogenesis. Furthermore, TGFβ signaling might be modulated by the TGFβ1 type 3 receptor (endoglin) that is expressed on activated endothelial cells, which enhances TGFβ1–ALK1 signaling while exerting inhibitory effects on TGFβ1–ALK5 signaling48. TGFβ pathway acts as a potent inducer of tumor angiogenesis in several assays.
Tumor angiogenesis is crucial for tumor growth and invasion, as blood vessels deliver nutrients and oxygen to the tumor cells and allow them to intravasate the blood system. Several models indicate the role of tumor cell–secreted TGF-β1 in tumor angiogenesis. Increased expression of TGF-β1 in transfected prostate carcinoma or hepatocellular carcinoma enhanced tumor angiogenesis, and local administration of neutralizing antibodies to TGF-β1 strongly reduced tumor angiogenesis49, 50. Intraperitoneal injection of TGFβ antibodies reduced angiogenesis and tumorigenicity of a renal carcinoma cell line that lacks TβRII, and is therefore not responsive to TGF-β51. TGFβ blockade significantly inhibited the expression of VEGF preventing abnormalization of diaphragm lymphatic vessels and completely abolished ascites formation52.
The mechanism of angiogenesis stimulation by TGFβ signaling also includes the induction of key angiogenic factors such as connective tissue growth factor (CTGF) and VEGFA which directly act on endothelial cells to stimulate cell proliferation and migration53, 54. In addition, TGFβ can induce the expression, secretion and activation of matrix metalloproteinase 2 (MMP2) and MMP9, and downregulate the expression of tissue inhibitor of metalloproteinase (TIMP) in tumor and endothelial cells28. These metalloproteinase activities result in the enhancement of migratory and invasive properties of endothelial cells, which are required for tumour angiogenesis. Indirect stimulation of angiogenesis by TGFβ may occur through the potent chemoattractant activity of TGFβ for monocytes, which release angiogenic cytokines55. Thus, TGFβ–induced changes in the microenvironment provide favorable conditions for endothelial cell migration and capillary formation.
TGFβ a master regulator of inflammation in the tumor microenvironment
Inflammatory cells are components of the microenvironment of normal tissues and organs, regulating various processes during development, including epithelial growth and branching and clearance of apoptotic cells56. Inflammation and the significant effect of the tumor microenvironment on tumor progression and aggressiveness are identified as the hallmarks of cancer57. There is a progressive change in the composition of the immune cell infiltrate with tumor stage to one that is more conducive to tumor progression58. Interactions between cancer cells and cells of the innate and adaptive immune system occur in the tumor organ and illustrate the complexity and dynamics of the tumor tissue. The tumor microenvironment comprises inflammatory cells, including Tie2+,VEGFR1+, CD11b+, and F4/80+ populations; innate and adaptive immune cells, including natural killer (T) cells, T cells, and B cells; as well as cancer-associated fibroblasts we discussed above59. These stromal cells collectively create an environment that promotes tumor progression by providing growth factors, pro-angiogenic factors, proteases, and adhesion molecules that facilitate tumor cell proliferation, angiogenesis, invasion, and metastasis60, 61. This dynamic tumor microenvironment provides a selective pressure for tumor cell variants that give rise to genomic instability, genomic heterogeneity, and epigenetic alteration62.
TGFβ controls immune responses and maintains immune homeostasis through its impact on proliferation, differentiation and survival of multiple immune cell lineages. Overproduction of TGFβ by tumor cells and tumor infiltrating leukocytes has an adverse effect on anti-tumor immunity and inhibits significantly host tumor immune surveillance63. TGFβ markedly and directly suppresses the cytotoxic program of CTLs and regulates both the clonal expansion of CD8+ T cells and CD8+ T-cell cytotoxicity in vivo64. TGFβ also has a significant impact on CD4+ T-cell differentiation and function. TGFβ induces Foxp3 and generates induced regulatory T cells (Tregs)65. Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of immature dendritic cells (DCs), macrophages, granulocytes and other myeloid cells in early stages of their differentiation and have properties similar to those that have been described for M2 macrophages. Increased levels of MDSC, marked by CD11b+/CD14+/CD15+/CD33+, were observed in the peripheral blood of advanced stage NSCLC patients (n =87) compared with healthy controls66. In late stages of tumor development, TAMs and MDSCs can produce TGFβ and are classically involved in cancer progression and metastasis67. It is not clear whether TGFβ is directly involved in converting TAM from an M1 to M2 phenotype or whether, TAM may contribute to the general immunosuppressive tumor microenvironment by producing large amounts of TGFβ.
