

Abstract
Although Lgr5+ intestinal stem cells have been expanded in vitro as organoids, homogeneous culture of these cells has not been possible thus far. Here we show that two small molecules, CHIR99021 and valproic acid, synergistically maintain self-renewal of mouse Lgr5+ intestinal stem cells, resulting in nearly homogeneous cultures. The colony-forming efficiency of cells from these cultures is ~100-fold greater than that of cells cultured in the absence of CHIR99021 and valproic acid, and multilineage differentiation ability is preserved. We made use of these homogeneous cultures to identify conditions employing simultaneous modulation of Wnt and Notch signaling to direct lineage differentiation into mature enterocytes, goblet cells and Paneth cells. Expansion in these culture conditions may be feasible for Lgr5+ cells from the mouse stomach and colon and from the human small intestine. These methods provide new tools for the study and application of multiple intestinal epithelial cell types.
A single layer of epithelial cells that actively self-renews and is organized into crypts and villi clothes the intestine. It has recently been shown that the renewal of intestinal epithelium is driven by Lgr5+ intestinal stem cells (ISCs) that reside at the bottom of crypts1. Lgr5+ stem cells can be isolated and cultured in vitro to form organoids containing crypt-villus structures that recapitulate the native intestinal epithelium2. Although stem cells can be readily expanded for multiple passages in the form of organoids, existing culture conditions provide little to no control over their self-renewal and differentiation. Typical cultures consist of heterogeneous cell populations including stem cells and differentiated cells2.
Paneth cells have been shown to be an important constituent of the Lgr5+ stem cell niche within intestinal crypts3-5. In particular, the self-renewal and proliferation of Lgr5+ stem cells both in vitro and in vivo are dependent on direct cell contact between Lgr5+ stem cells and Paneth cells6, which complicates the ability to control the fate of Lgr5+ stem cells in culture. This is evidenced by the inefficient culture of single Lgr5+ stem cells in the absence of Paneth cells3. Indeed, when cultured as organoids, ISCs spontaneously differentiate into all epithelial cell types, with stem cells being maintained only at the tip of crypts. The inability to efficiently expand Lgr5+ stem cells considerably limits the translation to therapies as well as the study of intestinal epithelial biology, given that differentiated progeny do not divide.
The self-renewal and differentiation of ISCs is controlled by the coordinated regulation of several signaling pathways, including Wnt, Notch and bone morphogenetic protein (BMP) pathways7-12. Here we identified small molecules that target these signaling pathways to maintain the self-renewal of Lgr5+ stem cells and to control their differentiation independently of cues provided by other cell types.
RESULTS
Maintenance of Lgr5+ stem cell self-renewal
For conventional intestinal organoid cultures, small-intestinal crypts isolated from Lgr5-EGFP-IRES-CreERT2 mice were embedded in Matrigel and cultured in the presence of epidermal growth factor (EGF), Noggin and R-spondin 1 (collectively, ENR) as described previously2. Lgr5-GFP was expressed in cells at the crypt bottoms in approximately half of the cultured crypts, a result consistent with the in vivo expression of the GFP reporter (Supplementary Fig. 1a). The growth factors used in the ENR condition provide essential, but not adequate, cues for sustaining the self-renewal of Lgr5+ stem cells when they lose contact with Paneth cells, leading to stem cell differentiation. We postulated that other factors are essential to maintain the self-renewal of ISCs in the absence of Paneth cells. To identify such factors, we tested selected small molecules that modulate signaling pathways (Wnt, Notch and BMP) known to be important in ISCs, performing experiments under the ENR condition and using the Lgr5-GFP reporter. We found that CHIR99021 (CHIR or C; ENR-C denotes addition of CHIR to the ENR condition), a glycogen synthase kinase 3β (GSK3β) inhibitor, promoted the proliferation of crypt cells, as indicated by increased cell numbers and a larger than average size of organoids compared to those observed with ENR-only cultures (Fig. 1a,b and Supplementary Fig. 1b,c). CHIR increased the percentage and relative GFP intensity of GFP+ cells in the culture, a result indicating increased self-renewal of stem cells (Fig. 1a,b). However, the organoids still contained a large number of GFP− cells (Fig. 1a). Valproic acid (VPA or V), a histone deacetylase (HDAC) inhibitor, which we selected for its role in Notch activation13,14, also markedly increased the GFP expression in these organoids (Fig. 1a). When CHIR and VPA were combined (CV), the total cell number as well as the percentage and relative intensity of GFP-expressing cells significantly increased (P < 0.0001, n = 3; Fig. 1a,b). Under CV conditions, we observed Lgr5-GFP reporter expression throughout the organoids (Fig. 1a), which indicated minimal differentiation and increased self-renewal of stem cells under these conditions.
Figure 1.
