

Abstract
A collection of αIIbβ3 integrin receptor antagonists possessing a unique MIDAS metal ion displacement mechanism of action is presented. Insight into these agents’ structure-activity relationships, binding modality, and pharmacokinetic and pharmacodynamic profiles highlight the potential of these small molecule ion displacement ligands as attractive candidates for clinical development.
The αIIbβ3 integrin receptor is an important mediator of platelet aggregation and to date three αIIbβ3 antagonists have been approved for human use by the U.S. Food and Drug Administration, including the small molecules eptifibatide and tirofiban (Figure 1).1-3 Integrin receptors are heterodimeric complexes that rely upon coordinated conformational changes to the α and β subunits to initiate ligand binding, and upon ligand binding to convey signals to within cells that ultimately alter a wide array of phenotypes ranging from cytoskeletal reorganization, to cellular differentiation, to specified immune responses.4 The use of αIIbβ3 antagonists to alter platelet aggregation results in clinical utility for these agents as antithrombotic therapies. The aforementioned small molecule αIIbβ3 antagonists are modeled after the Arg-Gly-Asp (RGD) motif found in some of the naturally occurring ligands.5 The binding of both eptifibatide and tirofiban are particularly reliant upon interactions with a conserved Asp (224) residue found in the αIIb subunit and span a defined binding pocket to also engage a Mg2+ ion found in the β3 subunit’s metal ion-dependent adhesion site (MIDAS) domain.5,6 Crystallographic analysis of these drugs (and other RGD mimetics) demonstrates that after these antagonists (and a related peptide from the ligand fibrinogen) bind, the β3 unit undergoes a ‘swing-out’ motion resulting in a major change in conformation.5-8 This conformational change has been theorized to contribute to the thrombocytopenia caused by the RGD mimetic agents by exposing neoepitopes for which some individuals have pre-formed antibodies.9 Attempts to develop oral RGD mimetic agents to inhibit αIIbβ3 failed because the agents produced thrombocytopenia and some were associated with a paradoxical increase in mortality.10,11 This latter effect has been theorized to be due to these agents’ ability to “prime” the receptor to bind fibrinogen as the conformational change induced by the agents leaves the receptor in a high affinity ligand binding state.3,11-15 Thus, while αIIbβ3 represents a validated drug target, there remains a need to identify small molecule αIIbβ3 antagonists that do not alter the β3 subunit conformation since these may have better safety profiles.
Figure 1.
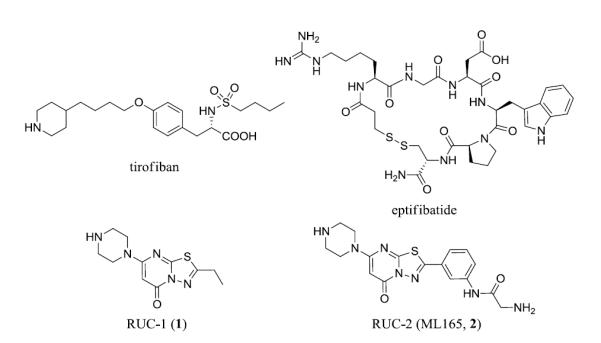
Chemical structures of tirofiban, eptifibatide, RUC1 (1) and RUC2 (ML165, 2).
In an attempt to identify a novel small molecule capable of retaining the advantageous physiological effects associated with αIIbβ3 receptor antagonism without the negative consequences of receptor ‘priming’ we screened and identified a novel 5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-5-one based small molecule antagonist.16 This agent, named RUC-1 (1, Figure 1), was found to inhibit adhesion of platelets to fibrinogen and block ADP-induced platelet aggregation at modest potencies. It was also noted that 1 was selective for the αIIbβ3 receptor over related integrins αVβ3 and α2β1 and for human αIIbβ3 over murine and rat αIIbβ3.16,17 The specificity for human αIIb/β3 resided in the αIIb subunit and thus its antithrombotic properties could be determined in a transgenic murine model in which mice express human αIIb in complex with murine β3 (hαIIb/mβ3).16,17 Intriguingly, the β3 domain of the receptor was shown to undergo little or no swing-out and the receptor did not undergo priming upon compound binding when compared with eptifibatide and tirofiban binding.16-18 An X-ray crystallographic analysis of this agent bound to the αIIbβ3 receptor confirmed molecular dynamic (MD) simulations suggesting that 1 bound exclusively to the αIIb domain of the receptor (PDB codes: 3NID, 3NIG, 3NIF).18 Pharmacokinetic studies conducted in dogs demonstrated rapid oral absorption (Tmax ≈ 0.5 hr), high oral bioavailability (~92%), and rapid elimination (t½ ≈ 2hr)(Table S1).
