

Abstract
Astrocytes react to CNS injury by building a dense wall of filamentous processes around the lesion. Stromal cells quickly take up residence in the lesion core and synthesize connective tissue elements that contribute to fibrosis. Oligodendrocyte precursor cells proliferate within the lesion and help to entrap dystrophic axon tips. Here we review evidence that this aggregate scar acts as the major barrier to regeneration of axons after injury. We also consider several exciting new interventions that allow axons to regenerate beyond the glial scar, and discuss the implications of this work for the future of regeneration biology.
Keywords: regeneration, spinal cord injury, axon growth cone, glial scar, hypertrophy, chondroitin sulfate proteoglycans
Introduction
Traumatic injury to the CNS elicits physical damage to vascular networks and neural circuit architecture. Intense local inflammation is an immediate consequence of injury, and causes progressive cavitation that exacerbates the extent of the primary lesion. In time, resident astrocytes become hypertrophic and form a dense scar that protects intact neural networks from further damage (Bush et al., 1999; Faulkner et al., 2004). For years we have thought of this astrocytic scar as the main impediment for regenerating axons attempting to reach their distal targets. However, we have recently come to recognize that the lesion is a complex system of interacting cell types (Barnabé-Heider et al., 2010; Busch et al., 2010; Göritz et al., 2011; Horn et al., 2008; Meletis et al., 2008; Sabelström et al., 2013; Soderblom et al., 2013). These cells react to injury in a stereotyped fashion, forming a mature lesion with two distinct components: The penumbra is composed of hypertrophic astrocytes, whereas the lesion core is composed of NG2 glia/oligodendrocyte precursor cells (OPCs), meningeal and/or vascular derived fibroblasts, pericytes, ependymal cells, and phagocytic macrophages (Fig. 1).
Fig. 1.

Anatomy of a contusive spinal cord lesion. Spinal cord lesions have two distinct components—the lesion penumbra is composed of hypertrophic astrocytes, and the lesion core is composed of NG2+ oligodendrocyte precursor cells, PDGFRβ+ fibroblasts, and macrophages/microglia. Dystrophic axons become entrapped within the lesion in close association with NG2 glia. The layered architecture of the glial scar is thought to reflect both the dynamic polarization of different populations of cells at distinct times after injury and segregation of distinct populations via chemorepulsion.
Regenerating axons attempting to navigate the mature lesion ultimately abort their mission and form dystrophic end bulbs that persist indefinitely (Ramón y Cajal, 1928). These dystrophic end bulbs have long been considered a hallmark of regeneration failure (Tom et al., 2004), and have been identified within a human spinal cord lesion 42 years after injury (personal communication with Ruschel, Sliwsinski, Blesch, Weidner, and Bradke). Here we will review the formation of the glial scar as a framework for understanding axonal dystrophy. Additionally, we will consider several active processes within the lesion that underlie regeneration failure—including inhibitory interactions between axons and extracellular matrix, and phagocytic processes that cause axonal dieback.
With our growing understanding of the processes that actively block regeneration of severed axons, strategies have emerged that allow axons to navigate through and beyond injury sites, or to circumvent the lesion altogether. These studies have advanced the idea that long distance regeneration of axons past sites of CNS trauma can restore significant function to denervated targets, providing new hope for translational success.
Glial Scar Formation after CNS Insult
Breakdown of the blood-brain barrier (BBB) and leakage of blood and serum elements into the CNS parenchyma is considered a key event in glial scar formation. As such, several molecules derived from the blood or produced via inflammation have been advanced as potential triggers of scar formation, including interleukin-1 (Giulian et al., 1988), transforming growth factor-β (TGFβ) isoforms (Asher et al., 2000; Lagord et al., 2002; Moon and Fawcett, 2001), and fibrinogen (Schachtrup et al., 2010). Recently, Schachtrup et al. (2010) linked the release of a blood-derived fibrinogen-TGFβ complex directly to astrogliosis. Genetic deletion of fibrinogen reduced the amount of active TGFβ at the lesion, with corresponding decreases in both astrocytic hypertrophy and production of chondroitin sulfate proteoglycans (CSPGs). Active TGFβ acts via a TGFβ-R/Smad2 dependent pathway in astrocytes, presumably turning on an intrinsic transcriptional program responsible for gliosis (Schachtrup et al., 2010). It appears that Smad2 translocation is a critical event in the induction of gliosis—blocking kinesin-dependent Smad2 translocation with the microtubule stabilizing agent taxol has been associated with reduced scarring at the lesion (Hellal et al., 2011).
Perhaps equally as important to the development of the lesion, injury causes leukocyte extravasation and accumulation of inflammatory cells in the lesion core (Fig. 2A, B). Recent work using 3D-imaging combined with genetic labeling of microglia and infiltrating monocyte-derived macrophages (CX3CR1-GFP) has revealed that the density of inflammatory cells increases by 40-fold in lesioned white matter and 9-fold in lesioned gray matter (Ertürk et al., 2011). This is partly due to rapid polarization of microglia toward sites of CNS injury (Davalos et al., 2005; Nimmerjahn et al., 2005), but probably largely reflects the recruitment and accumulation of blood-borne cells (Ajami et al., 2011). Indeed, activated macrophages/microglia markedly increase expression of matrix metalloproteases (MMPs) after injury, and this contributes to vascular permeability and accumulation of more inflammatory cells within the lesion. MMP inhibitors applied in the acute phase of injury enhance functional recovery (Noble et al., 2002). While these pools of activated macrophages/microglia are thought to be important for lesion debridement, they also drive secondary injury through inflammatory processes. Zymosan, a non-toxic but potent inflammatory agent, delivered through microneedles in a manner that causes minimal damage to the CNS parenchyma, induces an inflammatory response that is sufficient for eliciting secondary tissue damage and causes astrocytes to rapidly migrate away from the inflammatory epicenter (Fitch et al., 1999). More recent experiments have led to the surprising finding that activated macrophages are responsible for prolonged dieback of injured axons after injury (Horn et al., 2008) (Fig. 2D). The timing of axonal dieback correlates well with the accumulation of pro-inflammatory CD16/32+ or ED1+ macrophages at the lesion (Horn et al., 2008; Kigerl et al., 2009) (Fig. 2C, D), and macrophages are found in close association with dystrophic axon endings. While the nature of this association has been elusive, in vitro experiments indicate that macrophages initiate dieback through matrix metalloprotease activity and direct physical interaction with injured axons (Horn et al., 2008). In support of this, macrophage engulfment of axon fragments has been observed at sites of intense inflammation (Gensel et al., 2009) (Fig. 2E).
Fig. 2.

Inflammatory processes within the lesion core. (A) After injury, macrophages and microglia accumulate within the lesion core. (B) Recruitment of inflammatory cells occurs by extravasation of leukocytes from damaged blood vessels and migration of resident microglia to sites of CNS injury. Tissue macrophages and phagocytic microglia synthesize a contingent of cytokines that promote inflammation. (C) Accumulation of inflammatory cells within the lesion core reaches peak density by 30 days after injury (data adapted from Horn et al., 2008; Kigerl et al., 2009). (D) Dieback of injured axons occurs in two distinct phases: Acute axonal degeneration occurs via intracellular Ca2+-dependent cysteine proteases, whereas protracted axonal dieback occurs via direct interaction with inflammatory cells. Protracted axonal dieback correlates well with the accumulation of inflammatory cells within the lesion core (C). Data in (D) adapted from Kerschensteiner et al. (2005) and Horn et al. (2008). (E) Data republished from Gensel et al. (2009) with permission from the Society for Neuroscience; permission conveyed through the Copyright Clearance Center, Inc. EGFP labeled dorsal root ganglion neurons were microtransplanted at a site distant to zymosan injection. EGFP+ axons are observed growing toward the site of zymosan injection, where activated OX42+ macrophages (red) are observed engulfing DRG axon fragments.