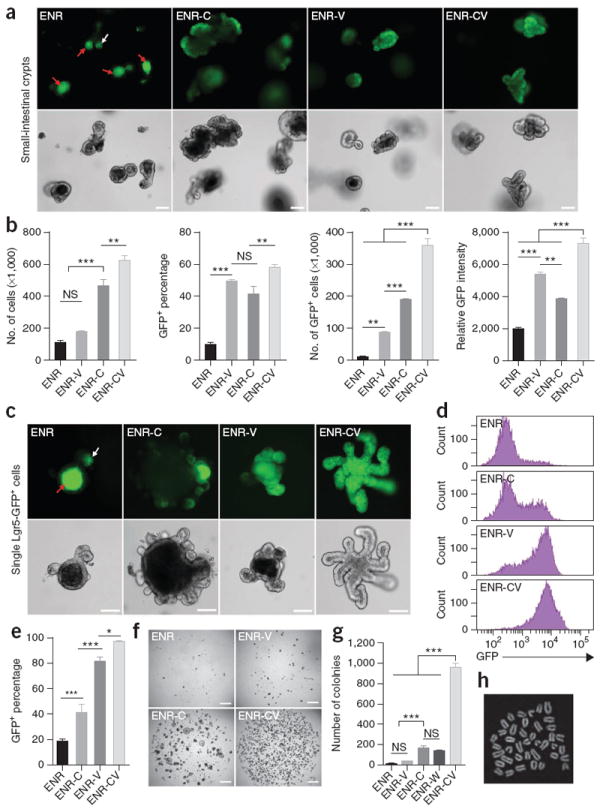
The combination of CHIR and VPA promotes self-renewal of Lgr5+ stem cells. (a) GFP fluorescence and bright-field images of small-intestinal crypts cultured for 6 d in the presence of EGF, Noggin and R-spondin 1 (ENR); ENR and VPA (ENR-V); ENR and CHIR (ENR-C); and ENR, VPA and CHIR (ENR-CV). Apoptotic cells are visible in the lumen with autofluorescence (red arrows), and white arrow indicates Lgr5-GFP at the bottom of crypts. (b) Quantification of cell proliferation (number of live cells) and GFP expression in crypts cultured for 6 d. Lgr5-GFP expression was measured by flow cytometry. (c) Fluorescence and bright-field images of single Lgr5-GFP cells after 9 d of culture in multiple conditions. (d,e) Flow cytometry analysis of GFP expression 7 d after seeding of single Lgr5-GFP+ cells in conditions as indicated. Error bars, s.d. (n = 3 wells). (f,g) Representative images of 4,000 FACS-isolated single Lgr5-GFP cells cultured under one of several conditions for 7 d (f) and quantification of colony numbers (g). (h) Metaphase spread of a cell cultured in the CV condition for 80 d. Scale bars, 100 μm (a,c) and 1 mm (f). ***P < 0.001; **P < 0.01; * P < 0.05; NS, not significant (P > 0.05). Error bars, s.d. (n = 3 wells). Experiments in b and e were performed in three biological replicates (n = 3 animals), with similar results (data not shown).
The level of GFP expression in the CV condition was similar to that of GFPhigh stem cells observed in freshly isolated crypts2, but CV cultures lacked the GFPlow population representing immediate stem cell daughter cells15 (Supplementary Fig. 1d). Notably, R-spondin 1 and Noggin were still required to maintain the self-renewal of Lgr5+ stem cells in CV conditions. Although EGF promoted the proliferation of crypts, it could be removed without affecting maintenance of the Lgr5+ cells (Supplementary Fig. 1e). Increasing the concentration of CHIR partially eliminated the need for R-spondin 1 promotion of GFP expression (Supplementary Fig. 1f), which is consistent with the role of R-spondin 1 in increasing Wnt–β-catenin signaling16.
We tested the effects of CHIR and VPA on self-renewal by assaying colony formation of single GFPhigh cells following FACS sorting of cells from dispersed crypts (which also allows us to circumvent mosaic GFP expression; Supplementary Fig. 1a,d). We observed low colony-forming efficiency (below 1%) in ENR, as previously reported2. The addition of CHIR increased colony formation by 20- to 50-fold (Fig. 1f,g and Supplementary Fig. 2b,c), as did the addition of recombinant Wnt3A (ENR-W; ref. 3 and Fig. 1g), but only moderately maintained GFP expression (Fig. 1c–e). In contrast, VPA promoted GFP expression (Fig. 1c–e) but only weakly increased colony-forming efficiency (Fig. 1f,g and Supplementary Fig. 2b,c). In the combined CV condition, we observed a significant increase in colony formation (P < 0.0001, n = 3; Fig. 1f,g and Supplementary Fig. 2b,c) from 25% to 40% of the total seeded cells and >90% of the live cells (Supplementary Fig. 2d); >97% of the cells were GFP+ (Fig. 1c–e and Supplementary Fig. 2a). Furthermore, cells cultured in the CV condition could be passaged as single cells for more than ten passages with a colony morphology, proliferative ability, colony-forming efficiency and stem cell potential similar to that of freshly isolated Lgr5-GFP+ cells (Supplementary Fig. 2e–h). The cells showed a normal karyotype after 15 passages (2n = 40 chromosomes; Fig. 1h).
We further examined the behavior of Lgr5− or Lgr5low cells in CV conditions. Owing to the mosaic expression of Lgr5-GFP reporter, it is technically challenging to obtain truly Lgr5− cells17. Instead, we isolated single Lgr5-GFPlow cells and cultured them in CV. In an outcome consistent with previous reports that Lgr5low cells can revert to the stem cell state if re-exposed to niche signals in vivo17,18, we found that, after culture in CV for 9 d, Lgr5-GFPlow cells were able to re-express the reporter at a level comparable to that of Lgr5-GFPhigh cells (Supplementary Fig. 2i–m), although they displayed a lower colony-forming efficiency than Lgr5-GFPhigh cells (Supplementary Fig. 2m).
We tested the effects of these conditions on Lgr5+ stem cells in other tissues and species. Culture in CV conditions or in VPA alone promoted GFP expression in organoids derived from stomach (Supplementary Fig. 3a–c) and colon (Supplementary Fig. 3d–f) of Lgr5-GFP transgenic mice. We also observed that colonic crypts cultured in CV expressed higher levels of mRNA transcripts of the stem cell marker Lgr5 and lower levels of differentiation markers than colonic crypts cultured in ENR-W19 or ENR-C (Supplementary Fig. 3g). The CV condition also substantially increased colony formation of cells from freshly isolated human small-intestinal crypts (Supplementary Fig. 3h–j); as in the mouse, we observed that human cells in CV expressed higher levels of LGR5 transcripts and similar or lower levels of differentiation markers than cells cultured in previously reported culture conditions (with the addition of Wnt3a, nicotinamide, A83-01, SB202190 and PGE2 to the ENR condition; Supplementary Fig. 3k)19,20. These results suggest that the combination of CHIR and VPA promotes the maintenance of the epithelial stem cell state in multiple tissues.