To optimize this agent a series of analogues were synthesized and examined for improved potency in the aforementioned platelet/fibrinogen adhesion and platelet aggregation assays. From these efforts, an analogue of 1 named RUC-2 (2, ML165, Figure 1) was identified that possessed increased affinity for the receptor and maintained the favorable lack of effect on receptor conformation as judged by several analyses, including electron microscopy of αIIbβ3 nanodiscs, Stokes radii measurements by gel filtration, exposure of ligand-induced binding site epitopes for monoclonal antibodies, and dynamic light scattering.19 Importantly, 2 also did not prime the receptor to fibrinogen binding nor in the majority of cases, induce recruitment to platelets of αIIbβ3-dependent antibodies from the serum of patients who developed thrombocytopenia when treated with eptifibatide or tirofiban.19 X-ray crystallographic studies revealed that 2 possessed a unique binding modality whereby the primary amine of the chemical structure of the ligand replaced the Mg2+ found within the MIDAS domain by binding to Glu220, one of the major Mg2+ coordinating residues (PDB code: 3T3M).19 This agent represents the first in a novel pharmacological class, which we term “ion displacement ligands” with remarkable selectivity and a lack of detrimental conformational events for the ligand-bound receptor. Here, we detail the structure-activity relationships (SAR) revealed during the optimization effort, highlight additional MD simulations that underscore the unique binding of this class of molecules and provide pharmacokinetic (PK) parameters for advanced analogues.
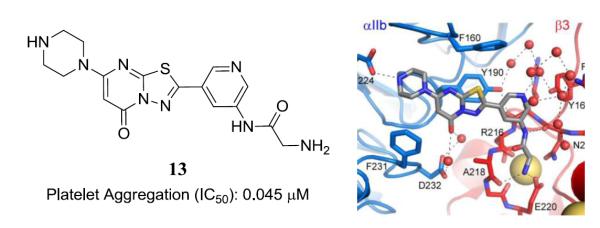
The optimization of 1 focused on structural alteration to the piperazine domain and the 2- and 6-positions of the core 5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-5-one heterocycle. Numerous analogues were synthesized and tested and a collection of representative SAR is presented in table 1. It rapidly became apparent that the secondary amine of the piperazine domain would not tolerate alteration as judged by conversions to piperidine (analogue 3), morpholine (analogue 4) and N-methyl-piperazine (analogue 5). These results were consistent with the crystallographic analysis that demonstrated the key interaction of this moiety with αIIb Asp224.18 All additional attempts to alter the piperazine ring system led to a loss in activity, including steric additions to the 2- and 6-positions (for instance 2,6-dimethylpiperazine), bicyclic analogues (for instance 2,5-diazabicyclo[2.2.2]octane), alternate diamines (for instance N-phenylazetidin-3-amine), and selected spirocyclic compounds (for instance 2,7-diazaspiro[4.4]nonane)(data not shown). Changes to the 2-position of the 5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-5-one were limited to fluorine and selected alkyl groups (R2 in Table 1). A methyl group at this domain remained active (slight loss in activity) while any larger substitution resulted in an inactive analogue (data not shown). The fluorine substitution (analogue 6) retained equivalent activity to that of 1. Changes to the 6-position of the heterocycles included modified alkyl and aryl groups. Changing from the ethyl in 1 to a tert-butyl (analogue 7) produced an agent with equivalent activity as did the phenyl analogue 8. Multiple ring substitutions on the phenyl ring were explored including carboxylic acids (analogues 9 and 10). In general, potencies for these agents were equivalent to that displayed by 1. Incorporation of a 2-aminoacetamide at the meta position of