Rapid migration of astrocytes away from the inflammatory epicenter initiates the formation of an astrocytic scar at the lesion penumbra (Fig. 3A). Astrocytes however only undergo modest proliferation in response to injury, and this proliferation is confined to a thin layer at the lesion margin (Faulkner et al., 2004). Measurements from 3D-imaging studies indicate that the density of astrocytes in lesioned tissue is only 2–4 fold that of naïve tissue (Ertürk et al., 2011). Reactive glial responses are instead chiefly characterized by astrocytic hypertrophy, where astrocytes swell in size and exhibit high expression of the intermediate filament proteins GFAP, vimentin, and nestin (Barrett et al., 1981; Bignami and Dahl, 1976; reviewed by Yang et al., 1994). Hypertrophic astrocytes undergo restructuring as a population into a mesh-like layer of entangled filamentous processes that acts as a major physical barrier to regenerating axons in long descending or ascending tracts (Wanner et al., 2013) (Fig. 3D, E). Additionally, astrocytes begin synthesizing and depositing CSPGs into the extracellular matrix within 24 hours after injury, and high concentrations of CSPGs persist throughout the lesion for months (Jones et al., 2003; McKeon et al., 1999; Tang et al., 2003).
Fig. 3.
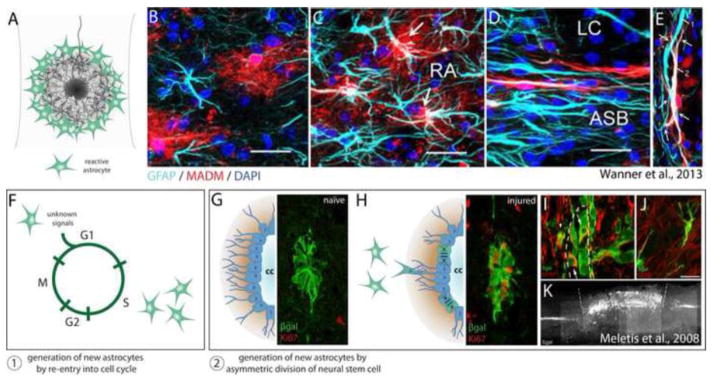
Astrocyte heterogeneity. (A) Astrocytes become hypertrophic in response to CNS insult, forming a dense wall of filamentous processes at the lesion penumbra. (B–E) Data republished from Wanner et al. (2013) with permission from the Society for Neuroscience; permission conveyed through the Copyright Clearance Center, Inc. A GFAP-Cre/MADM-reporter genetic mosaic mouse was used to sparsely label astrocytes with RFP, enabling visualization of fine astrocytic processes, independent astrocytic domains, and astrocytes only weakly immunoreactive for GFAP. (B) In the uninjured cord astrocytes occupy mutually exclusive domains, exhibit a bushy morphology with many fine processes, and express varying amounts of the intermediate filament protein GFAP. (C) 14 days after injury, reactive astrocytes (RA) more distal to the lesion core exhibit increased expression of GFAP, however independent domains and stellate morphology are largely maintained. (D) At the lesion penumbra (ASB), astrocytes no longer maintain a bushy appearance, but take on an elongated morphology with extensive overlap of individual territory. (E) Two different astrocytes (1, 2) form a mesh-like layer of entangled filamentous processes at the lesion penumbra. (F–K) Generation of new astrocytes after spinal cord injury results from at least two distinct mechanisms: 1) Generation of new astrocytes by re-entry of adult astrocytes into the cell cycle. 2) Generation of new astrocytes by asymmetric division of neural stem cells lining the central canal. (G–K) Fluorescent images reprinted from Meletis et al. (2008). (G) Upon tamoxifen administration, FoxJ1-CreER drives βgal reporter expression in a population of ependymal cells lining the central canal. These cells undergo only basal levels of division in the uninjured cord (note absence of Ki67+ cells). (H) After injury, several βgal+ ependymal cells undergo division, demonstrated by co-expression with the proliferation marker Ki67. (I) βgal+ neural stem cell progeny migrate away from the ependymal cell layer lining the central canal (marked by a dashed line), a majority of which become astrocytes (J). (K) Sagittal section demonstrating that βgal+ neural stem cell progeny form a major component of the glial scar one month after injury. Note the specificity of the FoxJ1-CreER line, where βgal expression is confined to the central canal of the spinal cord in regions rostral and caudal to the lesion.
The glial scar serves as a major barrier for regenerating axons, and therefore its emergence in evolution amongst higher vertebrates appears counterproductive (see review by Rolls et al., 2009). Converging evidence from targeted genetic manipulations in mice indicates that the astrocytic penumbra of the scar serves a primary role in confining inflammation to the lesion epicenter and protecting intact neural networks from uncontrolled damage (Faulkner et al., 2004; Herrmann et al., 2008; Okada et al., 2006; Wanner et al., 2013). Using mice engineered to express herpes simplex virus-derived thymidine kinase under the control of a GFAP promoter element, selective ablation of dividing astrocytes can be achieved by administration of ganciclovir (Bush et al., 1999). Faulkner et al. (2004) used this model to selectively ablate dividing astrocytes after spinal cord injury, and found that depletion of reactive astrocytes results in greatly expanded invasion of inflammatory cells beyond the lesion center resulting in a larger lesion volume and more extensive motor deficits. In a related set of experiments, Okada et al. (2006) showed that conditional deletion of Stat3 in nestin-positive cells prevents organization of the astrocytic penumbra of the glial scar after injury. This resulted in greater infiltration and spread of inflammatory cells, and increased motor deficits (Okada et al., 2006; see also Herrmann et al., 2008; Wanner et al., 2013). Taken together, these studies suggest that the glial scar prevents inflammatory processes from propagating to healthy tissue.
After injury a large population of fibroblasts takes up residence in the lesion core, forming a fibrotic scar replete with extracellular fibronectin, collagen, and laminin (see review by Shearer and Fawcett, 2001) (Fig. 4A). Penetrating injuries that disrupt the meninges are associated with meningeal fibroblast infiltration of the lesion core. Within the lesion, meningeal fibroblasts segregate from astrocytes via Ephrin-B2/EphB2 bidirectional signaling (Bundeson et al., 2003), and are thought to contribute to the inhibitory properties of the scar by expression of repulsive axon guidance molecules (Pasterkamp et al., 1999) and by promoting astrocytic reactivity (Wanner et al., 2008). Another perhaps more significant origin of the fibrotic scar has recently been revealed through careful genetic fate mapping studies (Göritz et al., 2011; Soderblom et al., 2013). Traumatic injury disrupts perivascular niches, causing type-A pericytes to delaminate from blood vessels (Göritz et al., 2011) (Fig. 4C, D). Genetic fate mapping reveals that these type-A pericytes proliferate and give rise to stromal cells, which contribute to the fibrotic scar through their deposition of extracellular matrix (Fig. 4E, F). Genetic abrogation of type-A pericyte progeny largely eliminates PDGFRβ+ stromal cells within the lesion, and unexpectedly, results in failure of the lesion to close after a penetrating spinal cord injury (Göritz et al., 2011). In another recent study, Soderblom et al. (2013) used genetic fate mapping to show that collagen-1α1/PDGFRβ/CD13 positive perivascular fibroblasts proliferate even in response to contusive injury where the meninges are largely intact, and form a major cellular component of the fibrotic scar. The fibrotic core of the glial scar appears in its mature form by 2 weeks post-injury (Göritz et al., 2011; Soderblom et al., 2013) (Fig. 4B).
Fig. 4.
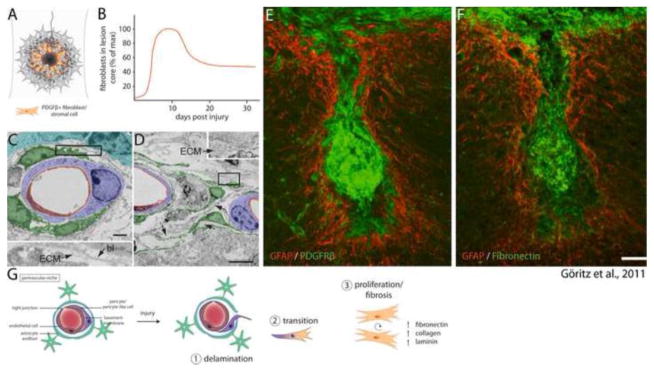
Fibrosis within a contusive lesion. (A) Fibroblasts take up residence within the lesion core, reaching their maximum density around 10 days post injury (B, data adapted from Göritz et al., 2011; Soderblom et al., 2013). (C–F) Data reprinted from Göritz et al. (2011) with permission from AAAS. (C) Electron micrograph of a perivascular niche 5 days post injury. Whereas macrostructure is largely maintained, Type-A pericytes (pseudocolored green) have detached from the surrounding basal lamina (bl) and deposited ECM within the basal lamina sheath. (D) 14 days after injury several type-A pericytes (green) have left their perivascular niche and exhibit extensive fibrosis. (E) 18 weeks post injury the lesion core is occupied by a large contingent of PDGFβ+ fibroblasts, which are associated with deposition of fibronectin in the lesion core (F). (G) Fibrosis within a contusive lesion is caused by perivascular progenitors, and occurs in three distinct phases: 1) Injury causes delamination of pericytes from the endothelial basement membrane. 2) Pericytes that have detached from the basement membrane undergo transition into a mesenchymal state. 3) Progenitors give rise to fibroblasts, which synthesize extracellular matrix that contributes to fibrosis.
