

Calcific aortic valve disease is common, affecting 25% of adults over the age of 65 years, with progression to severe valve obstruction resulting in more than 50 000 aortic valve replacements annually in the United States. “Degenerative” calcific aortic valve disease was thought for many years to be a passive accumulation of calcium binding to the aortic surface of the valve leaflet. Now, convincing data indicate aortic stenosis is an active disease process with a distinctive histological appearance, associated clinical factors, and variable disease progression, which suggests this disease may be amenable to medical therapy to prevent or slow disease progression.
Aortic valve disease is an active cellular process ranging from aortic sclerosis (a process similar to early atherosclerosis) to severe calcification with bone formation and valve obstruction.1–3 Ex vivo studies have defined the cellular markers and potential signaling pathways important in the progression of this disease. Early valvular sclerotic lesions demonstrate a chronic inflammatory cell infiltrate (macrophages and T lymphocytes), lipid accumulation (apolipoprotein [apo] B, apo(a), and apoE), and α-actin–expressing cells in the lesion and adjacent fibrosa. End-stage calcified valves contain mature lamellar bone2 with expression of specific bone markers important in the development of osteoblast bone formation.3 In addition, angiotensin-converting enzyme (ACE) and angiotensin II type 1 (AT1) and type 2 (AT2) receptors are present in stenotic aortic valves, implicating this signaling pathway in the disease process.4
These observations are analogous to the cellular findings in vascular atherosclerosis and corroborate epidemiological studies that showed similar associations of clinical risk factors with both atherosclerosis and aortic valve disease.5 Our understanding of the biological mechanisms that result in calcification of valve leaflets has led to the hypothesis that targeted drug therapy might prevent slow disease progression. (Figure) Two of the leading candidates for drug therapy are HMG-CoA reductase inhibitors (statins) and ACE inhibitors.
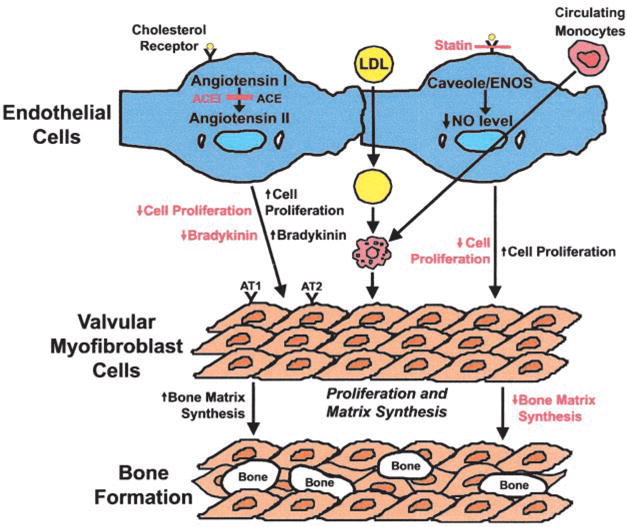
Important cellular pathways involved in development of aortic valve calcification, with potential targets for drug therapy outlined in red. ACEI indicates ACE inhibitor.
The hypothesis that lipids contribute to the development of valve calcification and stenosis can be tested in emerging experimental models.6,7 In addition to a lipid-lowering effect, statins have other beneficial effects in vascular atherosclerosis, including modification of endothelial function, decreased inflammation via inhibition of macrophage activation, up-regulation of endothelial nitric oxide synthase (eNOS), and inhibition of smooth muscle cell proliferation.8 Experimental models suggest that statins might have similar effects in valve tissue, with demonstration of a decrease in atherosclerotic effects, a reduction in osteoblast gene markers, and inhibition of calcification via upregulation of eNOS.9 Although laboratory models allow detailed evaluation of the effects of blocking these pathways in a controlled setting, determination of the long-term clinical effects of targeted medical therapy, in the milieu of the other clinical and genetic factors that may affect disease progression and clinical outcome, requires evaluation in patients with calcific valve disease.
In this issue of Circulation, Rosenhek and colleagues10 evaluate the effects of statin therapy in a retrospective study of 211 adults with varying degrees of aortic stenosis. The data convincingly demonstrate a significantly lower rate of disease progression in those treated with a statin compared with those not taking statin therapy. The magnitude of the effect of therapy is similar to that in previous studies, with all showing an ≈50% annual reduction in measurable disease progression, whether quantified by Doppler echocardiographic jet velocity or valve area or by electron-beam tomographic valve calcium scores (Table).11–15 One unique aspect of the present study is the inclusion of patients with severe stenosis and the demonstration that disease progression is reduced across the range of disease severity. Although encouraging, this is a somewhat surprising finding given the extensive valve calcification seen in end-stage disease, a process that appears less amenable to intervention than the early valve lesion.