Stem cell expansion through reduction of differentiation
As previously reported, cells in the ENR condition grew into organoids with crypt-villus structure containing all intestinal epithelial cell types2, including enterocytes (Alp+), goblet cells (Muc2+), enteroendocrine cells (ChgA+) (Supplementary Fig. 4), Paneth cells (Lyz+) and Lgr5-GFP+ stem cells (Fig. 2a). Ki67 and EdU staining revealed proliferating cells only within the crypt domains (Fig. 2b,c). In the CV condition, however, GFP+ stem cells were present throughout the colony; we observed few Paneth cells (Fig. 2a) and no other cells types. In line with this, Ki67+ and EdU+ cells were found throughout the colony (Fig. 2b,c).
Figure 2.
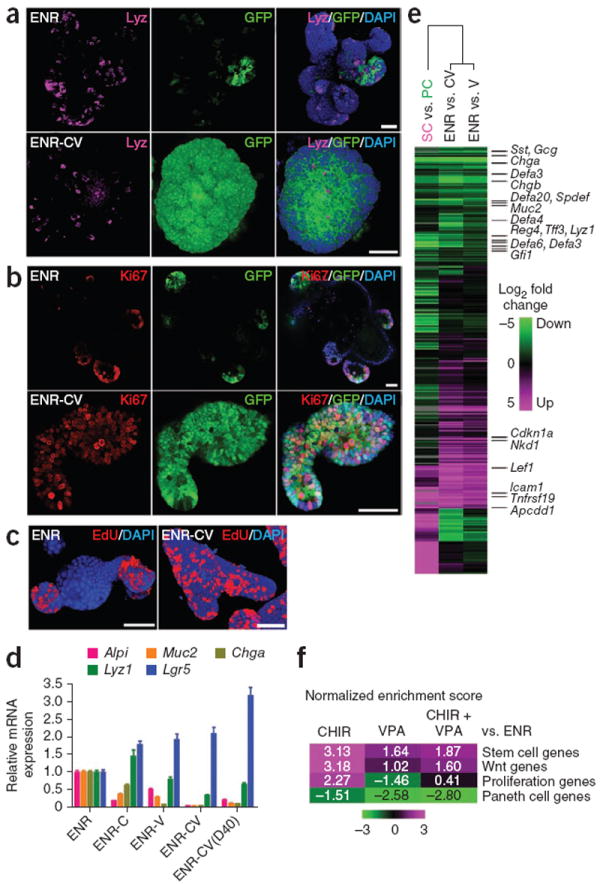
The combination of CHIR and VPA maintains the stem cell state of Lgr5+ stem cells. (a–c) Confocal images showing lysozyme (a), Ki67 (b) and EdU (c) staining of organoids cultured under the ENR and ENR-CV conditions. DAPI was used as a nuclear stain. (d) Quantitative real-time PCR analysis of relative mRNA expression of markers for intestinal epithelial cells (Alpi for enterocytes, Muc2 for goblet cells, Chga for enteroendocrine cells, Lyz1 for Paneth cells and Lgr5 for ISCs) cultured for 6 d under conditions as indicated. ENR-CV(D40) indicates cells that were cultured in the CV condition for 40 d. Error bars, s.d. (n = 3 wells). Experiments were performed in three biological replicates (n = 3 animals), with similar results (data not shown). (e) Microarray analysis of gene expression changes in organoids cultured for 6 d in the presence of ENR-V (V) or ENR-CV (CV) compared with ENR. The heat map shows clustering to previously reported array data of sorted Paneth cells (PC) versus Lgr5 stem cells (SC). Green and magenta indicate down- and upregulation, respectively. (f) Gene-set enrichment analysis (GSEA) comparing organoid microarray data with published gene expression signatures. Normalized enrichment scores are represented as a heat map to visualize the degree of enrichment in the up- or downregulated genes in organoids (magenta and green, respectively). Refer to Supplementary Figure 5 for detailed analysis. Scale bars, 50 μm.
On the basis of quantitative PCR measurements of marker transcript expression, we observed that treatment with CHIR alone reduced enterocyte differentiation and increased Paneth cell differentiation (Fig. 2d), consistent with a previous report5, whereas VPA alone decreased secretory cell differentiation (Fig. 2d). In combined CV conditions, we observed minimal levels of Alpi, Muc2 and Chga, moderate levels of Lyz1 and high levels of Lgr5 expression compared to levels in cells in the ENR-only condition. This expression pattern was maintained over multiple passages (Fig. 2d and Supplementary Fig. 2h).
We further analyzed global effects of CV culture on gene expression by microarray analysis. We visualized gene expression changes by performing a clustering analysis with our previously reported microarray data of sorted Paneth cells versus Lgr5+ stem cells3. As seen with the phenotype data, we observed a collective downregulation of Paneth cell markers and induction of stem cell markers (Fig. 2e). We validated these effects by gene-set enrichment analysis (GSEA)21, which showed upregulation of the Lgr5+ stem cell signature genes22 after CHIR and VPA treatments and strong downregulation of Paneth cell genes3 following VPA treatment (Fig. 2f and Supplementary Fig. 5). Wnt and R-spondin target-gene expression16 was induced by CHIR treatment, which also promoted expression of proliferation markers23, whereas VPA treatment did not induce such effects.