Similar to TAM, tumor-associated neutrophils (TAN) also have polarized functions. Although no published studies have investigated the role of TAN in human NSCLC, recent preclinical work by Fridlender and colleagues showed that in mouse models of lung cancer, TGFβ induces a population of TAN with protumor function (N2), whereas TGFβ blockade results in antitumor neutrophils68. Depletion of these N1 neutrophils resulted in increased tumor growth68. TGFβ also regulates production of IL-1 to control chronic inflammation, and the production of chemokines and chemokine receptors that recruit inflammatory cells69. These chemokines include CXCL12, a key regulator of hematopoietic stem and progenitor cell trafficking between the peripheral circulation and targeted tumor tissues69. SDF-1 mediates its effects on chemotaxis through its receptor, CXCR4, which is highly expressed on putative stem and progenitor cells60. CXCR4 is the receptor for CXCL12. In lung adenocarcinoma, tumor expression of CXCL12 has been shown to correlate with accumulation of CXCR4-expressing immune cells, 30% of which were protumor Treg70.
Inflammatory cells are another component of the tumor microenvironment. Brown and colleagues provided compelling evidence that ionizing radiation induced recruitment of tumor-protective BMDC, that promote blood vessel formation sufficient to support the growth of recurring tumors post-irradiation71. Suppression of BMDCs after irradiation with antibodies against CD11b greatly71 enhance the tumor radiation response in preclinical models. Conversely, inhibition of tissue resident macrophage before radiotherapy significantly protects against radiation-induced normal tissue damage72. Moreover, CAF promote further cell recruitment through secretion of chemokines such as SDF-1, which in turn enhance the recruitment of BMDCs involved in tumor vasculogenesis43. As discussion above, TGFβ activation is of great relevance, which includes the recruitment BMDCs to the tumor, aiding in the recovery of tumor growth by enhancing angiogenesis, supplying growth factors and creating a local immunosuppressive environment.
Alteration TGFβ transduction pathways augment radiation response, from “Molecular” to “Microenvironment”
The sensitivity of LTGFΒ activation in vivo13 and demonstration of a redox switch14, 16. suggested that it could have a role in orchestrating cell responses proximal to DNA damage. Consistent with this idea, epithelial cells of Tgfb1 knockout embryos did not undergo radiation-induced apoptosis or cell cycle arrest after dams were irradiated with 5 Gy 73. TGFβ depletion, by either gene knockout or by using TGFΒ neutralizing antibodies, also decreased p53 ser-18 phosphorylation in irradiated tissue. These data indicate that TGFβ is essential for p53-mediated cellular responses that mediates cell fate decisions in vivo73.
Studies in primary Tgfb1 null and wildtype mammary epithelial cultures established that the effects of TGFΒ on radiation-induced molecular events and cell fate decisions are cell intrinsic, and are compromised as a function of TGFΒ abundance (i.e. both heterozygote and null cells were compromised when compared to wildtype cells, and when compared to each other). Treatment with TGFβ restored the molecular and cell fate response. Moreover the mouse phenotype was replicated in human cells using a small molecule inhibitor the TGFΒ type I receptor (TBRIKI). In both models, TGFβ treatment prior to irradiation restored radiation-induced signaling events. Thus, although radiation elicits TGFβ activation through its redox sensor function, epithelial cells have severely compromised DNA damage response if TGFβ is not present before they are exposed. Inhibition of TGFβ signaling in normal epithelial cells, either by chemical or by genetic means, leads to increased radiosensitivity as measured by clonogenic survival74.
It is interesting to ask why extracellular TGFβ controls the intracellular DNA damage response, but it is also interesting to ask how. ATM is a nuclear sensor of DNA damage that initiates, recruits and activates a complex program of checkpoints for cell cycle, apoptosis and genomic integrity 75, 76. TGFβ1 depletion compromises radiation-induced ATM kinase activity and autophosphorylation, leading to reduced phosphorylation of critical DNA damage transducers γH2AX, Chk2, p53 and Rad1774. Likewise, TGFβ1 antisense was also demonstrated to block ATM kinase and DNA damage response in A549 lung cancer cells77. Recent studies further support the generality that TGFβ inhibition prior to radiation attenuates DNA damage responses and increases clonogenic cell death in human and murine breast cancer and glioma cells 78, 79.