Nonrandomized Retrospective Studies of Medical Therapy to Prevent Progression of Calcific Aortic Stenosis.
| First Author, Year | No. of Subjects | Mean Age (% Male) | Mean F/U Interval | Study Groups | Parameter Reported | P |
|---|---|---|---|---|---|---|
| EBT calcium | ||||||
| Pohle 200111 | 104 | 65 y (86) | 1.25 y | Increase in valve calcium (per year) | ||
| LDL ≤130 mg/dL | 9 ±22% | ≤0.001 | ||||
| LDL >130 mg/dL | 43 ±44% | |||||
| Shavelle 200212 | 65 | 67 y | 2.5 y | Increase in valve calcium (per year) | ||
| Statin therapy (43%) | 12.1% | 0.006 | ||||
| No statin therapy | 32% | |||||
| Echocardiography | ||||||
| Aronow 200114 | 180 | 82 y (31) | 2.8 y | Increase in peak gradient (mm Hg/y) | ||
| LDL ≥125 mg/dL, not on statin | 6.3±1.4 | <0.0001 | ||||
| LDL ≥125 mg/dL, on statin | 3.4±1.0 | |||||
| LDL <125 mg/dL, not on statin | 3.1 ±1.1 | |||||
| Novaro 200115 | 174 | 68 y (44) | 1.8 y | Decrease in valve area (cm2/y) | 0.03 | |
| Statin therapy (33%) | 0.06±0.16 | |||||
| No statin therapy | 0.11 ±0.18 | |||||
| Bellamy 200213 | 156 | 77 y (58) | 3.7 y | Decrease in valve area (cm2/y) | 0.04 | |
| Statin therapy (24%) | 0.04±0.15 | |||||
| No statin therapy | 0.09±0.17 | |||||
| Rosenhek 200410 | 211 | 70 y (51) | 2.0 y | Increase in aortic velocity (m · s−1·y−1) | ||
| Statin therapy | 0.10±0.41 | 0.0001 | ||||
| No statin therapy | 0.39±0.42 | |||||
| ACE inhibitor | 0.29±0.44 | 0.29 | ||||
| No ACE inhibitor | 0.35±0.44 |
EBT indicates electron-beam tomography.
The mechanism of the effect of statins on the disease process in the aortic valve remains unclear. The rate of disease progression was linked to serum LDL levels in only 2 of these 6 studies.11,14 The others postulate that the discordance between disease progression and serum lipid levels indicates that antiinflammatory effects of statins are more important than lipid-lowering effects in calcific valve disease. Clarification of the mechanism of the association between statin therapy and the rate of disease progression will be crucial to effectively tailor therapy for this disease process.
The study by Rosenhek et al10 also is the first large series to evaluate the effect of ACE inhibitor therapy on aortic stenosis progression. ACE inhibitors interfere with the renin-angiotensin system and exert beneficial actions on vascular tissues beyond their blood pressure–lowering effects. ACE inhibitors reduce atherogenesis in experimental models both by inhibiting the conversion of inactive angiotensin I to active angiotensin II and by decreasing bradykinin levels, which in turn releases nitric oxide, resulting in improved endothelial function and a decrease in smooth muscle cell proliferation.16
In the study by Rosenhek et al,10 there was no significant difference in disease progression in patients taking ACE inhibitor therapy compared with those not taking an ACE inhibitor, despite careful consideration of the effects of coexisting hypertension. Although these data are discouraging, it would be appropriate to evaluate ACE inhibitor therapy in other retrospective databases and in animal models before concluding this therapy is ineffective. Hemodynamic effects of ACE inhibitors might obscure the effects at the tissue level by changing the flow conditions across the valve.17 Other considerations include the relative tissue effects of different ACE inhibitors, timing of therapy in the disease process, and the sample size needed to demonstrate an effect that may be clinically significant over a longer follow-up interval. In addition, ACE inhibitors might impact long-term clinical outcome by other mechanisms, for example, by modulation of the left ventricular response to chronic pressure overload.18 In any case, the observation that 63% of these consecutive patients with aortic stenosis were already taking ACE inhibitor therapy has significant implications for design of future clinical trials.
We are making progress toward medical therapy for calcific valve disease, but the studies to date are not ideal. All of these publications are based on nonrandomized, retrospective databases, which have many well-known limitations. However, the impressively lower rate of disease progression in patients taking statin therapy in 6 different databases, even with these small, possibly biased patient groups, provides compelling justification for further investigation of this potential therapy. Ultimate proof of the efficacy of statin therapy in adults with aortic stenosis will depend on prospective clinical trials; these retrospective studies establish an association but not a cause-and-effect relationship.
At least 2 prospective, randomized, placebo-controlled multicenter studies of lipid-lowering therapy to prevent disease progression in aortic stenosis are in progress: the Aortic Stenosis Progression Observation: Measuring Effect of Rosuvastatin (ASTRONOMER) study in Canada and the Simvastatin and Ezetimide in Aortic Stenosis (SEAS) study in Europe (personal communication from K.L. Chan, 2004; and Rossebo et al19). We should await the results of these trials to determine whether it will become appropriate to prescribe statin therapy routinely in patients with calcific valve disease.