Lgr5+ stem cells remain multipotent
ISCs have the ability to self-renew as well as differentiate into all cell lineages of the intestinal epithelium. To test the differentiation capability of Lgr5+ stem cells cultured in the CV condition, we transferred colonies from the CV to the ENR condition, which permits spontaneous differentiation into the mature cell types of the intestine. After withdrawal of CHIR and VPA, the cultures regained the typical morphology of intestinal organoids with crypt-villus structure and Lgr5+ stem cells at crypt bottoms (Fig. 3a and Supplementary Fig. 4c). mRNA expression of Alpi, Muc2 and Chga increased, and these cells expressed a level of Lyz1 similar to that of cells continuously cultured in CV (Fig. 3b). Immunocytochemistry staining for these markers confirmed the existence of differentiated cell types in the culture (Fig. 3a), demonstrating that expanded Lgr5+ stem cells can differentiate into all lineages of the intestinal epithelium.
Figure 3.
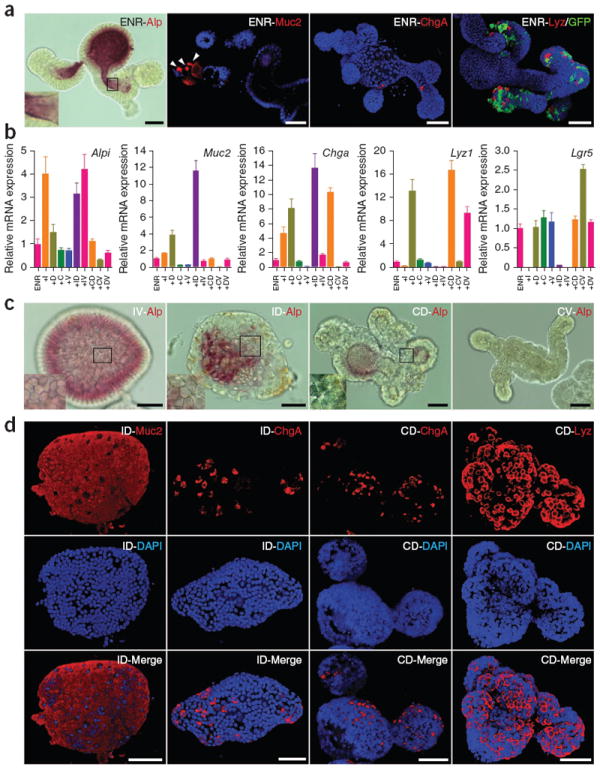
Differentiation of ISCs cultured under the CV condition. (a) Staining of the indicated differentiation markers of Lgr5-GFP cells transferred from the CV condition to ENR and cultured for 4 d. DAPI (blue) was used to stain nuclei, and GFP indicates the presence of stem cells. (b) Real-time reverse-transcription PCR analysis of relative mRNA expression of mature intestinal epithelial markers from cells initially cultured in CV (6 d) and then transferred to the indicated conditions. ENR was present in all conditions and mRNA expression was normalized to expression in ENR alone. I, IWP-2; D, DAPT; C, CHIR; V, VPA. Error bars, s.d. (n = 3 wells). (c) Alp staining of cells cultured under multiple conditions. The morphological change of cells in the ID and CD conditions reflects formation of goblet and Paneth cells, respectively. (d) Immunocytochemistry staining of differentiation markers. Cells cultured under the ID and CD conditions were used for Muc2, ChgA and Lyz staining. Three-dimensional reconstructed confocal images are shown. Scale bars, 50 μm.
Controlled differentiation of Lgr5+ stem cells
We attempted to direct the differentiation of Lgr5+ stem cells. As Wnt and Notch are two of the main signaling pathways that control the differentiation of ISC, we used the Wnt pathway inhibitor IWP-2 and Notch inhibitor DAPT to induce the differentiation of cultured Lgr5+ stem cells. Single stem cells were cultured in CV for 6 d to obtain a homogeneous starting population of stem cells that was then transferred into separate wells and cultured under ENR conditions in the presence of single or multiple inhibitors (Supplementary Fig. 4b). We observed that replacing CV with DAPT decreased Lgr5 expression and induced expression of differentiation markers Alpi, Muc2, Chga and Lyz1 (Fig. 3b), a result consistent with previous reports that Notch inhibition induces secretory cell differentiation24-27. Alternatively, Wnt inhibition with IWP-2 preferentially induced Alpi expression, with modestly elevated Muc2 and Chga expression and completely abolished Lyz1 and Lgr5 expression. This indicates that Wnt signaling is required to maintain ‘stemness’ and to suppress enterocyte differentiation yet is also required for Paneth cell differentiation. Notably, the presence of VPA produced a lower level of expression of Muc2, Chga and Lyz1 but not Alpi (Fig. 3b), indicating that VPA specifically suppressed secretory cell lineage differentiation. The combination of IWP-2 and VPA specifically induced enterocyte differentiation, presumably by combining the effects of both inhibitors, in which IWP-2 induced enterocyte differentiation while VPA suppressed the differentiation of Lgr5+ stem cells toward secretory cell types. Similarly, the combination of DAPT and CHIR mainly induced Paneth cell differentiation, and the combination of IWP-2 and DAPT primarily induced goblet cell differentiation. These conditions also induced clear morphological changes (Fig. 3c and Supplementary Fig. 4d), and we further confirmed our observations by staining for enterocyte, goblet cell and Paneth cell markers (Fig. 3c,d and Supplementary Fig. 4e,f). Under the various optimized direct differentiation conditions, at day 4 we observed either ~95% enterocytes, 90% goblet cells or 47% Paneth cells (which increased to 85% at day 7). The presence of IWP-2 or CHIR did not notably influence Chga expression, indicating that differentiation of enteroendocrine cells (~10%) is independent of Wnt signaling but may require alternative signaling activities.