Recent studies shed light on the importance of the tumor microenvironment in modulating the tumor response to radiotherapy and open new opportunities for the development of novel therapeutic strategies to synergize radiotherapy. Increasingly evidence shows that ionizing radiation induces modifications of the tumor microenvironment, which profoundly impacts tumor biology which is particularly relevant to radiation response71, 80. Strategies to improve radiotherapy are changing from “molecular” regulatory cancer cell intrinsic sensitivity to targeting of the “microenvironment” for better irradiation responses. As in normal organs, tumor vasculature is an important component in tumor stroma to ensure growth and development. Endothelial cell apoptosis precedes tumor cell apoptosis by 3 to 5 days, suggesting that tumor cells are dependent on endothelial cells for survival81. Tumor vascular endothelium plays a crucial role in tumor radiation response. Paris et al. also suggest that early phase microvascular endothelial apoptosis is mandatory for tumor cure82. Recent data indicate that radiation-induced endothelial cell apoptosis83 can lead to vascular destruction and secondary tumor cell death84.
TGFβ Biology in Non-Small Cell Lung Cancer
Human lung cancer can be divided into two main histopathological groups: non-small cell lung cancer (NSCLC) and small-cell lung cancer (SCLC). About 80% of lung cancers are NSCLC, which can be further subdivided into adenocarcinomas, squamous cell, bronchioalveolar, and large cell carcinomas85. Radiotherapy is a well-established therapeutic modality in NSCLC. However cancer recurrences after radiotherapy often develop into more aggressive disease that is difficult to treat and associated with poor prognosis.
NSCLC contain multiple cell types and extracellular matrix components and develop through complex interactions between these different components of the tissues using processes that often resemble those used by the lung organ. Distortion of TGFβ signaling is perhaps the most important prerequisite in tumor progression in NSCLC86, 87. Reduced TGFβRII expression was reported in 40-80% of NSCLC at the protein and/or mRNA level and decreased sensitivity to the growth inhibitory effects of TGF-β88, 89. It has been demonstrated that both microsatellite instability and promoter methylation are associated with TGFβR2 mutations in NSCLC89, 90. Moreover, TGFβRII deletion increases proliferation, local inflammation, and TGFβ ligand elaboration; TGFβRII knockdown in airway epithelial cells increases migration and invasion in both lung adenocarcinoma and squamous cell carcinoma indicating that TGFβRII loss plays a causal role in lung carcinogenesis87.
Reduced TGFβRII expression in human NSCLC is associated with specific histologic subtypes, more aggressive tumor behavior, or reduced patient survival. Genetic variations in the TGFβ pathway were also predictors of survival in advanced NSCLC86. Interestingly, TGFβRII down-expression is more common in lung squamous carcinoma patients of males and smokers, and both are at higher risk of NSCLC87. Long-term cigarette smoke reduced treatment reduced apoptosis, increased cell viability, decreased TGFβ-mediated growth suppression through down-regulation of Smad3 and enhanced tumorigenicity in human lung adenocarcinoma. These data provide evidence that cigarette smoking promotes NSCLCS tumorigenicity partly by abrogating TGF-β-mediated growth inhibition and apoptosis by reducing expression of Smad324.
One of the most consistent histological features of NSCLC invasion is the appearance of desmoplasia: stromal changes characterized by the activation of stromal fibroblasts into CAF, increased matrix protein disposition, new blood vessel formation, and immune cell infiltration. The cell surface serine protease fibroblast activation protein-a (FAP-a) is selectively expressed on CAF in lung tumors as well as granulation tissue and in fibrotic lesions91. TGFβ is a powerful factor inducing FAP expression in NIH3T3 fibroblasts40. The induction of fibroblasts and FAP by TGFβ is thought to convert fibroblasts to a proinvasive state92. Targeting FAP-α inhibits tumor cell proliferation indirectly, increases accumulation of collagen, decreases myofibroblast content, and decreases blood vessel density in NSCLC91. Moreover, FAP-expressing cells are an immune-suppressive component of the tumor microenvironment in preclinical NSCLCs model. Depletion of FAP-expressing cells in established Lewis lung carcinomas, caused rapid hypoxic necrosis of both cancer and stromal cells in immunogenic tumors by a process involving interferon-γ and tumor necrosis factor–α93.
A recent study showed CAF significantly increases the invasiveness of co-cultured NSCLC cells compared with normal fibroblasts while enhancing tumorigenicity of A549 NSCLC cells in vivo94. Forty-six differentially expressed genes were identified between CAFs and NFs from 15 patients. These genes are involved in signal transduction (14/46), response to stress (11/46), cell adhesion (7/46), and angiogenesis (3/46). Fourteen genes differentially expressed between CAFs and NFs were also commonly differentially expressed in NSCLC tumor stroma compared with normal lung parenchyma. Importantly, 7 of the 14 overlap genes were reported as transcriptional targets of the TGFβ signaling pathway. There was prognostic significance of the CAF-associated gene-expression signature in multiple independent cohorts of NSCLC patients94.