the phenyl ring (analogue 2), however, produced a striking increase in potency in both the adhesion assay (83% at 1 μM) and the aggregation assay (0.096 μM). The aforementioned crystallographic analysis identified the novel interaction between 2 and the MIDAS domain19 and we endeavored to fully explore the SAR of this new binding modality. Interestingly, a single methyl group on the alkyl chain [(S)-2-aminopropanamide](analogue 12) results in a significant loss in activity. This result conforms to the crystallographic analysis that demonstrates a limited amount of free space as the amide NH forms a key hydrogen bond with the backbone Asn215 carbonyl, resulting in proximal binding to Asn215 limiting the required space for the incorporated methyl group. Furthermore, additional bulk placed upon the primary amine (data not shown) eliminated activity. Again, crystallographic analysis of 2 highly suggests that the replacement of the Mg2+ ion by the primary amine allows for a nearly identical conformation for the amino acid side-chains that define the MIDAS domain and mitigates any alteration to this key pharmacophore. Modification of the phenyl ring (substitutions X2 and X3 in Table 1) maintained similar activity and selected analogues incorporating ring nitrogens [13 and 14] provided slight increases in potency and improved key physicochemical properties (see below). Based upon the modest potency enhancement demonstrated by 13 and on selected pharmacokinetic and pharmacodynamic studies with this agent, we further confirmed that 13 neither primes the receptor for fibrinogen binding nor changes the β3 conformation as judged by the binding of a conformation-specific monoclonal antibody; it also does not possess any activity versus the αVβ3 receptor suggesting a high degree of selectivity for αIIbβ3 akin to our studies with 2 (manuscript in preparation).19
Table 1.
Platelet adhesion data as % inhibition at a set concentration and platelet aggregation data as IC50’s.
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Entry | R1 | R2 | XI | R3 | X2 | X3 | Adhesion (% inhibition)a | Aggregation (IC50)b |
| tirofiban | NA | NA | NA | NA | NA | NA | 88% (10 μM) | NP |
| eptifibatide | NA | NA | NA | NA | NA | NA | NP | 0.012 μM |
| 1 | Ethyl | H | NH | NA | NA | NA | 51% (20 μM) | 4.0-10 μM |
| 3 | Ethyl | H | CH2 | NA | NA | NA | 23%(100 μM) | >100 μM |
| 4 | Ethyl | H | 0 | NA | NA | NA | 20%(100 μM) | 49 μM |
| 5 | Ethyl | H | NMe | NA | NA | NA | 35%(100 μM) | >100 μM |
| 6 | Ethyl | F | NH | NA | NA | NA | 41%(100 μM) | 8.3 μM |
| 7 | tert-Butyl | H | NH | NA | NA | NA | 35% (50 μM) | 4.0 μM |
| 8 | NA | NA | NA | H | CH | CH | 56%(100 μM) | 3.0 μM |
| 9 | NA | NA | NA | COOH | CH | CH | 37% (50 μM) | 80 μM |
| 10 | NA | NA | NA | H | C-COOH | CH | 49% (50 μM) | 6.5 μM |
| 11 | NA | NA | NA | 2-acetamidoacetic acid | CH | CH | 78% (50 μM) | 2.0 μM |
| 12 | NA | NA | NA | (S)-2-aminopropanamide | CH | CH | 66% (100 μM) | 5.87 μM |
| 2 (RUC-2) | NA | NA | NA | 2-aminoacetamide | CH | CH | 83% (1 μM) | 0.096 μM |
| 13(RUC-4) | NA | NA | NA | 2-aminoacetamide | CH | N | 93% (1 μM) | 0.045 μM |
| 14 (RUC-3) | NA | NA | NA | 2-aminoacetamide | N | CH | 98% (1 μM) | 0.033 μM |
| 15 | NA | NA | NA | 2-aminoacetamide | CH | C-OH | 69% (1 μM) | 0.1 μM |
| 16 | NA | NA | NA | 2-aminoacetamide | CH | C-CH2OH | 89% (1 μM) | 0.075 μM |
Adhesion assays were performed in triplicate at concentrations of 100 μM, 50 μM, 30 μM, 10 μM and/or 1 μM.
Aggregation assays performed in triplicate using adjusted dose ranges governed by potency. NA = not applicable. NP = not performed.