Toward elucidating the mechanism of CHIR and VPA
CHIR is a highly specific GSK3 inhibitor that inhibits GSK3β-mediated β-catenin degradation28. We tested other Wnt–β-catenin pathway activators, including lithium and Wnt3a, and found that they also increased proliferation, as indicated by increased colony size and cell numbers compared with those of the ENR-only condition (Fig. 4a,b). Colonies in these conditions showed a cyst-like morphology (Fig. 4a), as previously reported3. Similarly, we tested the effects of other HDAC inhibitors including pan-HDAC inhibitors and type-specific inhibitors (Supplementary Fig. 6). We found that pan-HDAC inhibitor trichostatin A (TSA) as well as HDAC6-specific inhibitors tubastatin A and compound 7 showed a similar effect of promoting GFP expression, as did VPA (Fig. 4c–e and Supplementary Fig. 6a,b). At higher concentrations, TSA and VPA showed a marked inhibitory effect on proliferation, but Lgr5-GFP expression was maintained (Fig. 4d).
Figure 4.
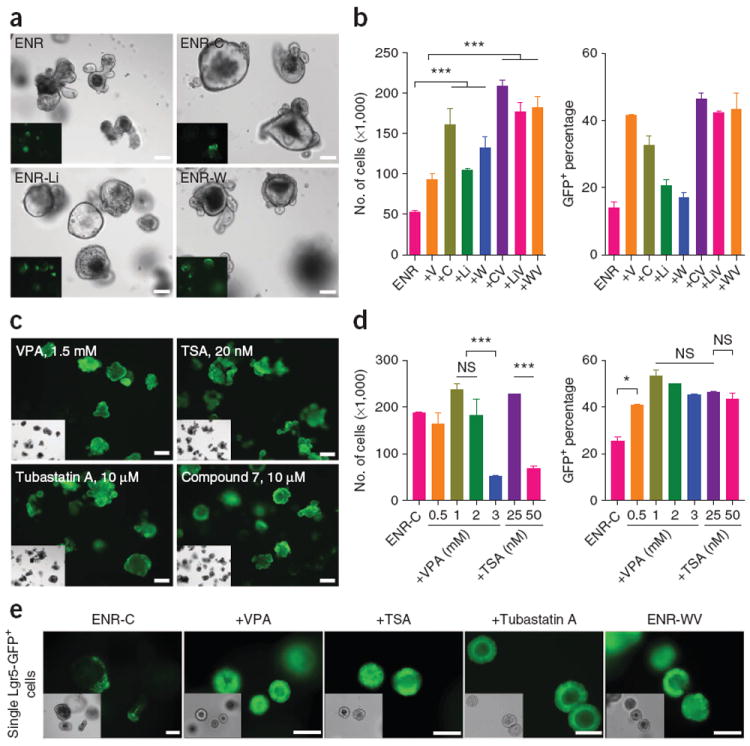
Exploring the mechanism of action for CHIR and VPA. (a) Morphology and Lgr5-GFP expression of crypts cultured in multiple conditions for 6 d. C, CHIR; Li, LiCl; W, Wnt3a; V, VPA. (b) Cell numbers and percentage of GFP+ cells for 6-d crypt cultures. The data are representative of three independent experiments. Error bars, s.d. (n = 3 wells). (c) 6-d cultures of crypts in the ENR-C (control) condition or with histone deacetylase (HDAC) inhibitors. See Supplementary Figure 6 for complete analysis. TSA, trichostatin A. (d) Effects of VPA and TSA on cell proliferation and GFP expression at multiple concentrations. Error bars, s.d. (n = 2 wells). Experiments were performed in three biological replicates (n = 3 animals), with similar results (data not shown). (e) Effects of other HDAC inhibitors and recombinant Wnt protein. Morphology and GFP expression of single Lgr5-GFP cells cultured for 6 d are shown. TSA concentration is 25 nM; tubastatin A is 10 μM. Scale bars, 100 μm. ***P < 0.001; *P < 0.05; NS, not significant (P > 0.05).
DISCUSSION
Pluripotent stem cells, including embryonic stem cells and induced pluripotent stem cells, can be cultured homogeneously in vitro. However, adult stem cells in vivo are generally thought to depend on a specialized cellular niche supporting their self-renewal. Ex vivo expansion of pure populations of adult stem cells has shown limited success29. Here we have shown that, when provided with essential niche signals, Lgr5+ ISCs can be cultured homogeneously with rapid proliferation kinetics and long-term self-renewal ability.
The ability to expand high-purity Lgr5+ stem cells in vitro permits further investigation of conditions for directing the differentiation of Lgr5+ stem cells toward mature cell types, which are otherwise difficult to obtain given that mature gut epithelial cells do not proliferate. This was not easily achievable using cells cultured in the previously described (ENR) conditions owing to the existence of mixed cell populations in organoid cultures, the low survival rate of pure single stem cells following isolation by cell sorting, and the low efficiency of colony formation. We used our culture system to reveal that differentiation of each cell lineage in the intestinal epithelium requires a distinct combination of signals (Fig. 5). Particularly, we showed that although Notch inhibition leads the specification of Lgr5+ stem cells toward secretory cell types, simultaneous activation with the Wnt pathway causes the differentiation of Paneth cells, whereas Wnt inhibition gives rise to goblet cells. This result provides insight into the ongoing discussion about whether Notch inhibition results in the generation of only goblet cells26,30,31 or all secretory cell types24,32-34.
Figure 5.