We also need to keep an open mind about what therapies might be most effective for calcific valve disease. Inhibition of lipid accumulation and the effects of ACE inhibitors in the valve tissue are the first pathways to be studied; however, other more specific therapies targeting endothelial disruption, inflammation, or tissue calcification may be more effective. Therapy may need to be tailored to the stage of the disease process; some interventions may prevent initiation of the disease process, whereas others may be more effective in slowing calcium accumulation in end-stage disease. Experimental models are critical to define the disease process and timing of therapy and for development of future clinical trials for aortic valve disease. In the meanwhile, we should continue to evaluate cardiac risk profiles in all our patients, particularly those with calcific valve disease, and institute appropriate primary and secondary preventative measures in accordance with national and international clinical guidelines.
This is a commentary on article Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110(10):1291-5.
Footnotes
The opinions expressed in this editorial are not necessarily those of the editors or of the American Heart Association.
Disclosure: Dr Rajamannan is an inventor on a patent assigned to the Mayo Clinic titled “Method for Slowing Heart Valve Degeneration.” Dr Otto is coinventor on a patent application by the University of Washington titled “Methods of Inhibition of Stenosis and Sclerosis of the Aortic Valve.” Dr Otto has formally relinquished all financial interest in the patent.
References
- 1.O'Brien KD, Reichenbach DD, Marcovina SM, et al. Apolipoproteins B, (a) and E accumulate in the morphologically early lesion of “degenerative” valvular aortic stenosis. Arterioscler Thromb. 1996;16:523–532. doi: 10.1161/01.atv.16.4.523. [DOI] [PubMed] [Google Scholar]
- 2.Mohler ER, III, Gannon F, Reynolds C, et al. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 3.Rajamannan NM, Subramaniam M, Rickard D, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Brien KD, Shavelle DM, Caulfield MT, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–2230. doi: 10.1161/01.cir.0000035655.45453.d2. [DOI] [PubMed] [Google Scholar]
- 5.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 6.Rajamannan NM, Subramaniam M, Springett M, et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–2665. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drolet MC, Arsenault M, Couet J. Experimental aortic valve stenosis in rabbits. J Am Coll Cardiol. 2003;41:1211–1217. doi: 10.1016/s0735-1097(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 8.Laufs U, Liao JK. Isoprenoid metabolism and the pleiotropic effects of statins. Curr Atheroscler Rep. 2003;5:372–378. doi: 10.1007/s11883-003-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajamannan NM, Subramaniam M, Springett M, et al. Atorvastatin inhibits aortic valve calcification in an experimental model of chronic hypercholesterolemia via nonlipid lowering effects. J Am Coll Cardiol. 2003;41(suppl A):842. [Google Scholar]
- 10.Rosenhek R, Rader F, Loho N, et al. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–1295. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- 11.Pohle K, Maffert R, Ropers D, et al. Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation. 2001;104:1927–1932. doi: 10.1161/hc4101.097527. [DOI] [PubMed] [Google Scholar]
- 12.Shavelle DM, Takasu J, Budoff MJ, et al. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002;359:1125–1126. doi: 10.1016/S0140-6736(02)08161-8. [DOI] [PubMed] [Google Scholar]
- 13.Bellamy MF, Pellikka PA, Klarich KW, et al. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol. 2002;40:1723–1730. doi: 10.1016/s0735-1097(02)02496-8. [DOI] [PubMed] [Google Scholar]
- 14.Aronow WS, Ahn C, Kronzon I, et al. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001;88:693–695. doi: 10.1016/s0002-9149(01)01821-5. [DOI] [PubMed] [Google Scholar]
- 15.Novaro GM, Tiong IY, Pearce GL, et al. Effect of hydroxymethylglutaryl coenzyme A reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001;104:2205–2209. doi: 10.1161/hc4301.098249. [DOI] [PubMed] [Google Scholar]
- 16.Lonn E. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in atherosclerosis. Curr Atheroscler Rep. 2002;4:363–372. doi: 10.1007/s11883-002-0074-7. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien KD, Zhao XQ, Shavelle DM, et al. Hemodynamic effects of the angiotensin-converting enzyme inhibitor, ramipril, in patients with mild to moderate aortic stenosis and preserved left ventricular function. J Investig Med. 2004;52:185–191. doi: 10.1136/jim-52-03-33. [DOI] [PubMed] [Google Scholar]
- 18.Routledge HC, Townend JN. ACE inhibition in aortic stenosis: dangerous medicine or golden opportunity? J Hum Hypertens. 2001;15:659–667. doi: 10.1038/sj.jhh.1001260. [DOI] [PubMed] [Google Scholar]
- 19.Rossebo A, Pedersen T, Skjaerpe T, et al. Design of the Simvastatin and Ezetimide in Aortic Stenosis (SEAS) Study. Atherosclerosis. 2003;170(suppl 4):253. Abstract. [Google Scholar]