Increased TGFβ expression correlates with poor prognosis, increased tumor growth and angiogenesis in NSCLCs95. TGFβ level was found significantly higher in patients with lymph node metastasis and advanced stage NSCLC95. It has been confirmed that anti-VEGF therapy inhibits tumor angiogenesis and subsequent tumor growth and metastases in NSCLCs96. A randomized trial demonstrated that adding bevacizumab to carboplatin and paclitaxel improved survival in advanced NSCLCs97.
Inflammation and tumor microenvironment are critical players in NSCLC cancer progression. Cytotoxic T lymphocytes (CTLs), mostly CD8+ were associated with prolonged survival in squamous cell carcinomas in a investigation of 1,290 NSCLC tumors98. Stromal CD4+ T-cell and co-localization of stromal CD8+ T cells and CD4+ T-cell have all shown association with improved NSCLC survival99, 100. The protumor association of Treg tumor infiltration was examined in a study of 100 stage I to III NSCLC tumors showing that tumor-infiltrating Foxp3+ Tregs increased tumor recurrence101. Antitumor tumor-associated macrophages (TAM) in NSCLC are of the M1 phenotype and accumulate intratumorally, whereas protumor TAMs of the M2 phenotype accumulate in the stroma and express interleukin-8 (IL-8), IL-10, and trigger receptor expressed on myeloid cells (TREM-1).
IL-8 is an angiogenic factor, and the angiogenic role of TAM in NSCLC has been shown by correlating macrophage density with intratumor microvessel counts and poor patient outcomes102. IL-10 is an immunosuppressive cytokine, and its expression by TAM has been observed more commonly in stages II, III, and IV NSCLCs, thus correlating with decreased overall survival (26). TAM expression of TREM-1, which can initiate and amplify an inflammatory response, is increased in malignant pleural effusions of NSCLC patients103. Furthermore, in 68 stage I to III NSCLC patients, an increased level of TREM-1 high TAM in resected specimens was an independent predictor of shorter overall survival103.
In patients with early stage NSCLC who are unable to tolerate surgical resection because of medical co-morbidities, conventional radiotherapy remains the standard therapy. However local recurrence is still the main cause of NSCLC radiotherapy failure, with local failure of up to 50% and poor long-term survival of 15–30%41, 104. There are essentially two approaches to improve local control with radiation therapy - either use a higher radiation dose or use more effective radiation sensitization.
Hypofractionated stereotactic body radiation therapy (SBRT) allows escalation of the fractional dose, which is important to improve the local control rate of tumors and overall survival for medically inoperable patients with early-stage NSCLC 105. However, there are still many issues to be elucidated. Firstly, radiation dose and fractionation vary because of a lack of biologically effective dose (BED) guidelines. Importantly, radiation induced normal tissue damage is still the biggest obstacle to limit the dose escalation of SBRT in NSCLC 106. Another approach that has enormous appeal involves the development of nontoxic, yet effective molecularly targeted radiosensitizers. If a drug selectively sensitizes the tumor response to radiation, one could use selective escalation of the effective biologic dose to the tumor, and therefore improve local control without increasing morbidity.
We hypothesize that the microenvironment regulates many tumor responses to radiation, thus providing novel routes for manipulating the response to ionizing radiation. Although little is known how TGFβ modulates the irradiated tumor microenvironment, given its pleiotropic roles in NSCLC it seems reasonable that TGFβ inhibition could both increase tumor cell radiosensitivity and shift microenvironment to augment NSCLC response to radiotherapy.
Preclinical studies support this conjecture that tumor growth, host cells and the composition of the microenvironment are highly interdependent (Du and Barcellos-Hoff, unpublished data). As found for brain and breast tumors78, 79, most murine and human lung cancer cells were sensitized by TGFβ inhibition prior to radiation as measured by in vitro clonogenic assays. Using the Lewis lung cancer syngeneic subcutaneous tumors, tumor growth control was significantly improved by use of TGFβ neutralizing antibodies concurrent with single or fractionated radiation treatment. Notably, even though irradiated tumors treated with TGFβ inhibition were significantly smaller at experiment termination, hypoxia was higher and vessel density was also significantly decreased, compared with non-irradiated, bigger tumors. Martin Brown has shown that hypoxia promotes mobilization of CD11b+ monocytes producing the pro-angiogenic factor MMP9 into the tumor microenvironment, which in preclinical GBM is necessary for tumor regrowth – and that blockade of this crucial event prevents tumor recurrence71. The combined treatment of radiation and TGFβ inhibition decreased CD11b+/MMP9 monocytes, consistent with Brown and colleagues but suggestive that TGFβ is necessary for the recruitment of the CD11b+/MMP9 cells and therefore, tumor regrowth.