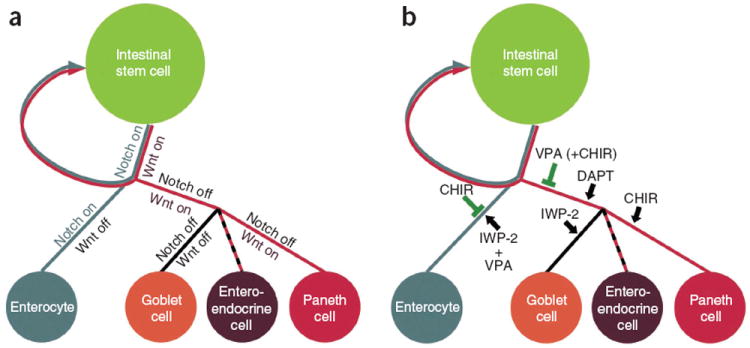
Model for ISC self-renewal and differentiation. (a) Under physiological conditions, the self-renewal and differentiation of ISCs is controlled by the cooperation of Wnt and Notch pathways. (b) When Lgr5+ stem cells are cultured in vitro, CHIR activates the Wnt pathway and inhibits enterocyte differentiation, whereas VPA alone or together with CHIR suppresses secretory cell specification. The combination of CHIR and VPA maintains ISCs in an undifferentiated, self-renewing state. Additional combinations of inhibitors promote various intestinal cell fates as shown.
We have shown that CHIR alone (ENR-C) is not sufficient to suppress the differentiation of stem cells, whereas further addition of VPA allows maintenance of the stem cell state. Strong Wnt activation alone, such as by ENR-C, ENR-W or loss of the tumor suppressor adenomatous polyposis coli (APC) mutation3, caused cells to form spherical colonies (Figs. 1f and 4a, Supplementary Fig. 2b and ref. 3). These colonies grew much faster and to larger sizes than colonies cultured in ENR or ENR-CV, a result suggesting the presence of fast-cycling transit-amplifying cells. Indeed, when single stem cells were cultured in CV, we initially observed a spherical morphology (Fig. 4e), but cells adopted a columnar shape with higher apical-basal length than that seen in the aforementioned ENR-C, ENR-W or APC deficiency cultures. We speculate that this columnar epithelium more easily folds passively into nonspherical structures and that addition of VPA may result in this columnar cell shape by inhibiting generation of transit-amplifying cells. Indeed, Lgr5 stem cells in vivo also have a columnar phenotype, which suggests that CV-treated cells resemble cells of normal morphology.
Efficient activation of the Wnt–β-catenin pathway can be achieved by administration of GSK3 inhibitors, Wnt proteins or other agonists, although these agents may influence other signaling components beyond β-catenin stabilization, including Notch, Myc and Jun, which may also play a role in the observed phenotype. However, pharmacologic activation of the Notch pathway remains challenging with the limited agonists available such as soluble Notch ligands Dll1, Dll4 or Jag1 (ref. 35). The activity of soluble Notch ligands is difficult to reconcile and usually requires clustering or immobilization to induce signaling but may otherwise show antagonist activity35. It is no question that HDAC inhibition will have multiple cellular effects, but here we showed that the primary effect of HDAC inhibition in the intestinal epithelium is homogeneous expansion of Lgr5+ stem cells. This effect does not depend on a general block of differentiation but rather on a specific block of secretory differentiation, as enterocyte differentiation did occur normally. Instead we have shown that VPA is epistatic to secretory lineage differentiation after γ-secretase inhibition (Supplementary Fig. 7 and Supplementary Results). We have demonstrated that homogeneous stem cells can be obtained in the absence of the juxtacrine Notch signals provided by Paneth cells or global activation of the Notch ligand (Supplementary Fig. 7 and Supplementary Results). Thus, we introduce a new concept to modulate the Notch pathway in the intestine by chemical modulation using HDAC inhibition. This strategy could be applied to other biological systems that involve spontaneous Notch signaling.
Our methods to generate high-purity cultures of Lgr5+ ISCs and their progenies provide a potentially unlimited source of intestinal epithelial cell types that should facilitate study of intestinal epithelial biology and the identification of agents that modulate this biology via high-throughput screens. These methods will ultimately help accelerate the development of therapeutics for multiple diseases.
ONLINE METHODS
Mice
Heterozygous Lgr5-EGFP-IRES-CreERT2 mice were obtained from Jackson Labs, and 6- to 12-week-old mice were used for crypts isolation. Animal experimental procedures were approved by the Committee on Animal Care (CAC) at MIT. Dll1EGFP-IRES-CreERT2 mice have been described18. The conditional alleles Atoh1fl (ref. 36), Rbpjfl (ref. 37) and Apcfl (ref. 38) have been described. For gene-ablation experiments, 6- to 8-week-old mice were injected intraperitoneally with a single dose of 5 mg tamoxifen dissolved in sunflower oil according to guidelines and reviewed by the animal ethics committee of the Royal Netherlands Academy of Arts and Sciences (KNAW).
Crypt and single-cell isolation
Crypts and single Lgr5-GFP cells were isolated as previously described2. Briefly, the proximal half of the small intestine was harvested, opened longitudinally and washed with cold PBS to remove luminal content. The tissue was then cut into 2- to 4-mm pieces with scissors and further washed 5–10 times with cold PBS by pipetting up and down using a 10-ml pipette. Tissue fragments were incubated with 2 mM EDTA in PBS for 30 min on ice. After removal of EDTA, tissue fragments were washed with PBS to release crypts. Supernatant fractions enriched in crypts were collected, passed through a 70-μm cell strainer and centrifuged at 300g for 5 min. The cell pellet was resuspended with cell culture medium without growth factors and centrifuged at 150g to remove single cells. Crypts were then cultured or used for single-cell isolation. Single cells were obtained by incubating crypts in culture medium for 45 min at 37 °C and triturating with a glass pipette. Dissociated cells were passed through a 20-μm cell strainer and negative stained with propidium iodide, and single viable GFPhigh cells were sorted by flow cytometry (FACS Aria, BD) as previously described2. Stomach and colon crypts were isolated as previously described19,39. Studies relevant to human materials were approved by the Committee On the Use of Humans as Experimental Subjects (COUHES) at MIT. Deidentified human small-intestinal tissue was obtained through the Massachusetts General Hospital Tissue Repository. Human small-intestinal crypts were isolated from resected normal small-intestinal specimens as previously described19,20. Briefly, the muscle layer and submucosa were removed under stereomicroscope. The mucosal layer was further cut into small pieces and washed with cold PBS. Samples were then incubated with 8 mM EDTA in PBS with slow rotation for 60 min at 4 °C and further washed with PBS. Supernatant enriched in crypts was collected for further experiments.