Adding to the considerable complexity of the lesion environment are several cell types that undergo migration and proliferation when their usual niche is disturbed due to trauma. For example, injury disrupts progenitor niches. After damage, a large and heterogeneous population of immature cells expressing markers associated with progenitors (including nestin, vimentin, and NG2) invade the lesion core (Busch et al., 2010; Lytle et al., 2006; Zai and Wrathall, 2005). In vivo imaging has demonstrated that NG2 oligodendrocyte progenitors rapidly extend processes and migrate toward CNS lesions (Hughes et al., 2013). This migration is preceded by microglia temporally, and it is thought that dynamic polarization of different populations of cells at distinct times after injury orchestrates the formation of a structurally layered scar (Hughes et al., 2013). At least a subset of invading progenitors, NG2 glia, seems to act as a highly attractive substrate for dystrophic axons, helping to stabilize them within the hostile lesion environment (McTigue et al., 2006; Busch et al., 2010). Additionally, careful genetic fate mapping of Sox9/FoxJ1/vimentin positive ependymal cells, which normally line the central canal and constitute a small population of neural stem cells within the adult spinal cord, has demonstrated that these cells give rise to new astrocytes after injury (Meletis et al., 2008; Barnabé-Heider et al., 2010; Sabelström et al., 2013) (Fig. 3G–K). Genetic abrogation of this population reveals that neural stem cell progeny are required for proper organization of the glial scar, and for restricting secondary tissue damage and neuronal loss (Sabelström et al., 2013).
Altogether we recognize the glial scar as a highly complex system of interacting cell types (Fig. 1). Ultimately the glial scar serves as a vital structure that preserves intact circuit elements from damage by intense inflammation at the lesion core. However, the formation of the glial scar creates a physical and molecular barrier that entraps dystrophic axons and severely limits their ability to regenerate over long distances.
Response of Axons to the Glial Scar
The glial scar serves as the primary barrier to regenerating axons. Compelling evidence of its pivotal role in halting advancing growth cones comes from microtransplantation studies in which adult dorsal root ganglion neurons were grafted into white matter tracts of the CNS with or without prior injury (Davies et al., 1997; Davies et al., 1999). Dissociated adult neurons initiated axons, and these axons grew rapidly (1 mm per day) over long distances through intact as well as degenerating white matter. Regenerating axons, however, entered into but then halted abruptly within CSPG-rich territory of the lesion core, and growth cones took on the bulbous morphology of dystrophic endings (Davies et al., 1999) (Fig. 5A). Dystrophic growth cones were first described as “sterile clubs” by Ramon y Cajal, who also noted their persistence in a stable and quiescent state (Ramón y Cajal, 1928) (Fig. 5B, C). Thus, when confronted with the inhibitory environment of the glial scar, growth cones undergo dramatic changes that prevent their continued growth.
Fig. 5.
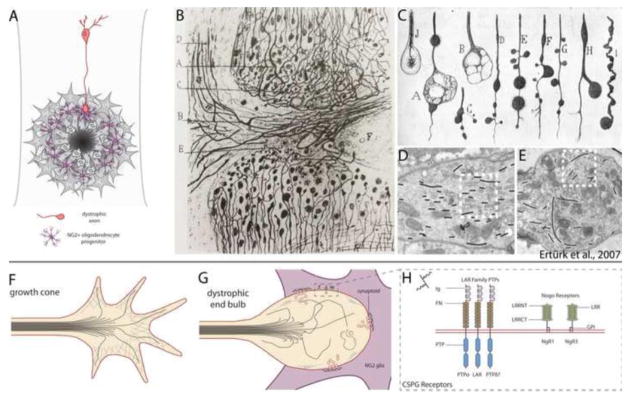
The glial scar acts as the primary barrier to regenerating axons. (A) Regenerating axons halt abruptly at the glial scar in close association with NG2 glia, and axon endings take on a state of dystrophy. (B, C) Reprinted from Cajal’s Degeneration and Regeneration of the Nervous System (1928) by permission of Oxford University Press, USA. (B) Drawing by Ramón y Cajal of the edges of a complete transection lesion to the spinal cord. Cajal noted that adjacent to the lesion, fine axons terminate in rings or little clubs while larger axons end in voluminous clubs. (C) Several drawings by Cajal of the various appearances of retraction clubs at the lesion. (D, E) Data republished from Ertürk et al. (2007) with permission from the Society for Neuroscience; permission conveyed through the Copyright Clearance Center, Inc. (D) Electron micrograph of a growth cone in the lesioned sciatic nerve. Growth cones exhibit highly parallel arrays of microtubules (traced with black lines). (E) Electron micrograph of a retraction bulb from a lesioned central axon of a dorsal root ganglion neuron. Retraction bulbs exhibit disorganized or splayed microtubules. (F) Illustration of a growth cone. Microtubules are bundled and oriented toward the direction of the growing axon. Growth cones also have a highly organized F-actin network and several filopodia. (G) Dystrophic end bulbs have disorganized microtubules and a disrupted F-actin network. Large membrane blebs or inclusions can be observed within retraction clubs. Dystrophic axons associate with NG2 glia in the outer margin of the lesion core, forming “synaptoids” that exhibit similar properties to a mature synapse, with numerous presynaptic vesicles and omega structures, and a post-synaptic density at the NG2 glial membrane. (H) Several recently identified CSPG receptors act to signal inhibition, and may entrap axons on the surface of NG2 glia. These receptors fall into two classes: the LAR family of transmembrane protein tyrosine phosphatases (which includes PTPσ, LAR, and PTPδ), and the Nogo receptors NgR1 and NgR3. LAR family receptor protein tyrosine phosphatases have high sequence similarity and contain three N-terminal Ig domains, 8 fibronectin repeats, and two intracellular tandem phosphatase domains. The first Ig domain of PTPσ, LAR, and PTPδ contains a canonical GAG binding motif (BB-X-BB, where B is lysine or arginine). NgR1 and NgR3 have several leucine-rich repeats, are GPI-anchored, and contain a cluster of basic residues in the C-terminal stalk required for binding CSPGs.
Recent work has revealed that dystrophic growth cones are more active than previously thought (Tom et al., 2004), and chronically injured axons retain the potential to regenerate (Houle et al., 1991; Kwon et al., 2002; Li and Raisman, 1995). Our laboratory has characterized dystrophic growth cones using an in vitro assay that reproduces the gradient of proteoglycans found at the glial scar after injury (Tom et al., 2004). Live imaging of growth cones that stall and become dystrophic in a gradient of CSPGs has revealed that—despite their abnormal appearance and lack of forward movement—dystrophic endings can be extremely dynamic at least for a few days in vitro. These endings continually endocytose membrane, and can sometimes send out short filopodia that undergo repeated cycles of advancement and retraction (Tom et al., 2004). Additional in vivo studies on dystrophic growth cones have indicated no overt mitochondrial deficiencies or trans-Golgi network abnormalities that might contribute to their stagnancy. Instead, disorganized microtubules underlie the formation of the dystrophic growth cone (Ertürk et al., 2007) (Fig. 5D–G). Ertürk et al. (2007) showed that destabilization of microtubules with nocodazole was sufficient for conversion of growth cones from an active to a dystrophic state, and that stabilization of microtubules using taxol helped to prevent the formation of dystrophic growth cones in vivo. Further, Hellal et al. (2011) showed that moderate stabilization of microtubules with taxol allows axon regeneration and functional recovery after contusive lesion. Together, these studies have led us to believe that end bulbs are not sterile clubs, but dynamic endings that retain the potential to be coaxed into a state of growth.