Given that radiation induced immunity is critical for long term benefit107, we also studied the response to the combined treatment of fractionated radiation and TGFβ inhibition on the peripheral anti-tumor immune response. Analysis of monocyte maturation and activation markers CD11b and F4/80 in tumors suggest that distinct BMDC are recruited as a function of treatment: the F4/80+ macrophage population is more differentiated, while CD11b is more immature. TGFβ inhibition concurrent with radiation treatment also affects systemic maturation as evidenced by analysis of cells from spleens of treated mice. These preliminary data taken together suggest that TGFβ inhibition concurrent with fractionated radiation treatment may cooperate in directing both the microenvironment and the immune system towards an anti-tumor response, which could lead not only to better control of the primary tumor growth but also to abrogation of relapse.
Summary
Although the radiation response of tumor cells has been far better characterized than that of the microenvironment, there is growing appreciation that the two are inextricably intertwined. The therapeutic effects of radiotherapy are traditionally considered as due to the induction of double strand DNA breaks in cancer cells causing cell cycle arrest, senescence or apoptosis108. Accordingly, efforts aimed at understanding and improving the therapeutic efficacy of radiotherapy largely concentrated on the molecular mechanisms of DNA damage and repair. When cancer is treated with radiation, death of tumor cells coupled with changes in tumor microenvironment leads to tumor regression. It is likely that additional avenues of benefit may accrue by augmenting radiation effects on host cells, including innate and adaptive immune cells, CAF and endothelial cells.
As normal organ wound healing, the remaining tumor organ sends tissue damage signals to initiate remodeling and repair. TGFβ is a key factor in normal organ wound healing following damage. Among all radiation-induced cytokines in tumor microenvironment, TGFβ activation could potentially mediate enhancement of DNA damage repair, recruitment of BMDCs to the tumor, aiding in the recovery of tumor growth by enhancing angiogenesis, supplying growth factors and creating a local immunosuppressive environment which all contribute to tumor relapse following treatment.
There are several TGFβ inhibitors in clinical trials, as recently comprehensively reviewed by Akhurst and Hata109. We believe the therapeutic benefit from TGFβ inhibitors will be best exploited in the context of radiation therapy; first because of its control of the DNA damage response leads to greater tumor cell kill and second, as occurs during normal tissue wound recovery, because it mediates many of the microenvironmental responses that promote tumor regrowth The excellent safety profiles demonstrated in these clinical trials, as well as the possibility of protection from late complications in irradiated normal tissues110, provide further motivation for assessing TGFβ inhibitors as an adjunct to radiation therapy of carcinomas.
Acknowledgments
The authors acknowledge research support from NIH Integrative Cancer Biology Program, Varian Medical Systems and Genzyme, Inc..
Abbreviations
- TGFβ
transforming growth factor β
- ECM
extracellular matrix
- NSCLC
non-small cell lung cancer
- EMT
epithelial to mesenchymal transition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakasone E, Askautrud H, Kees T, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce J, Pollard J. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delaney G, Jacob S, Featherstone C, et al. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–1137. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 5.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGF{beta} activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nature Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 7.Rifkin D. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 8.Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Gen. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 9.Glick A, Kulkarni A, Tennenbaum T, et al. Loss of expression of transforming growth factor beta in skin and skin tumors is associated with hyperproliferation and a high risk for malignant conversion. Proc Natl Acad Sci U S A. 1993;90:6076–6080. doi: 10.1073/pnas.90.13.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanford L, Ormsby I, Gittenberger-de GA, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown PD, Wakefield LM, Levinson AD, et al. Physiochemical activation of recombinant latent transforming growth factor-beta’s 1, 2, and 3. Growth Factors. 1990;3:35–43. doi: 10.3109/08977199009037500. [DOI] [PubMed] [Google Scholar]
- 12.Barcellos-Hoff MH. A novel redox mechanism for TGF-beta activation. Mol Biol Cell. 1994;5:139a. [Google Scholar]
- 13.Barcellos-Hoff MH. Radiation-induced transforming growth factor β and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res. 1993;53:3880–3886. [PubMed] [Google Scholar]