Cell culture
Isolated crypts or single cells were cultured as previously described2 with minimal modification. Briefly, crypts or single cells were entrapped in Matrigel and plated at the center of wells in a 24-well plate. Following polymerization of Matrigel (growth factor reduced; BD Bioscience), 500 μl of culture medium (Advanced DMEM/F12 (Life Technologies)) was added, containing growth factors including EGF (50 ng/ml, Life Technologies), Noggin (100 ng/ml, PeproTech) and R-spondin 1 (500 ng/ml, R&D) and small molecules including CHIR99021 (3 μM, Stemgent) and valproic acid (1 mM, Sigma-Aldrich). (See Supplementary Table 1 for details.) For comparison of different culture conditions, small molecules or growth factors were added to freshly isolated crypts immediately after plating in Matrigel to test the ability to minimize potential differentiation of the ISCs within the crypts and thus sustain crypt cultures. Cell culture medium was changed every other day. For single-cell culture, cells were embedded in Matrigel containing Jagged-1 peptide (1 μM; AnaSpec), and Y-27632 (10 μM; Tocris) was added for the first 2 d. Cells were passaged either as cell colonies as previously described2 or as single cells. For single-cell passage, cell culture medium was removed and Accutase (Life Technologies) was added. After incubation at 37 °C for 10–20 min, cell colonies were dissociated into single cells by pipetting. Cells were then washed, embedded in fresh Matrigel and plated into 24-well plates. Cells cultured in the CV condition were passaged every 6 d at a 1:20 split ratio.
Flow cytometry analysis
Cell culture medium was removed and Accutase was added. After incubation at 37 °C for 10–20 min, organoids or cell colonies were dissociated into single cells, and live-cell numbers were counted using a hemocytometer. The cells were then stained with propidium iodide (PI) and analyzed using a flow cytometer (FACSCalibur; BD). For comparisons of GFP expression, PI-negative cells were gated as GFP+ and GFP− populations and analyzed using BD FACSDiva software.
Chromosome analysis
For karyotyping, cell colonies were harvested and dissociated into single cells using Accutase. After centrifugation, cell pellets were resuspended with 0.56% KCl and incubated for 15 min at room temperature. Cells were then centrifuged, fixed in methanol:acetic acid (3:1) on ice and dropped onto glass slides. Air-dried cells were then stained with Giemsa staining, and metaphase spreads were imaged and analyzed. A total of ten metaphases were counted.
RNA extraction, qPCR and microarray experiments
RNA was isolated from cultured cells (RNeasy Mini Kit; Qiagen) according to the manufacturer’s protocol. Quantitative real-time PCR was performed with QuantiTect Probe PCR kit (Qiagen) using commercially available primers and TaqMan probes (Life Technologies). Microarray analysis was performed in duplicates (two independent organoid cultures) using 500 ng of whole RNA that was labeled using Quick Amp Labeling Kit, two color (Agilent Technologies), with Cy5 and Cy3 following the manufacturer’s guidelines. Hybridization of cRNA from control or treated organoids (Cy5 labeled) versus control organoids (Cy3 labeled, as internal reference) was done on 4X44K Agilent Whole Mouse Genome dual-color Microarrays (G4122F). Data were retrieved and normalized using Feature Extraction (v.9.5.3, Agilent Technologies), and data analysis was performed using Excel (Microsoft). Features were included only if signal intensities for the Cy5 channel were twofold higher than the background signal in at least one condition in both biological replicates. After log2 transformation, expression ratios were subtracted from the control ratio (control Cy5 versus control Cy3) to correct for dye bias in labeling. Average gene expression changes were compared to published array data of sorted Paneth cells versus stem cells (ref. 3, GSE25109) using the software Cluster and TreeView (M. Eisen, Stanford). Array probes that show at least one greater-than-threefold (log2) expression were included, and arrays were mean centered before hierarchical clustering. GSEA was performed using the freely available software (v.2.0, Broad Institute; ref. 21) using preranked lists of mean expression changes and input signatures that were derived from published microarray data3,16,22. GEO data sets accession numbers were GSE23672 and GSE25109 for Lgr5 stem cells and Paneth cells, respectively (sorted cells from crypts), and GSE28265 for genes downregulated in organoids 1 d after withdrawal of R-spondin. For these microarrays (same platform, G4122F) the 200 most differentially regulated probes were used as input. For analysis of proliferation genes, the MSigDB gene set “YU_MYC_TARGETS_UP” was used, which includes 44 experimentally verified Myc targets23. The number of permutations was 1,000.