We previously reported the synthesis of 2 from an 5-(3-nitrophenyl)-1,3,4-thiadiazol-2-amine (see reference 19). Analogues reported here-in, including 13 were synthesized utilizing the same protocol from commercially available materials [for instance, 5-nitronicotinic acid for the production of 13 (Scheme S1)]. To facilitate the multiple follow-up studies undertaken with 13 we optimized this route utilizing 5-bromonicotinic acid (Scheme 1). Conversion to the acid chloride followed by addition of hydrazinecarbothioamide yielded the required acyl-hydrazinecarbothioamide (17) in greater than 50% yield over two steps. Addition of 17 to PPA at 100 °C cleanly formed the 1,3,4-thiadiazol-2-amine 18 which was converted to the 7-chloro-5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-5-one 19 by condensing with methyl-3-chloro-3-oxopropanoate followed by chlorination via POCl3 treatment. Nucleophilic displacement of the 3-chloro moiety with 1-Boc-piperazine was followed by a Buchwald-Hartwig amination using Boc-protected 2-aminoacetamide which proceeded with an 80% yield. Treatment with TFA afforded deprotection of both Boc protecting groups.
Scheme 1.
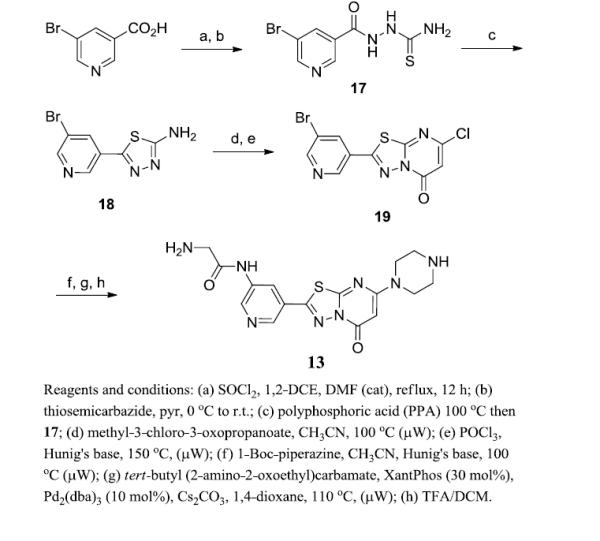
Synthesis of 13.
To better understand the subtle improvement in potency found through incorporation of the ring nitrogens into the core of 2 we carried out molecular dynamic simulations of the integrin αIIbβ3 headpiece bound to 2, 13, or analogue 14. Starting from the crystallographic pose of 2, each agent’s initial geometry was optimized ab initio using a restricted Hartree-Fock (RHF) calculation with a 6-31G (d) basis set, as implemented in the Gaussian 09 program,20 and the resulting geometries used to calculate electrostatic potential-derived (ESP) point charges employing the RESP methodology, as implemented in the AMBER suite of programs,21 and reported previously.19 Ligands were then docked in an equivalent geometric orientation within the binding pocket to that seen in the crystal structure of the 2-αIIbβ3 headpiece. All three resulting αIIbβ3 headpiece-drug systems, including the SyMBS and ADMIDAS Ca2+ metal ions and the crystallographic water molecules found in all metal binding sites, were immersed in truncated octahedral boxes of ~22,000 TIP3P water molecules and neutralized by the addition of 7 Na+ counter-ions at random locations. Each system was simulated for 20 ns by all-atom, standard MD simulations, using the general AMBER force field (GAFF) for the ligands and the ff12SB force field for the protein, within the AMBER12.0 suite of programs.21
Detailed analysis of representative snapshots from the 20 ns simulation trajectories of 2-αIIbβ3 (Figure 2, panel A), 14-αIIbβ3 (Figure 2, panel B), and 13-αIIbβ3 (Figure 2, panel C) shows that both 14 and 13 maintain a similar geometry to the pose adopted by 2 during dynamics. Stable interactions monitored during simulations included the piperazine-Asp224 hydrogen bond, the primary amine interactions with Glu220 and the backbone carbonyl oxygen of Asp218, and a hydrogen bond between the phenylacetamide and the backbone carbonyl oxygen of β3 Asn215. We additionally noted a stable π-π stacking interaction between each of the compounds’ fused ring and the αIIb Tyr190 aromatic ring and a water-mediated hydrogen bond between the carbonyl group in each of the compounds’ fused ring and the side chain carboxyl group of αIIb Asp232. The average distances between the aforementioned atoms and protein residues monitored during simulation are reported in Supplemental Table 2. There are no direct interactions between residues of the αIIbβ3 headpiece and 13 and 14 that are not also present in 2, making understanding of the slight potency enhancement for these agents not obvious. There are additional water-mediated interactions between the inserted ring nitrogen in 13 and Tyr166 that may differentiate the binding of this congener from that of 2 (Figure 2). Similarly, the inserted nitrogen in 14 appears to support additional water-mediated interactions with Tyr166 and Arg214 (Figure 2B). These additional water-mediated interactions with the β3 subunit may account for the slightly higher affinity of 13 and 14 relative to 2.