We recently sought to better understand interactions between dystrophic axons and cells of the lesion environment, and found that severed sensory axons form stable contacts with NG2 glia (Busch et al., 2009; see also McTigue et al., 2006). NG2 glia initially appear to act as a supportive substrate that prevents axon dieback caused by inflammatory cells; however, this interaction leads to long lasting entrapment of dystrophic axons at the NG2 glial cell surface. In vitro and in vivo studies demonstrate that these stabilizations exhibit several properties of axodendritic synaptic specializations (unpublished data, Filous and Silver) (Fig. 5G). Given that axons form synapses with NG2 glia in a number of brain regions under homeostatic conditions (Bergles et al., 2000; Chittajulla et al., 2004; Lin et al., 2005), it is possible that synaptic-like interactions between dystrophic axons and NG2 glia emerge in the context of injury and act to further constrain axon growth. These observations are consistent with the hypothesis that regenerating axons form synapse-like terminals with reactive glia, which was originally advanced by Carlstedt (1985).
Advances in imaging have allowed live in vivo studies of acute and chronically injured axons in the lesion environment (Di Maio et al., 2011; Farrar et al., 2012; Kerschensteiner et al., 2005; Ylera et al., 2009). Ylera et al. (2009) recently used in vivo imaging to demonstrate that chronically injured axons can, indeed, be aroused into a robust regenerative state. Using a two-photon laser to transect individual GFP-labeled sensory axons without causing any significant damage to the surrounding parenchyma, axons exhibited modest yet continuous growth. When this minimal lesion was followed with peripheral conditioning, a manipulation known to promote the growth of central axons (Neumann and Woolf, 1999; Richardson and Issa, 1984), axon growth became vigorous, allowing several axons to grow right through the miniature CNS lesion (Ylera et al., 2009). These results strongly support the notion that environmental factors associated with the glial scar play a critical role in regeneration failure. Further, these results predict that manipulations promoting the intrinsic ability of axons to grow might allow robust regeneration of injured axons—especially if mitigation of inhibitory environmental factors can be achieved concomitantly.
Proteoglycan Inhibitors and Their Cognate Receptors
CSPGs comprise a diverse family of extracellular matrix proteins which includes neurocan, aggrecan, brevican, phosphacan, versican, and NG2 (Margolis and Margolis, 1993). These proteins undergo post-translational modification with complex glycosaminoglycan chains which vary in both length and sulfation pattern. Extensive work has implicated proteoglycans as barriers to growing axons in the developing nervous system (Brittis et al., 1992; Cole and McCabe, 1991; Snow et al., 1990b; Wu et al., 1998), and CSPGs are highly inhibitory to growing axons in culture (Snow et al., 1990a). The first evidence that CSPGs might have a causative role in regeneration failure emerged in the early 1990s when it was shown that reactive astrocytes synthesize CSPGs in response to small lesions at the dorsal root entry zone (Pindzola et al., 1993). Indeed, astrocytes rapidly assemble and secrete CSPGs as they migrate away from the core of central lesions (Jones et al., 2003; McKeon et al., 1999; Tang et al., 2003). Furthermore, removal of the glycosaminoglycan chains of lesion-induced proteoglycans leads to regeneration and functional recovery after spinal cord injury (Bradbury et al., 2002).
While the contribution of CSPGs to the inhibitory nature of the glial scar has been known for many years (see review by Silver and Miller, 2004), mechanistic explanation as to how CSPGs inhibit advancing growth cones was lacking—until recently. For years it was posited that CSPGs exert inhibition through relatively nonspecific mechanisms such as substrate occlusion (McKeon et al., 1995), or presentation of a negatively charged boundary that repels growing axons (Gilbert et al., 2005). This view has changed considerably with the discovery of several receptors that directly bind sulfated glycosaminoglycan moieties (Dickendesher et al., 2012; Fisher et al., 2011; Shen et al., 2009) (Fig. 5H).
Receptor protein tyrosine phosphatase sigma (PTPRS, or PTPσ) was the first receptor identified with the ability to both bind CSPGs and convey axonal growth inhibition (Shen et al., 2009). PTPσ is a member of the class IIa/Leukocyte common antigen-related (LAR) family of receptor tyrosine phosphatases, along with its sister proteins PTPδ and LAR. As all three LAR family receptors exhibit high sequence similarity and contain a cluster of lysine residues that comprise a canonical glycosaminoglycan binding motif (Duan and Giger, 2010; Hileman et al., 1998), it came as no surprise when LAR was also shown to bind CSPG (Fisher et al., 2011). PTPσ knockouts exhibit increased axonal regeneration after peripheral nerve injury (McLean et al., 2002), optic nerve crush (Sapieha et al., 2005), dorsal column lesion (Shen et al., 2009), corticospinal tract injury (Fry et al., 2010), and cardiac ischemia reperfusion injury (Gardner and Habecker, 2013). In contrast, the role of LAR in regeneration failure remains uncertain, as several studies report either increased or severely impaired regeneration through manipulation of LAR signaling (Fisher et al., 2011; Van der Zee et al., 2003; Xie et al., 2001).
In addition to LAR family phosphatases, the NOGO receptors NgR1 and NgR3 have also been identified as receptors for CSPGs (Dickendesher et al., 2012). This discovery established a link between the two major families of inhibitory molecules associated with CNS injury—myelin derived inhibitors and proteoglycans. Dickendesher et al. (2012) showed that NgR1−/−;NgR2−/−;NgR3−/− mutants exhibited significant axonal regeneration after optic nerve crush, and synergistic axonal growth was observed in animals that also lacked PTPσ. Whether signaling by NgR1/3 and PTPσ converges on a common intracellular effector (e.g. cytoskeleton, see review by Yiu and He, 2006) remains a major question as intracellular signaling pathways are worked out in greater detail.
Heparan sulfate proteoglycans (HSPGs) also act as ligands for LAR family receptors (Aricescu et al., 2002; Coles et al., 2011; Fox and Zinn, 2005), and promote axon extension (Coles et al., 2011). Therefore, a significant question in LAR family receptor biology is how HSPGs and CSPGs exert opposite phenotypic changes at the growth cone through the same receptor. A recent report has offered a mechanistic explanation to this conundrum (Coles et al., 2011). Both HSPGs and CSPGs bind to the same extracellular motif of PTPσ. This binding site exhibits conformational flexibility at the structural level, and can accommodate various glycosaminoglycans with similar affinities. However, HSPG and CSPG ligands differ in their ability to cause clustering of the PTPσ ectodomain—HSPG ligands cause ectodomain aggregation in a size exclusion chromatography assay, and addition of CSPG diminishes this effect. Additionally, PTPσ localization in axons grown on an HSPG substrate is confined to small puncta (Coles et al., 2011). Together these results indicate that the oligomerization status of PTPσ acts to switch axonal endings between states of growth and inactivity. Therapeutically, this characteristic of PTPσ offers a unique point of intervention; drugs that mimic HSPG binding might augment the regenerative potential of neurons while mitigating the inhibitory environment of the glial scar. Indeed, the complex nature of chondroitin sulfate biology—synthesis, macromolecular organization, and signaling—suggests multiple candidate targets for intervention (Brown et al., 2012; Carulli et al., 2010; Grimpe and Silver, 2004; Hur et al., 2011; Takeuchi et al., 2013; Wang et al., 2008).
Astrocyte Heterogeneity
Astrocytes are a heterogeneous population of cells that exhibit a spectrum of phenotypic changes in response to injury (reviewed by Sofroniew, 2009). These changes are heavily influenced by proximity to the lesion, but may also be deterministic based on ontological identity. While reactive astrogliosis is caused largely by autogenous cell hypertrophy and division by re-entry into the cell cycle (Fig. 3F), at least a subset of astrocytes within the glial scar is derived from adult neural stem cells (Meletis et al., 2008; Barnabé-Heider et al., 2010; Sabelström et al., 2013) (Fig. 3G–K). Astrocytes derived from neural stem cells appear to express less GFAP relative to vimentin, consistent with a more immature state. These ependymal-derived astrocytes, although present within the penumbra of the glial scar, do not synthesize CSPGs and are permissive to axonal sprouting (Meletis et al., 2008). The lesion penumbra is therefore composed of at least two functionally and developmentally distinct populations of astrocytes.