Image analysis
Cell culture medium was removed, and samples were washed with PBS. Organoids or cell colonies cultured in Matrigel were fixed by directly adding 4% PFA and incubating for 10–30 min at room temperature. Matrigel was then mechanically disrupted, and cells were transferred into BSA-coated Eppendorf tubes. Samples were washed with PBS, permeabilized with 0.25% Triton X-100 for 30 min and stained with primary and secondary antibodies. The following antibodies were used: anti-Ki67 (ab16667, Abcam, 1:200), anti-Muc2 (sc-15334, Santa Cruz, 1:100), anti-chromogranin A (sc-13090, Santa Cruz, 1:200) and anti-lysozyme (A0099, Dako, 1:400). DyLight 650 conjugated antibodies (Abcam, 1:200) were used as secondary antibodies. 5-ethynyl-2-deoxyuridine (EdU) staining followed the manufacturer’s protocol (Click-IT; Invitrogen). Images were acquired either by confocal microscopy (Zeiss LSM 710) or by inverted microscope (EVOS; Advanced Microscopy Group). For RBP-J/GFP/Lyz coimmunofluorescence, 4-μm paraffin sections were incubated after citrate-mediated antigen retrieval using the following. Chicken anti-GFP (GFP-1020, Aves Labs, 1:250) signals were detected by anti-chicken biotin (703-065-155, Jackson ImmunoResearch, 1:500) and streptavidin–Alexa 568 (S-11226, Molecular Probes, 1:500); goat anti-lysozyme (sc-27958, Santa Cruz, 1:250) was detected using anti-goat C5 (705-175-147, Jackson ImmunoResearch, 1:400); and rat anti–RBP-J (clone T6708, Cosmo Bio, 1:100) signals were detected using rabbit anti-rat antibody (6180-01, SouthernBiotech, 1:400) followed by anti-rabbit HRP conjugate (BrightVision from Immunologic) and TSA–Alexa 488 detection (Life Technologies, 1:500) on a Leica Sp5 confocal microscope.
Statistical analysis
Data are expressed as mean ± s.d. Differences between groups were determined using one-way analysis of variance (ANOVA) and Tukey’s post hoc method of multiple comparisons. The level of significance (P value) is indicated in each experiment.
Supplementary Material
Acknowledgments
This research was supported by US National Institutes of Health (NIH) grant DE013023 to R.L. and a Harvard Institute of Translational Immunology/Helmsley Trust Pilot Grant in Crohn’s Disease to J.M.K. H.F.F. was supported by an EMBO long-term fellowship. We also thank D. Breault, R. Montgomery and R. Shivdasani for critically reviewing the manuscript; W. Cho, M. Haraguchi and Q. Wang for helpful discussions; W. Salmon and N. Watson of the W.M. Keck Biological Imaging Facility at the Whitehead Institute for assistance with imaging; G. Paradis, X. Song and M. Jennings of the MIT Flow Cytometry Core Facility for assistance with flow cytometry and CCSG NIH grant CA014051 for support; A. Bhan, V. Yajnik, M. Miri and A. Brunelle at Massachusetts General Hospital for providing human tissue; and all of the staff of the MIT animal care facility.
Footnotes
AUTHOR CONTRIBUTIONS
X.Y., H.F.F., H.C., R.L. and J.M.K. conceived of and designed the experiments. X.Y. and H.F.F. performed the experiments. J.H.v.E. provided Dll1EGFP-IRES-CreERT2, Atoh1 knockout organoids and Rbpj/Apc knockout-mice intestine. X.Y., H.F.F., J.H.v.E., H.C., R.L. and J.M.K. analyzed the data. X.Y., H.F.F., R.L. and J.M.K. wrote the manuscript.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Accession codes. NCBI Gene Expression Omnibus: microarray data are deposited under accession number GSE51539.
References
- 1.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 2.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 3.Sato T, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yilmaz ÖH, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–495. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farin HF, van Es JH, Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology. 2012;143:1518–1529. doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 6.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Scoville DH, Sato T, He XC, Li L. Current view: intestinal stem cells and signaling. Gastroenterology. 2008;134:849–864. doi: 10.1053/j.gastro.2008.01.079. [DOI] [PubMed] [Google Scholar]
- 8.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 9.Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7:349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- 10.Stanger BZ, Datar R, Murtaugh LC, Melton DA. Direct regulation of intestinal fate by Notch. Proc Natl Acad Sci USA. 2005;102:12443–12448. doi: 10.1073/pnas.0505690102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zecchini V, Domaschenz R, Winton D, Jones P. Notch signaling regulates the differentiation of post-mitotic intestinal epithelial cells. Genes Dev. 2005;19:1686–1691. doi: 10.1101/gad.341705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. 2011;73:213–237. doi: 10.1146/annurev.physiol.70.113006.100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenblatt DY, et al. Valproic acid activates Notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;12:942–951. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- 14.Stockhausen MT, Sjölund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer. 2005;92:751–759. doi: 10.1038/sj.bjc.6602309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Flier LG, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 16.de Lau W, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476:293–297. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 17.Wang F, et al. Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology. 2013;145:383–395. doi: 10.1053/j.gastro.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Es JH, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–1104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 20.Jung P, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muñoz J, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu D, Cozma D, Park A, Thomas-Tikhonenko A. Functional validation of genes implicated in lymphomagenesis: an in vivo selection assay using a Myc-induced B-cell tumor. Ann NY Acad Sci. 2005;1059:145–159. doi: 10.1196/annals.1339.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milano J, et al. Modulation of notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 25.Wong GT, et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 26.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 27.Fre S, et al. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 28.Bain J, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conti L, et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pellegrinet L, et al. Dll1- and Dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011;140:1230–1240. doi: 10.1053/j.gastro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riccio O, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9:377–383. doi: 10.1038/embor.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kazanjian A, Noah T, Brown D, Burkart J, Shroyer NF. Atonal homolog 1 is required for growth and differentiation effects of notch/gamma-secretase inhibitors on normal and cancerous intestinal epithelial cells. Gastroenterology. 2010;139:918–928. doi: 10.1053/j.gastro.2010.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TH, Shivdasani RA. Genetic evidence that intestinal Notch functions vary regionally and operate through a common mechanism of Math1 repression. J Biol Chem. 2011;286:11427–11433. doi: 10.1074/jbc.M110.188797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanDussen KL, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene. 2008;27:5148–5167. doi: 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shroyer NF, et al. Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology. 2007;132:2478–2488. doi: 10.1053/j.gastro.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 37.Han H, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- 38.Shibata H, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 39.Barker N, et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.