- 14.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Molec Endocrin. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 15.Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species active TGF-β1. Lab Invest. 2004;84:1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 16.Jobling MF, Mott JD, Finnegan M, et al. Isoform specificity of redox-mediated TGF-β activation. Radiat Res. 2006;166:839–848. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 17.Barcellos-Hoff MH, Derynck R, Tsang ML-S, et al. Transforming growth factor-β activation in irradiated murine mammary gland. J Clin Invest. 1994;93:892–899. doi: 10.1172/JCI117045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulder K, Levine A, Hernandez X, et al. Modulation of c-myc by transforming growth factor-beta in human colon carcinoma cells. Biochem Biophys Res Commun. 1988;150:711–716. doi: 10.1016/0006-291x(88)90449-4. [DOI] [PubMed] [Google Scholar]
- 19.Chen C, Kang Y, Siegel P, et al. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 20.Sandhu C, Garbe J, Bhattacharya N, et al. Transforming growth factor beta stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol Cell Biol. 1997;17:2458–2467. doi: 10.1128/mcb.17.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battegay E, Raines E, Seifert R, et al. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. 1990;63:515–524. doi: 10.1016/0092-8674(90)90448-n. [DOI] [PubMed] [Google Scholar]
- 22.Bruna A, Darken R, Rojo F, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Matsuyama S, Iwadate M, Kondo M, et al. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res. 2003;63:7791–7798. [PubMed] [Google Scholar]
- 24.Samanta D, Gonzalez A, Nagathihalli N, et al. Smoking attenuates transforming growth factor-β-mediated tumor suppression function through downregulation of Smad3 in lung cancer. Cancer Prev Res (Phila) 2012;5:3453–3463. doi: 10.1158/1940-6207.CAPR-11-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park K, Kim S, Bang Y, et al. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc Natl Acad Sci U S A. 1994;91:8772–8776. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 27.Vincent F, Nagashima M, Takenoshita S, et al. Mutation analysis of the transforming growth factor-beta type II receptor in human cell lines resistant to growth inhibition by transforming growth factor-beta. Oncogene. 1999;15:117–122. doi: 10.1038/sj.onc.1201166. [DOI] [PubMed] [Google Scholar]
- 28.Derynck R, Akhurst R, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 29.Reiss M. Transforming growth factor-beta and cancer: a love-hate relationship. Oncol Res. 1997;9:9447–9457. [PubMed] [Google Scholar]
- 30.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dvorak H. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 32.Border W, Noble N. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 33.Morty R, Königshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:607–613. doi: 10.1513/pats.200908-087RM. [DOI] [PubMed] [Google Scholar]
- 34.Schor S, Ellis I, Jones S, et al. Migration-stimulating factor: a genetically truncated onco-fetal fibronectin isoform expressed by carcinoma and tumor-associated stromal cells. Cancer Res. 2003;63:8827–8836. [PubMed] [Google Scholar]
- 35.Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 36.Quante M, Tu S, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Wever O, Demetter P, Mareel M, et al. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123:2229–2238. doi: 10.1002/ijc.23925. [DOI] [PubMed] [Google Scholar]
- 38.Tuxhorn J, McAlhany S, Yang F, et al. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res. 2002;62:6021–6025. [PubMed] [Google Scholar]
- 39.Rønnov-Jessen L, Petersen O. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1996;68:696–707. [PubMed] [Google Scholar]
- 40.Chen H, Yang W, Wen Q, et al. TGF-beta induces fibroblast activation protein expression; fibroblast activation protein expression increases the proliferation, adhesion, and migration of HO-8910PM. Exp Mol Pathol. 2009;87:189–194. doi: 10.1016/j.yexmp.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Valianou M, Cheng J. Identification and characterization of the promoter of fibroblast activation protein. Front Biosci (Elite Ed) 2010;2:1154–1163. doi: 10.2741/e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojima Y, Acar A, Eaton E, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;46:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orimo A, Gupta P, Sgroi D, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 44.Elenbaas B, Weinberg R. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 45.ten Dijke P, Arthur H. Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 46.Goumans M, Valdimarsdottir G, Itoh S, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 47.Oh S, Seki T, Goss K, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebrin F, Goumans M, Jonker L, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mazzocca A, Fransvea E, Lavezzari G, et al. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50:1140–1151. doi: 10.1002/hep.23118. [DOI] [PubMed] [Google Scholar]