Figure 2.
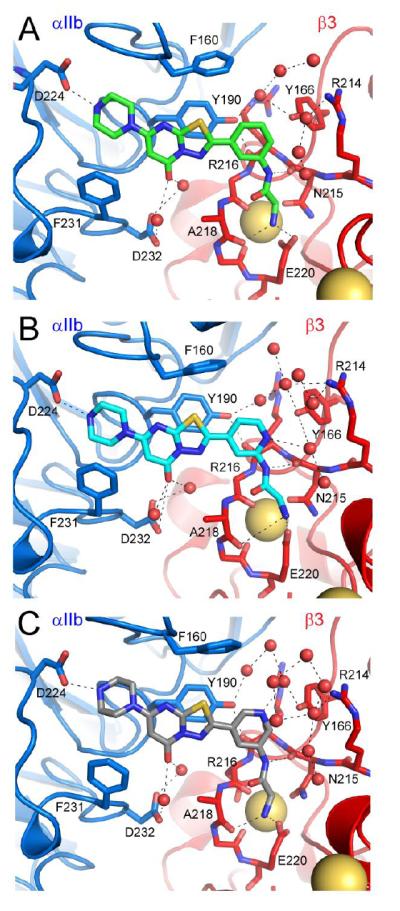
Visualization of MD simulations following 20 ns simulation trajectories of 2-αIIbβ3 (panel A), 14-αIIbβ3 (panel B), and 13-αIIbβ3 (panel C).
Table 2.
Selected physicochemical and in vitro ADME profiles and pharmacokinetic analysis of 2 and 13.
|
| ||||||
|---|---|---|---|---|---|---|
| Entry | Biological Stabilitya |
Kinetic Solubilityb |
Thermodynamic Solubilityb |
hERG Inhibitionc | CLint (μL/min/mg
protein)d |
t1/2 (min.) |
|
| ||||||
| 2 (RUC-2) | 97.5 (15 min.) | 35.8 (pH 6.6.) | 41.8 (pH 6.6.) | < 5% @ 25 μM | 0.684 (mouse) | > det. limit (mouse) |
| 93.3 (60 min.) | 34.6 (pH 7.4) | 28.0 (pH 7.4) | 2.83 (rat) | > det. limit (rat) | ||
| 84.5 (120 min.) | 36.1 (pH 8.5) | 46.2 (pH 8.5) | −1.16 (human) | > det. limit (human) | ||
| 43.4 (pH 12) | >400 (pH 12) | |||||
| 13(RUC-4) | 100 (15 min.) | >57.8 (pH 6.6.) | 340.4 (pH 6.6.) | < 5% @ 25 μM | 89.0 (mouse) | 15.6 (mouse) |
| 99.5 (60 min.) | >57.8 (pH 7.4) | 239.5 (pH 7.4) | 54.9 (rat) | 25.3 (rat) | ||
| 91.7 (120 min.) | >57.8 (pH 8.5) | >400 (pH 8.5) | 23.2 (human) | 58.9 (human) | ||
| >57.8 (pH 12) | >400 (pH 12) | |||||
|
| |||||||
|
2 (RUC-2)
Exposure Profile (IP) |
Exposure Profile (IV) |
13 (RUC-4)
Exposure Profile (IP) |
Exposure Profile (IV) | ||||
|
| |||||||
| Av. Weight (g) | 27 | Av. Weight (g) | 27 | Av. Weight (g) | 27 | Av. Weight (g) | 27 |
| Dose (mg) | 0.03 | Dose (mg) | 0.03 | Dose (mg) | 0.03 | Dose (mg) | 0.03 |
| Cmax (μM/mL) | 0.222 | CO (μg/mL) | 1.354 | Cmax (μg/mL) | 0.405 | CO (μg/mL) | 1.145 |
| Tmax (hr) | 0.08 | Tmax (hr) | NA | Tmax (hr) | 0.08 | Tmax (hr) | NA |
| AUC (μg-hr/mL) | 0.045 | AUC (ng-hr/mL) | 0.123 | AUC (μg-hr/mL) | 0.087 | AUC (μg-hr/mL) | 0.137 |
| t1/2(hr) | NA | t1/2 (hr) | 0.07 | t1/2(hr) | NA | t1/2 (hr) | 0.07 |
| % Bioavailabilitye | 36.8% | % Bioavailabilitye | 63.5% | ||||
|
| |||||||
Biological Stablity assays were conducted in mouse plasma in triplicate.