Astrocytes of adult mammals react to injury by walling off lesioned tissue, forming a so-called glial “scar” that acts as a major barrier to regenerating axons (Silver and Miller, 2004; Wanner et al., 2013). Yet lesions to immature mammals or cold-blooded vertebrates are not associated with such fate. Immature reactive astrocytes of mammalian fetuses are capable of supporting regeneration of adult central axons (Reier et al., 1986; Saunders et al., 1988). By implanting nitrocellulose into the brains of immature or mature animals, it is possible to collect scar tissue that forms on these implants for subsequent in vitro investigations. When hippocampal neurons were cultured on explanted scar tissue, axon growth was much more vigorous on scar tissue derived from young animals than that of mature animals (Rudge and Silver, 1990). Similar results have been obtained in studies of reactive astrocytes from the lesioned optic nerve (Bahr et al., 1995). In vivo studies further suggest that inherent differences between immature and mature astrocytes contribute to regeneration failure in adults (Smith and Miller, 1991; Smith et al., 1986; Smith and Silver, 1988). Importantly, immature astrocytes do not rapidly synthesize CSPGs in response to injury (Dow et al., 1994). Immature, but not mature, astrocytes transplanted near an injury site can migrate to the lesion and form a permissive bridge for regenerating axons (Davies et al., 2006; Filous et al., 2010). Interestingly, when rat or human fetal glial-restricted progenitors were differentiated into immature astrocytes and transplanted at sites of dorsal column lesion, sensory axons were able to regenerate (Haas and Fischer, 2013; Haas et al., 2012). Thus, astrocytes undergo developmental changes that predispose their response to injury in adulthood and contribute to the inhibitory nature of the glial scar.
Glial cells of lower vertebrate species are similar to mammalian neonatal astrocytes in that they too are capable of supporting axon growth. After spinal cord transection in lower vertebrates, meningeal and glial cells migrate and interact to form a “glial bridge” which allows regenerating axons to grow through the lesion (Butler and Ward, 1967; Goldshmit et al., 2012; Zukor et al., 2011). In Xenopus tadpoles, severed axons can regenerate directly through astrocytic scar tissue (Reier, 1979; Reier and de Webster, 1974). Thus, reactive glial cells in lower vertebrates exhibit differences to their mammalian counterparts in that they are not overtly repulsive to growing axons and are capable of organizing growth-permissive “bridges” that allow efficient regeneration of axons across a lesion. It would be very provocative to know if at least some of the well described pro-regenerative features of mammalian neonatal astrocytes or ependymo-glial cells in cold blooded species are retained within the subpopulation of astrocytes that is derived from ependymal cells after injury (Singer et al., 1979).
While the genetic program underlying growth inhibitory reactive gliosis of adult astrocytes is incompletely understood (Zamanian et al., 2012), several studies have attempted to alter the course of this program by manipulating growth factor signaling in astrocytes (Okada et al., 2006; White et al., 2009). Recent work has implicated acidic fibroblast growth factor (aFGF) signaling in promoting astrocyte migration and morphogenesis conducive to axon regeneration after zebrafish spinal cord injury (Goldshmit et al., 2012). A proteomics approach to studying how aFGF signaling attenuates astrocytic pathology suggested that downregulation of proteins involved in the process of secondary injury and glial scar formation might play a role (Tsai et al., 2008). In a mammalian transection model of spinal cord injury, aFGF in combination with chondroitinase can facilitate long distance regeneration through peripheral nerve grafts, and bring about lower urinary tract recovery after injury (Lee et al., 2013). In aFGF treated rats, elongated astrocytes align and integrate along the graft/cord interface creating a “glial bridge” into and out of the graft, in stark contrast to the dense network of glial processes normally observed at the glial scar (Lee et al., 2013). Indeed, “bridge building” by astrocytes appears to be an anatomical predictor of axon regeneration (Hurtado et al., 2011; Iseda et al., 2004; Liu et al., 2010; Ma et al., 2004; Zukor et al., 2013). Several studies with successful axon regeneration into or through grafts also exhibit migration and integration of astrocytes at the graft/cord interface (Guest et al., 1997; Hurtado et al., 2011; Joosten et al., 1995; Spilker et al., 2001), whereas lack of astrocyte integration is associated with regeneration failure (Hurtado et al., 2011; Xu et al., 1995). Thus, reactive astrocytes are a highly plastic population of cells, altering their growth permissive or inhibitory properties in a manner that is finely tuned to constraints of the complex lesion environment. Further studies identifying factors that contribute to diverse astrocytic responses may uncover new routes of therapeutic intervention.
Restoring Function to Denervated Targets
While our understanding of the lesion environment continues to mature, several strategies have emerged that allow axons to grow through or circumvent the lesion altogether (Alilain et al., 2011; Alto et al., 2009; De Lima et al., 2012; Lu et al., 2012; Sun et al., 2011). While these studies have advanced hope for translational success by demonstrating robust and lengthy regeneration of axons past sites of CNS trauma, they also point to an entirely new set of challenges that axons must overcome to successfully reinnervate their targets following injury.
First, overcoming the physical and chemical barriers of the scar is becoming possible. Tello demonstrated in a classic set of experiments performed over a century ago that central neurons can extend axons into pieces of sciatic nerve grafted into the cerebral cortex (Ramón y Cajal, 1928). Since then, grafting techniques have allowed some of the most convincing demonstrations that regeneration of adult central axons over long distances can promote functional recovery (Alilain et al., 2011; Houle et al., 2006; Keirstead et al., 1989; Lee et al., 2013; Lu et al., 2012; Sauvé et al., 2001; Tom et al., 2009). Indeed, Alilain et al. (2011) recently showed that a tibial nerve autograft can be used to “bridge” a C2 lateral hemisection of the adult spinal cord, allowing serotonergic and other bulbospinal axons to bypass the lesion and innervate phrenic motor neurons in spinal cord levels C3-5/6. Anatomical evidence of regeneration was accompanied by electrophysiological studies indicating remarkable restoration of function to the paralyzed hemidiaphragm. Furthermore, recording from the graft during respiratory challenge indicated that at least some of the fibers within the graft relay respiratory drive from medullary respiratory centers. Finally, transection of the peripheral nerve graft eliminated patterned activity in the ipsilateral diaphragm (Alilain et al., 2011). Tom et al. (2009) used a similar grafting approach to demonstrate that chronically injured axons can grow through a peripheral nerve “bridge” and restore a small amount of function in the chronically injured spinal cord. So, although many barriers underlie the failure of CNS axons to regenerate without exogenous intervention, CNS axons can, nonetheless, regenerate over great distances and make functionally meaningful connections with intact neural circuitry (see review by Horner and Gage, 2000).
Second, intrinsic properties of neurons can now be manipulated in ways that increase the growth of injured axons. Recent studies have identified two distinct neuronal signaling pathways that regulate axonal growth (Park et al., 2008; Smith et al., 2009). Deletion of PTEN (a negative regulator of mTOR signaling) or SOCS3 (a negative regulator of JAK/STAT signaling) promotes lengthy regeneration of retinal ganglion cell axons after optic nerve crush (Park et al., 2008; Smith et al., 2009). It has additionally been shown that knockdown or genetic deletion of PTEN in adult corticospinal projection neurons allows for regeneration of corticospinal axons after injury (Liu et al., 2010; Zukor et al., 2013). Interestingly, co-deletion of both PTEN and SOCS3 allows sustained regeneration of retinal ganglion cell axons through the length of the optic nerve, and frequently into and past the optic chiasm (Luo et al., 2013; Sun et al., 2011). mTOR and JAK/STAT signaling pathways appear to operate independently in the context of axon regeneration, and stimulating both pathways allows synergetic growth of axons (Sun et al., 2011).