- 50.Stearns M, Garcia F, Fudge K, et al. Role of interleukin 10 and transforming growth factor beta1 in the angiogenesis and metastasis of human prostate primary tumor lines from orthotopic implants in severe combined immunodeficiency mice. Clin Cancer Res. 1999;5:3711–3720. [PubMed] [Google Scholar]
- 51.Ananth S, Knebelmann B, Grüning W, et al. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 1999;59:2210–2216. [PubMed] [Google Scholar]
- 52.Liao S, Liu J, Lin P, et al. TGF-beta blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clin Cancer Res. 2011;17:1415–1424. doi: 10.1158/1078-0432.CCR-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang Y, Siegel P, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 54.Sánchez-Elsner T, Botella L, Velasco B, et al. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 55.Song C, T X, Gelehrter T. Glucocorticoid receptor inhibits transforming growth factor-beta signaling by directly targeting the transcriptional activation function of Smad3. Proc Natl Acad Sci U S A. 1999;96:11776–11781. doi: 10.1073/pnas.96.21.11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pollard J. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 58.Andreu P, Johansson M, Affara N, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–134. doi: 10.1016/j.ccr.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang L, Pang Y, Moses H. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Du R, Lu K, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balkwill F, Coussens L. Cancer: an inflammatory link. Nature. 2004;431:405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 62.Bristow R, Hill R. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 63.Wahl S, Wen J, Moutsopoulos N. TGF-beta: a mobile purveyor of immune privilege. Immunol Rev. 2006;213:213–227. doi: 10.1111/j.1600-065X.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- 64.Thomas D, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 65.Ghiringhelli F, Ménard C, Terme M, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202:1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu C, Wang Y, Wang C, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14-/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136:35–45. doi: 10.1007/s00432-009-0634-0. [DOI] [PubMed] [Google Scholar]
- 67.Ruffell B, Affara N, Coussens L. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fridlender Z, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu T, Tian L, Han Y, et al. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104:4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wald O, Izhar U, Amir G, et al. CD4+CXCR4highCD69+ T cells accumulate in lung adenocarcinoma. J Immunol. 2006;177:6983–6990. doi: 10.4049/jimmunol.177.10.6983. [DOI] [PubMed] [Google Scholar]
- 71.Kioi M, Vogel H, Schultz G, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du S, Qiang M, Zeng Z, et al. Inactivation of kupffer cells by gadolinium chloride protects murine liver from radiation-induced apoptosis. Int J Radiat Oncol Biol Phys. 2010;76:1225–1234. doi: 10.1016/j.ijrobp.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 73.Ewan KB, Henshall-Powell RL, Ravani SA, et al. Transforming growth factor-β1 mediates cellular response to DNA damage in situ. Cancer Res. 2002;62:5627–5631. [PubMed] [Google Scholar]
- 74.Kirshner J, Jobling M, Pajares M, et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 2006;66:10861–10869. doi: 10.1158/0008-5472.CAN-06-2565. [DOI] [PubMed] [Google Scholar]
- 75.Abraham RT. Checkpoint signaling: epigenetic events sound the DNA strand-breaks alarm to the ATM protein kinase. Bioessays. 2003;25:627–630. doi: 10.1002/bies.10310. [DOI] [PubMed] [Google Scholar]
- 76.Kastan MB, Lim DS, Kim ST, et al. Multiple signaling pathways involving ATM. Cold Spring Harb Symp Quant Biol. 2000;65:521–526. doi: 10.1101/sqb.2000.65.521. [DOI] [PubMed] [Google Scholar]
- 77.Wiegman EM, Blaese MA, Loeffler H, et al. TGFbeta-1 dependent fast stimulation of ATM and p53 phosphorylation following exposure to ionizing radiation does not involve TGFbeta-receptor I signalling. Radiother Oncol. 2007;83:289–295. doi: 10.1016/j.radonc.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 78.Bouquet F, Pal A, Pilones K, et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–6765. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hardee M, Marciscano A, Medina-Ramirez C, et al. Resistance of Glioblastoma-Initiating Cells to Radiation Mediated by the Tumor Microenvironment Can Be Abolished by Inhibiting Transforming Growth Factor-β. Cancer Res. 2012;72:4119–4129. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahn G, Brown J. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Browder T, Butterfield C, Kräling B, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- 82.Paris F, Fuks Z, Kang A, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–297. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 83.Czarnota G, Karshafian R, Burns P, et al. Tumor radiation response enhancement by acoustical stimulation of the vasculature. Proc Natl Acad Sci U S A. 2012;109:E2033–2041. doi: 10.1073/pnas.1200053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 85.Hoffman P, Mauer A, Vokes E. Lung cancer. Lancet. 2000;355:479–485. doi: 10.1016/S0140-6736(00)82038-3. [DOI] [PubMed] [Google Scholar]