Kinetic solublity assays were performed in triplicate using both CLND and IHPLC methods (CLND methods are shown). Data was recorded in both (μg/mL and μM (μg/mL is shown).
hERG channel inhibition was measured at 0.008 μM, 0.04 μM, 0.2 μM, 1.0 μM, 5.0 μM and 25.0 μM. Values were < 5% at all tested concentrations.
Microsomal stability data (hepatocyte data not shown).
Data represents measurements from 0 to 0.5 hours.
The novel binding modality demonstrated by these agents make them ideal probe molecules for further study of ion-displacement antagonists as a novel means of pharmacological intervention. We were also interested in examining 2 and its structural congeners for potential translation into clinical reagents. Given the limitations associated with the current approved drugs there is a need to develop novel αIIbβ3 antagonists that possess the appropriate combination of attributes to establish them as new drugs. As such, we examined key analogues for selected physicochemical and preclinical ADME properties. From the outset, we prioritized 2, 13 and 14 for advanced study. Prior to ADME analysis we noted that 14 possessed poor chemical stability as a solution (both DMSO-based and aqueous) and consequently this analogue was deprioritized. Initial assessments included an analysis of the aqueous solubility and biological stability of 2 and 13 (Table 2). Both analogues were highly stable in mouse plasma and each agent possessed a positive solubility profile across a range of pH values. Neither eptifibatide nor tirofiban possess oral bioavailability, which may be attributed to their having formal positively and negatively charged elements in their chemical structure. Likewise, 2 and 13 are charged drugs at physiological pH (pKa values for 2 were experimentally determined at 6.41 and 8.08) and thus, as expected, PAMPA and Caco2 permeability analyses indicated that it was unlikely that either agent possesses high oral bioavailability (data not shown). However, the intended clinical application for this agent is in the early, pre-hospital treatment of patients with myocardial infarction in hopes of ameliorating the mortality and morbidity by decreasing the resulting tissue death and long-term cardiac dysfunction. Several studies have explored the use of αIIbβ3 antagonists in a pre-hospital setting and present evidence that such therapeutic intervention translates into both acute and long-term benefit.22-24 To apply novel αIIbβ3 antagonists in such a setting requires a facile administration route and method of delivery. The anticipated use of autoinjector technology to provide a bolus dose of drug via IM injection would provide a means to deliver an anticipated dose (~ 1 mg/kg) of drug. Autoinjectors are limited in their volume of delivery and therefore require the API to possess significant solubility in the delivery vehicle (a primarily aqueous formulation). Both 2 and 13 are soluble small organic molecules as judged by both kinetic and thermodynamic solubility assessments, but 13 is much more soluble as judged by chemiluminescent nitrogen detection (CLND)(Table 2) and, in fact, many of the measured values exceeded the detection limit of these methods. An LC method was also performed and correlated well with the data shown in Table 2. Additional studies have demonstrated that 13 can be solubilized long-term up-to and exceeding levels of 60 mg/mL, which far exceeds the level that can be achieved for 2. As a result, for the ideal delivery mechanism, 13 represents a strong candidate agent.
Effort was also put forth to examine several predictive in vitro toxicology markers. Both 2 and 13 were further profiled for hERG inhibition with neither agent presenting activity versus this common target at all concentrations tested. As these agents represent cationic, amphiphilic drugs we also examined the capacity of 13 to produce a drug-induced phospholipidosis event.25 No appreciable induction of phospholipidosis was noted nor did 13 alter nuclear size or induce stenosis (data not shown). The fraction unbound (fu) for 13 in both whole blood and plasma protein was high (>85% and >30%, respectively; data not shown).