Although we are able to entice axons to grow either through or around a lesion, directing these axons to their appropriate targets remains a topic of study. Recently, using 3D-imaging combined with a tissue clearing technique, Luo et al. (2013) confirmed that PTEN/SOCS3 deletion within retinal ganglion cells allows axons to regenerate long distances; however, regenerating axons exhibit a variety of aberrant phenotypes. With respect to their course along the optic nerve, 40% of regenerating axons make a U-turn back toward the optic nerve head in areas proximal to the lesion, and 10% of axons make U-turns in the more distal nerve. The authors showed that high amounts of turning correlate with intense astroglial activation, indicating that reactive astrocytes well away from the injury site may misguide regenerating axons in the optic nerve. During development, axons reach a decision point at the optic chiasm, where roughly equal numbers of axons cross the optic chiasm and course within the contralateral optic tract as project to the ipsilateral optic tract. Luo et al. (2013) showed that regenerating axons are more likely to project to the ipsilateral optic tract than cross the chiasm, and regenerating axons often (~10–20% of cases) project to the intact contralateral optic nerve. None reached their appropriate targets in the thalamus or tectum. Thus, the precise molecular cues that once regulated axon pathfinding during development of the visual system are apparently not preserved into adulthood. Certain types of bulbospinal projections controlling more primitive functions such as micturition, respiration, and crude locomotion do appear to establish functional connections once beyond the glial scar. However, to promote appropriate target re-innervation of highly complex and topographically organized systems (such as visual and corticospinal systems), we may need to consider methods that encourage proper guidance and synaptic specification of axons that regenerate beyond the glial scar (Alto et al., 2009).
Conclusions and Perspective
As a field we have made significant progress in understanding the nature of central nervous system lesions, providing in recent years some of the most exciting evidence of functional recovery in experimental models of injury. Building on our knowledge of how lesions develop over time, we may eventually begin to expand these interventions to states of chronic injury. Undoubtedly, combination strategies that account for multiple obstacles to axon regeneration are likely to have the most success.
Acknowledgments
The authors thank Teresa Evans for her critical review of the manuscript. J.M.C. was supported by NSF grant DGE-0951783. J.S. received research funding from NINDS/NS025713, the Brumagin Memorial Fund, Unite to Fight Paralysis, and SCIS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto LT, Havton LA, Conner JM, Hollis ER, II, Blesch A, Tuszynski M. Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat Neurosci. 2009;12:1106–1113. doi: 10.1038/nn.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, McKinnell IW, Halfter W, Stocker AW. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol. 2002;22:1881–1892. doi: 10.1128/MCB.22.6.1881-1892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in the injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr M, Przyrembel C, Bastmeyer M. Astrocytes from adult rat optic nerves are nonpermissive for regenerating retinal ganglion cell axons. Exp Neurol. 1995;131:211–220. doi: 10.1016/0014-4886(95)90043-8. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Göritz C, Sabelström H, Takebayashi H, Pfrieger FW, Meletis K, Frisén J. Origin of new glial cells in intact and injured adult spinal cord. Cell Stem Cell. 2010;7:470–482. doi: 10.1016/j.stem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Barrett CP, Guth L, Donati EJ, Krikorian JG. Astroglial reaction in the gray matter lumbar segments after midthoracic transection of the adult rat spinal cord. Exp Neurol. 1981;73:365–377. doi: 10.1016/0014-4886(81)90272-7. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. The astroglial response to stabbing. Immunofluorescence studies with antibodies to astrocyte-specific protein (GFA) in mammalian and submammalian vertebrates. Neuropathol Appl Neurobiol. 1976;2:99–110. [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Brittis PA, Canning DR, Silver J. Chondroitin sulfate as a regulator of neuronal patterning in the retina. Science. 1992;255:733–736. doi: 10.1126/science.1738848. [DOI] [PubMed] [Google Scholar]
- Brown JM, Xia J, Zhuang B, Cho KS, Rogers CJ, Gama CI, Rawat M, Tully SE, Uetani N, Mason DE, Tremblay ML, Peters EC, Habuchi O, Chen DF, Hsieh-Wilson LC. A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc Natl Acad Sci USA. 2012;109:4768–4773. doi: 10.1073/pnas.1121318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundeson LQ, Scheel TA, Bregman BS, Kromer LF. Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci. 2003;23:7789–7800. doi: 10.1523/JNEUROSCI.23-21-07789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Cuascut FX, Hawthorne AL, Bai L, Miller RH, Silver J. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J Neurosci. 2010;30:255–265. doi: 10.1523/JNEUROSCI.3705-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Butler EG, Ward MB. Reconstitution of the spinal cord after ablation in adult Triturus. Dev Biol. 1967;15:464–486. doi: 10.1016/0012-1606(67)90038-3. [DOI] [PubMed] [Google Scholar]
- Carlstedt T. Regenerating axons form nerve terminals at astrocytes. Brain Res. 1985;347:188–191. doi: 10.1016/0006-8993(85)90911-4. [DOI] [PubMed] [Google Scholar]
- Carulli D, Pizzorusso T, Kwok JC, Putignano E, Poli A, Forostyak S, Andrews MR, Deepa SS, Glant TT, Fawcett JW. Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain. 2010;133:2331–2347. doi: 10.1093/brain/awq145. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol. 2004;561:109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GJ, McCabe CF. Identification of a developmentally regulated keratin sulfate proteoglycan that inhibits cell adhesion and neurite outgrowth. Neuron. 1991;7:1007–1018. doi: 10.1016/0896-6273(91)90345-z. [DOI] [PubMed] [Google Scholar]
- Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science. 2011;332:484–488. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Davies JE, Huang C, Proschel C, Noble M, Mayer-Proschel M, Davies SJ. Astrocytes derived from glial-restricted precursors promote spinal cord repair. J Biol. 2006;5:7. doi: 10.1186/jbiol35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Goucher DR, Doller C, Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;209:426–445. doi: 10.1523/JNEUROSCI.19-14-05810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lima S, Koriyama Y, Kurimoto T, Oliveira JT, Yin Y, Li Y, Gilbert HY, Fagiolini M, Martinez AM, Benowitz L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci USA. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickendesher TL, Baldwin KT, Mironova YA, Koriyama Y, Raiker SJ, Askew KL, Wood A, Geoffroy CG, Zheng B, Liepmann CD, Katagiri Y, Benowitz LI, Geller HM, Giger RJ. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat Neurosci. 2012;15:703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio A, Skuba A, Himes BT, Bhagat SL, Kyun JK, Tessler A, Bishop D, Son YJ. In vivo imaging of dorsal root regeneration: rapid immobilization and presynaptic differentiation at the CNS/PNS border. J Neurosci. 2011;31:4569–4582. doi: 10.1523/JNEUROSCI.4638-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow KE, Ethell DW, Steeves JD, Riopelle RJ. Molecular correlates of spinal cord repair in the embryonic chick: heparan sulfate and chondroitin sulfate proteoglycans. Exp Neurol. 1994;128:233–238. doi: 10.1006/exnr.1994.1132. [DOI] [PubMed] [Google Scholar]
- Duan Y, Giger RJ. A new role for RPTPsigma in spinal cord injury: signaling chondroitin sulfate proteoglycan inhibition. Sci Signal. 2010;3:pe6. doi: 10.1126/scisignal.3110pe6. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Mauch CP, Hellal F, Förstner F, Keck T, Becker K, Jährling N, Steffens H, Richter M, Hübener M, Kramer E, Kirchhoff F, Dodt HU, Bradke F. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2011;18:166–171. doi: 10.1038/nm.2600. [DOI] [PubMed] [Google Scholar]
- Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, Schaffer CB. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Meth. 2012;9:297–302. doi: 10.1038/nmeth.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Miller JH, Coulson-Thomas YM, Horn KP, Alilain WJ, Silver J. Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev Neurobiol. 2010;70:826–841. doi: 10.1002/dneu.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D, Xing B, Dill J, Li H, Hoang HH, Zhao Z, Yang XL, Bachoo R, Cannon S, Longo FM, Sheng M, Silver J, Li S. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J Neurosci. 2011;31:14051–14066. doi: 10.1523/JNEUROSCI.1737-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AN, Zinn K. The heparin sulfate proteoglycan Syndecan is an in vivo ligand for the Drosophila LAR receptor tyrosine phosphatase. Curr Biol. 2005;15:1701–1711. doi: 10.1016/j.cub.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Fry EJ, Chagnon MJ, López-Valez R, Tremblay ML, David S. Corticospinal tract regeneration after spinal cord injury in receptor protein tyrosine phosphatase sigma deficient mice. Glia. 2010;58:423–433. doi: 10.1002/glia.20934. [DOI] [PubMed] [Google Scholar]
- Gardner RT, Habecker BA. Infarct-derived chondroitin sulfate proteoglycans prevent sympathetic reinnervation after cardiac ischemia-reperfusion injury. J Neurosci. 2013;33:7175–7183. doi: 10.1523/JNEUROSCI.5866-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG. Macrophages promote axon regeneration with concurrent neurotoxicity. J Neurosci. 2009;29:2956–3968. doi: 10.1523/JNEUROSCI.3992-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RJ, McKeon RJ, Darr A, Clabro A, Hascall VC, Bellamkonda RV. CS-4,6 is differentially upregulated in glial scar and is a potent inhibitor of neurite extension. Mol Cell Neurosci. 2005;29:545–558. doi: 10.1016/j.mcn.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB. Interleukin-1 injected into the mammalian brain stimulates astrogliosis and neovascularization. J Neurosci. 1988;8:2485–2490. doi: 10.1523/JNEUROSCI.08-07-02485.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y, Sztal TE, Jusuf PR, Hall TE, Nguyen-Chi M, Currie PD. Fgf-dependent glial cell bridges facilitate spinal cord regeneration in zebrafish. J Neurosci. 2012;32:7477–7492. doi: 10.1523/JNEUROSCI.0758-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- Grimpe B, Silver J. A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci. 2004;24:1393–1397. doi: 10.1523/JNEUROSCI.4986-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest JD, Hesse D, Schnell L, Schwab ME, Bunge MB, Bunge RP. Influence of IN-1 antibody and acidic FGF-fibrin glue on the response of injured corticospinal tract axons to human Schwann cell grafts. J Neurosci Res. 1997;50:888–905. doi: 10.1002/(SICI)1097-4547(19971201)50:5<888::AID-JNR24>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Haas C, Fischer I. Human astrocytes derived from glial restricted progenitors support regeneration of the injured spinal cord. J Neurotrauma. 2013;30:1035–1052. doi: 10.1089/neu.2013.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas C, Neuhuber B, Yamagami T, Rao M, Fischer I. Phenotypic analysis of astrocytes derived from glial restricted precursors and their impact on axon regeneration. Exp Neurol. 2012;233:717–732. doi: 10.1016/j.expneurol.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- Houle JD. Demonstration of the potential for chronically injured neurons to regenerate axons into intraspinal peripheral nerve grafts. Exp Neurol. 1991;113:1–9. doi: 10.1016/0014-4886(91)90139-4. [DOI] [PubMed] [Google Scholar]
- Houle JD, Tom VJ, Mayes D, Wagoner G, Phillips N, Silver J. Combining an autologous peripheral nervous system “bridge” and matrix modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. J Neurosci. 2006;26:7405–7415. doi: 10.1523/JNEUROSCI.1166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci. 2013;16:668–676. doi: 10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Yang IH, Kim DH, Byun J, Saijilafu, Xu WL, Nicovich PR, Cheong R, Levchenko A, Thakor N, Zhou FQ. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci USA. 2011;108:5057–5062. doi: 10.1073/pnas.1011258108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Cregg JM, Wang HB, Wendell DF, Oudega M, Gilbert RJ, McDonald JW. Robust CNS regeneration after complete spinal cord transection using aligned poly-L-lactic acid microfibers. Biomaterials. 2011;32:6068–6079. doi: 10.1016/j.biomaterials.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseda T, Nishio T, Kawaguchi S, Yamanoto M, Kawasaki T, Wakisaka S. Spontaneous regeneration of the corticospinal tract after transection in young rats: a key role of reactive astrocytes in making favorable and unfavorable conditions for regeneration. Neuroscience. 2004;126:365–374. doi: 10.1016/j.neuroscience.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399–411. doi: 10.1016/s0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- Joosten EA, Bär PR, Gispen WH. Collagen implants and cortico-spinal axonal growth after mid-thoracic spinal cord lesion in the adult rat. J Neurosci Res. 1995;41:481–490. doi: 10.1002/jnr.490410407. [DOI] [PubMed] [Google Scholar]
- Keirstead SA, Rasminsky M, Fukuda Y, Carter DA, Aguayo AJ, Vidal-Sanz M. Electrophysiologic responses in hamster superior colliculus evoked by regenerating retinal axons. Science. 1989;246:255–257. doi: 10.1126/science.2799387. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BK, Liu J, Messerer C, Kobayashi NR, McGraw J, Oschipok L, Tetzlaff W. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci USA. 2002;99:3246–3251. doi: 10.1073/pnas.052308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagord C, Berry M, Logan A. Expression of TGFβ2 but not TGFβ1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol Cell Neurosci. 2002;20:69–92. doi: 10.1006/mcne.2002.1121. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lin CY, Jiang HH, DePaul M, Lin VW, Silver J. Nerve regeneration restores supraspinal control of bladder function after complete spinal cord injury. J Neurosci. 2013;33:10591–10606. doi: 10.1523/JNEUROSCI.1116-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Raisman G. Sprouts from cut corticospinal axons persist in the presence of astrocytic scarring in long-term lesions of the adult rat spinal cord. Exp Neurol. 1995;134:102–111. doi: 10.1006/exnr.1995.1041. [DOI] [PubMed] [Google Scholar]
- Lin SC, Huck JH, Roberts JD, Macklin WB, Somogyi P, Bergles DE. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron. 2005;46:773–785. doi: 10.1016/j.neuron.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Wang Y, Graham L, McHale K, Gao M, Wu D, Brock J, Blesch A, Rosenzweig ES, Havton LA, Zheng B, Conner JM, Marsala M, Tuszynski M. Long-distance growth and connectivity of neural stem cells after severe spinal cord injury. Cell. 2012;150:1264–1273. doi: 10.1016/j.cell.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Salgueiro Y, Beckerman SR, Lemmon VP, Tsoulfas P, Park KK. Three-dimensional evaluation of retinal ganglion cell axon regeneration and pathfinding in whole mouse tissue after injury. Exp Neurol. 2013;247:653–662. doi: 10.1016/j.expneurol.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle JM, Vicini S, Wrathall JR. Phenotypic changes in NG2+ cells after spinal cord injury. J Neurotrauma. 2006;23:1726–1738. doi: 10.1089/neu.2006.23.1726. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol. 2004;474:469–486. doi: 10.1002/cne.20149. [DOI] [PubMed] [Google Scholar]
- Margolis RK, Margolis RU. Nervous tissue proteoglycans. Experientia. 1993;49:429–446. doi: 10.1007/BF01923587. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Höke A, Silver J. Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol. 1995;136:32–43. doi: 10.1006/exnr.1995.1081. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MK, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J, Batt J, Doering LC, Rotin D, Bain JR. Enhanced rate of nerve regeneration and directional errors after sciatic nerve injury in receptor protein tyrosine phosphatase sigma knock-out mice. J Neurosci. 2002;22:5481–5491. doi: 10.1523/JNEUROSCI.22-13-05481.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Tripathi R, Wei P. NG2 colocalizes with axons and is expressed by a mixed cell population in spinal cord lesions. J Neuropathol Exp Neurol. 2006;65:406–420. doi: 10.1097/01.jnen.0000218447.32320.52. [DOI] [PubMed] [Google Scholar]
- Meletis K, Barnabé-Heider F, Carlén M, Evergren E, Tomilin N, Shupliakov O, Frisén J. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008;6:1494–1507. doi: 10.1371/journal.pbio.0060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon LDF, Fawcett JW. Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFβ1 and β2. Eur J Neurosci. 2001;14:1667–1677. doi: 10.1046/j.0953-816x.2001.01795.x. [DOI] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, Verhaagen J. Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol Cell Neurosci. 1999;13:143–166. doi: 10.1006/mcne.1999.0738. [DOI] [PubMed] [Google Scholar]