- 86.Lin M, Stewart D, Spitz M, et al. Genetic variations in the transforming growth factor-beta pathway as predictors of survival in advanced non-small cell lung cancer. Carcinogenesis. 2011;32:1050–1056. doi: 10.1093/carcin/bgr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malkoski S, Haeger S, Cleaver T, et al. Loss of transforming growth factor beta type II receptor increases aggressive tumor behavior and reduces survival in lung adenocarcinoma and squamous cell carcinoma. Clin Cancer Res. 2012;18:2173–2183. doi: 10.1158/1078-0432.CCR-11-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu J, Bao Y, Liu X, et al. Defective expression of transforming growth factor beta type II receptor (TGFBR2) in the large cell variant of non-small cell lung carcinoma. Lung Cancer. 2007;58:36–43. doi: 10.1016/j.lungcan.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 89.Zhang H, Chen X, Wang M, et al. Defective expression of transforming growth factor beta receptor type II is associated with CpG methylated promoter in primary non-small cell lung cancer. Clin Cancer Res. 2004;10:2359–2367. doi: 10.1158/1078-0432.ccr-0959-3. [DOI] [PubMed] [Google Scholar]
- 90.Kim W, Park C, Jung Y, et al. Reduced transforming growth factor-beta type II receptor (TGF-beta RII) expression in adenocarcinoma of the lung. Anticancer Res. 1999;9:301–306. [PubMed] [Google Scholar]
- 91.Santos A, Jung J, Aziz N, et al. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Denys H, Derycke L, Hendrix A, et al. Differential impact of TGF-beta and EGF on fibroblast differentiation and invasion reciprocally promotes colon cancer cell invasion. Cancer Lett. 2008;266:263–274. doi: 10.1016/j.canlet.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 93.Kraman M, Bambrough P, Arnold J, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 94.Navab R, Strumpf D, Bandarchi B, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2011;108:7160–7165. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hasegawa Y, Takanashi S, Kanehira Y, et al. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91:964–971. [PubMed] [Google Scholar]
- 96.Gridelli C, Maione P, Rossi A, et al. The role of bevacizumab in the treatment of non-small cell lung cancer: current indications and future developments. Oncologist. 2007;12:1183–1193. doi: 10.1634/theoncologist.12-10-1183. [DOI] [PubMed] [Google Scholar]
- 97.A S, Gray R, Perry M, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 98.Ruffini E, A S, Filosso P, et al. Clinical significance of tumor-infiltrating lymphocytes in lung neoplasms. Ann Thorac Surg. 2009;87:365–371. doi: 10.1016/j.athoracsur.2008.10.067. [DOI] [PubMed] [Google Scholar]
- 99.Hiraoka K, Miyamoto M, Cho Y, et al. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer. 2006;94:275–280. doi: 10.1038/sj.bjc.6602934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Al-Shibli K, Donnem T, Al-Saad S, et al. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res. 2008;14:5220–5227. doi: 10.1158/1078-0432.CCR-08-0133. [DOI] [PubMed] [Google Scholar]
- 101.Shimizu K, Nakata M, Hirami Y, et al. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol. 2010;5:585–590. doi: 10.1097/JTO.0b013e3181d60fd7. [DOI] [PubMed] [Google Scholar]
- 102.Chen J, Yao P, Yuan A, et al. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res. 2003;9:729–737. [PubMed] [Google Scholar]
- 103.Ho C, Liao W, Wang C, et al. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am J Respir Crit Care Med. 2008;177:763–770. doi: 10.1164/rccm.200704-641OC. [DOI] [PubMed] [Google Scholar]
- 104.Qiao X, Tullgren O, Lax I, et al. The role of radiotherapy in treatment of stage I non-small cell lung cancer. Lung Cancer. 2003;41:11–11. doi: 10.1016/s0169-5002(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 105.Baumann F, Leukel P, Doerfelt A, et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009;11:368–380. doi: 10.1215/15228517-2008-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Asai K, Shioyama Y, Nakamura K, et al. Radiation-Induced Rib Fractures After Hypofractionated Stereotactic Body Radiation Therapy: Risk Factors and Dose–Volume Relationship. International Journal of Radiation Oncology*Biology*Physics. 2012;84:768–773. doi: 10.1016/j.ijrobp.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 107.Formenti SC, Demaria S. Systemic effects of local radiotherapy. The Lancet Oncology. 2009;10:718–726. doi: 10.1016/S1470-2045(09)70082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Begg A, Stewart F, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 109.Akhurst R, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anscher MS, Thrasher B, Zgonjanin L, et al. Small Molecular Inhibitor of Transforming Growth Factor-[beta] Protects Against Development of Radiation-Induced Lung Injury. Int J Radiat Oncol Biol Phys. 2008;71:829–837. doi: 10.1016/j.ijrobp.2008.02.046. [DOI] [PubMed] [Google Scholar]