An analysis of the microsomal stability for 2 and 13 suggested that 2 was highly resistant to metabolism in three species (mouse, rat and human microsomes) while 13 possessed a more modest stability profile. Hepatocyte stability (mouse) was also assessed for 13 and found to be negligible (>95% remaining after 120 minutes). The exposure (PK) profile for each agent (both IV and IP administration route) suggested that each agent has a rapid onset and a limited time of exposure following a dose of 0.03 mgs. These data are generally contradictory to the microsomal stability results, suggesting an alternate route of metabolism/clearance. The limited exposure times are beneficial for use in the proposed pre-hospital infarction indication where a drug-induced antiplatelet effect is desired for a limited time interval, thus allowing first responders to administer drug without preventing the physicians in the receiving hospital from instituting whichever drugs and interventions they think best.
In summary, we report the discovery and optimization of a novel class of αIIbβ3 antagonists that demonstrate a novel ion displacement mechanism of action. This binding modality allows our lead agents 2 and 13 to inhibit receptor function without significant β3 conformational reorganization, thus avoiding receptor priming and perhaps decreasing the likelihood of developing thrombocytopenia. Our results suggest that advanced analogues 13 and 14 bind to the protein in a similar fashion as 2 with additional water-mediated interactions that potentially account for modest affinity gains. Neither 2 nor 13 possess any predictive toxicology liabilities and 13 possesses both the required aqueous solubility and chemical properties for the specified clinical applications. Pharmacodynamic analysis of both 2 and 13 in an engineered mouse model and 13 in a non-human primate model (manuscript in preparation), support the advancement of this class of αIIbβ3 antagonists into advanced preclinical studies.
Supplementary Material
Acknowledgments
The authors thank Jim Bougie, Thomas Daniel and William Leister for compound purification, as well as Paul Shinn, Danielle VanLeer for assistance with compound management. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research grant U54HG005033 and the Intramural Research Program of the National Human Genome Research Institute at the National Institutes of Health. This work was further supported, in part, by grants HL19278, HL13629, and HL48675, and from the National Heart, Lung, and Blood Institute, and CTSA grant ULRR024143 from the National Center for Research Resources, NIH. Computations were run, in part, on resources available through the Scientific Computing Facility at Icahn School of Medicine at Mount Sinai and in part on TeraGrid advanced computing resources provided by Texas Advanced Computing Center through TG-MCB080077.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Supplementary data associated with this article can be found, in the online version, at…
References
- 1.Bledzka K, Smyth SS, Plow EF. Integrin αIIbβ3: From discovery to efficacious therapeutic target. Circ. Res. 2013;112:1189–1200. doi: 10.1161/CIRCRESAHA.112.300570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang SH, Manning N, Armstrong N, Misso K, Allen A, Di Nisio M, Kleijnen J. Treatment with tirofiban for acute coronary syndrome (ACS): a systematic review and network analysis. Curr. Med. Res. Opin. 2012;28:351–370. doi: 10.1185/03007995.2012.657299. [DOI] [PubMed] [Google Scholar]
- 3.Coller BS, Shatil SJ. The GPIIb/IIIa (integrin αIIbβ3) odyssey: A technology-driven saga of a receptor with twists, turns and even a bend. Blood. 2008;112:3011–3025. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bi-directional, allosteric, signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibronectin-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen by the platelet integrin aIIbb3. J. Cell Biol. 2008;182:791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 9.Bougie DW, Wilker PR, Uuitschick ED, Curtins BR, Malik M, Levine S, Lind RN, Pereira J, Aster RH. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood. 2002;100:2017–2076. [PubMed] [Google Scholar]