- Pindzola RR, Doller C, Silver J. Putative inhibitory extracellular matrix molecules at the dorsal root entry zone of the spinal cord during development and after root and sciatic nerve lesions. Dev Biol. 1993;156:34–48. doi: 10.1006/dbio.1993.1057. [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S. Degeneration and regeneration of the nervous system. Hafner; New York: 1928. [Google Scholar]
- Reier PJ. Penetration of grafted astrocytic scars by regenerating optic nerve axons in Xenopus tadpoles. Brain Res. 1979;164:61–68. doi: 10.1016/0006-8993(79)90006-4. [DOI] [PubMed] [Google Scholar]
- Reier PJ, Bregman BS, Wujek JR. Intraspinal transplantation of embryonic spinal cord tissue in neonatal and adult rats. J Comp Neurol. 1986;247:275–296. doi: 10.1002/cne.902470302. [DOI] [PubMed] [Google Scholar]
- Reier PJ, de Webster HF. Regeneration and remyelination of Xenopus tadpole optic nerve fibers following transection or crush. J Neurocytol. 1974;3:591–618. doi: 10.1007/BF01097626. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurons. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Silver J. Inhibition of neurite outgrowth on astroglial scars in vitro. J Neurosci. 1990;10:3594–3603. doi: 10.1523/JNEUROSCI.10-11-03594.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabelström H, Stenudd M, Réu P, Dias DO, Elfineh M, Zdunek S, Damberg P, Göritz C, Frisén J. Resident neural stem cells restrict tissue damage and neuronal loss after spinal cord injury in mice. Science. 2013;342:637–640. doi: 10.1126/science.1242576. [DOI] [PubMed] [Google Scholar]
- Sapieha PS, Duplan L, Uetani N, Joly S, Tremblay ML, Kennedy TE, Di Polo A. Receptor protein tyrosine phosphatase sigma inhibits axon regrowth in the adult injured CNS. Mol Cell Neurosci. 2005;28:625–635. doi: 10.1016/j.mcn.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Kitchener P, Knott GW, Nicholls JG, Potter A, Smith TJ. Development of walking, swimming and neuronal connections after complete spinal cord transection in the neonatal opossum, Monodelphis domestica. J Neurosci. 1988;18:339–355. doi: 10.1523/JNEUROSCI.18-01-00339.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvé Y, Sawai H, Rasminsky M. Topological specificity in reinnervation of the superior colliculus by regenerated retinal ganglion cell axons in adult hamsters. J Neurosci. 2001;21:951–960. doi: 10.1523/JNEUROSCI.21-03-00951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J Neurosci. 2010;30:5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer MC, Fawcett JW. The astrocyte/meningeal cell interface – a barrier to successful nerve regeneration? Cell Tissue Res. 2001;305:267–273. doi: 10.1007/s004410100384. [DOI] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPsigma is a receptor for chondroitin sulfate proteoglycan, and inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Nordlander RH, Egar M. Axonal guidance during embryogenesis and regeneration in the spinal cord of the newt: the blueprint hypothesis of neuronal pathway patterning. J Comp Neurol. 1979;185:1–22. doi: 10.1002/cne.901850102. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith GM, Miller RH. Immature type-1 astrocytes suppress glial scar formation, are motile and interact with blood vessels. Brain Res. 1991;543:111–122. doi: 10.1016/0006-8993(91)91054-5. [DOI] [PubMed] [Google Scholar]
- Smith GM, Miller RH, Silver J. Changing role of forebrain astrocytes during development, regenerative failure, and induced regeneration upon transplantation. J Comp Neurol. 1986;251:23–43. doi: 10.1002/cne.902510103. [DOI] [PubMed] [Google Scholar]
- Smith GM, Silver J. Transplantation of immature and mature astrocytes and their effect on scar formation in the lesioned central nervous system. Prog Brain Res. 1988;78:353–361. doi: 10.1016/s0079-6123(08)60304-0. [DOI] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow DM, Lemmon V, Carrino DA, Caplan AI, Silver J. Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol. 1990;109:111–130. doi: 10.1016/s0014-4886(05)80013-5. [DOI] [PubMed] [Google Scholar]
- Snow DM, Steindler DA, Silver J. Molecular and cellular characterization of the glial roof plate of the spinal cord and optic tectum: a possible role for a proteoglycan in the development of an axon barrier. Dev Biol. 1990;138:359–376. doi: 10.1016/0012-1606(90)90203-u. [DOI] [PubMed] [Google Scholar]
- Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, Lee JK. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci. 2013;33:13882–13887. doi: 10.1523/JNEUROSCI.2524-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilker MH, Yannas IV, Kostyk SK, Norregaard TV, Hsu HP, Spector M. The effects of tubulation on healing and scar formation after transection of the adult rat spinal cord. Restor Neurol Neurosci. 2001;18:23–38. [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Yoshioka N, Onaga SH, Watanabe Y, Miyata S, Wada Y, Kudo C, Okada M, Ohko K, Oda K, Sato T, Yokoyama M, Matsushita N, Nakamura M, Okano H, Sakimura K, Kawano H, Kitagawa H, Igarashi M. Chondroitin sulphate N-acetylgalactosaminyl-transferase-1 inhibits recovery from neural injury. Nat Comm. 2013;4:2740. doi: 10.1038/ncomms3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJ. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–444. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Sandrow-Feinberg HR, Miller K, Santi L, Connors T, Lemay MA, Houlé JD. Combining peripheral nerve grafts and chondroitinase promotes functional axonal regeneration in the chronically injured spinal cord. J Neurosci. 2009;29:14881–14890. doi: 10.1523/JNEUROSCI.3641-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24:6531–6539. doi: 10.1523/JNEUROSCI.0994-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Shen LF, Kuo HS, Cheng H, Chak KF. Involvement of acidic fibroblast growth factor in spinal cord injury repair processes revealed by a proteomics approach. Mol Cell Proteomics. 2008;7:1668–1687. doi: 10.1074/mcp.M800076-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Zee CE, Man TY, Van Lieshout EM, Van der Heijden I, Van Bree M, Hendriks WJ. Delayed peripheral nerve regeneration and central nervous system collateral sprouting in leucocyte common antigen-related protein tyrosine phosphatase-deficient mice. Eur J Neurosci. 2003;17:991–1005. doi: 10.1046/j.1460-9568.2003.02516.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Katagiri Y, McCann TE, Unsworth E, Goldsmith P, Yu ZX, Tan F, Santiago L, Mills EM, Wang Y, Symes AJ, Geller HM. Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J Cell Sci. 2008;121:3083–3091. doi: 10.1242/jcs.032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner IB, Deik A, Torres M, Rosendahl A, Neary JT, Lemmon VP, Bixby JL. A new in vitro model of the glial scar inhibits axon growth. Glia. 2008;56:1691–1709. doi: 10.1002/glia.20721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, Rao M, Gensel JC, McTigue DM, Kaspar BK, Jakeman LB. Transforming growth factor α transforms astrocytes to a growth-supportive phenotype after spinal cord injury. J Neurosci. 2009;31:15173–15187. doi: 10.1523/JNEUROSCI.3441-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Schneider GE, Silver J, Poston M, Jhaveri S. A role for tectal midline glia in the unilateral containment of retinocollicular axons. J Neurosci. 1998;18:8344–8355. doi: 10.1523/JNEUROSCI.18-20-08344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Yeo TT, Zhang C, Yang T, Tisi MA, Massa SM, Longo FM. The leukocyte common antigen-related protein tyrosine phosphatase receptor regulates regenerative neurite outgrowth in vivo. J Neurosci. 2001;21:5130–5138. doi: 10.1523/JNEUROSCI.21-14-05130.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Guénard V, Kleitman N, Aebischer P, Bunge MB. A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol. 1995;134:261–272. doi: 10.1006/exnr.1995.1056. [DOI] [PubMed] [Google Scholar]
- Yang HY, Lieska N, Shao D, Kriho V, Pappas GD. Proteins of the intermediate filament cytoskeleton as markers for astrocytes and human astrocytomas. Mol Chem Neuropathol. 1994;21:155–176. doi: 10.1007/BF02815349. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylera B, Ertürk A, Hellal F, Nadrigny F, Hurtado A, Tahirovic S, Oudega M, Kirchhoff F, Bradke F. Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr Biol. 2009;19:930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukor K, Belin S, Wang C, Keelan N, Wang X, He Z. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci. 2013;33:15350–15361. doi: 10.1523/JNEUROSCI.2510-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukor KA, Kent DT, Odelberg SJ. Meningeal cells and glia establish a permissive environment for axon regeneration after spinal cord injury in newts. Neural Dev. 2011;6:1–23. doi: 10.1186/1749-8104-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]