- 10.Chew DP, Bhatt DL, Topol EJ. Oral glycoprotein IIb/IIIa inhibitors: why don’t they work? Am. J. Cardiovasc. Drugs. 2001;1:421–428. doi: 10.2165/00129784-200101060-00002. [DOI] [PubMed] [Google Scholar]
- 11.Cox D. Oral GPIIb/IIIa antagonists: what went wrong? Curr. Pharm. Des. 2004;10:1587–1596. doi: 10.2174/1381612043384673. [DOI] [PubMed] [Google Scholar]
- 12.Du XP, Plow EF, Frelinger AL, 3rd, O’Toole TE, Loftus JC, Ginsberg MH. Ligands ‘activate’ integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 13.Hantgan RR, Stahle MC. Integrin priming dynamics: mechanisms of integrin antagonist-promoted alphaIIbbeta3:PAC-1 molecular recognition. Biochemistry. 2009;48:8355–8365. doi: 10.1021/bi900475k. [DOI] [PubMed] [Google Scholar]
- 14.Jones ML, Harper MT, Aitken EW, Williams CM, Poole AW. RGD-ligand mimetic antagonists of integrin alphaIIbbeta3 paradoxically enhance GPVI-induced human platelet activation. J. Thromb. Haemost. 2010;8:567–576. doi: 10.1111/j.1538-7836.2009.03719.x. [DOI] [PubMed] [Google Scholar]
- 15.Bassler N, Loeffler C, Mangin P, Yuan Y, Schwarz M, Hagemeyer CE, Eisenhadt SU, Ahrens I, Bode C, Jackson SP, Peter K. A mechanistic model for the paradoxical platelet activation by ligand-mimetic αIIbβ3 (GPIIb/IIIa) antagonists. Arterioscler. Thromb. Vasc. Biol. 2007;27:e9–e15. doi: 10.1161/01.ATV.0000255307.65939.59. [DOI] [PubMed] [Google Scholar]
- 16.Blue R, Murcia M, Karan C, Jirousková M, Coller BS. Application of high-throughput screening to identify a novel αIIb-specific small-molecle inhibitor of αIIbβ3-mediated platelet interaction with fibrinogen. Blood. 2008;111:1248–1256. doi: 10.1182/blood-2007-08-105544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blue R, Kowalska MA, Hirsch J, Murcia M, Janczak CA, Harington M, Jirousková M, Li J, Fuentes R, Thornton MA, Filizola M, Poncz M, Karan C, Coller BS. Structural and therapeutic insights from the species specificity and in vivo antithrombotic activity of a novel αIIb-specific αIIbβ3 antagonist. Blood. 2009;114:195–201. doi: 10.1182/blood-2008-08-169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Zhu J, Negri A, Provasi D, Filizola M, Coller BS, Springer TA. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood. 2010;116:5050–5059. doi: 10.1182/blood-2010-04-281154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J, Choi W-S, McCoy JG, Negri A, Zhu J, Naini S, Li J, Shen M, Huang W, Bougie D, Rasmussen M, Aster R, Thomas CJ, Filizola M, Springer TA, Coller BS. Structure-guided design of a high-affinity platelet integrin αIIbβ3 receptor antagonist that disrupts Mg2+ binding to the MIDAS. Science Trans. Med. 2012;4:125ra32. doi: 10.1126/scitranslmed.3003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JAJ, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador. P.; Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 21. http://ambermd.org/
- 22.DE Luca G, Bellandi F, Huber K, Noc M, Petronio AS, Arntz HR, Maioli M, Gabriel HM, Zorman S, DE Carlo M, Rakowski T, Gyongyosi M, Dudek D. Early glycoprotein IIb-IIIa inhibitors in primary angioplasty-abciximab long term results (EGYPT-ALT) cooperation: individual patient’s data meta-analysis. J. Thromb. Haemost. 2011;9:2361–2370. doi: 10.1111/j.1538-7836.2011.04513.x. [DOI] [PubMed] [Google Scholar]
- 23.Xu Q, Yin J, Si LY. Efficacy and safety of early versus late glycoprotein IIb/IIIa inhibitors for PCI. Int. J. Cardiol. 2013;162:210–219. doi: 10.1016/j.ijcard.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Hassan AK, Liem SS, van der Kley F, Bergheanu SC, Wolterbeek R, Bosch J, Bootsma M, Zeppenfeld K, van der Laarse A, Atsma DE, Jukema JW, Schalij MJ. In-ambulance abciximab administration in STEMI Patients prior to primary PCI is associated with smaller infarct size, improved LV function and lower incidence of heart failure: results from the Leiden MISSION! Acute myocardial infarction treatment optimization program. Catheter Cardiovasc. Interv. 2009;74:335–343. doi: 10.1002/ccd.21980. [DOI] [PubMed] [Google Scholar]
- 25.Anderson N, Borlak J. Drug-induced phospholipidosis. FEBS Lett. 2006;580:5533–5540. doi: 10.1016/j.febslet.2006.08.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.