

Abstract
Tumor necrosis factor-alpha (TNF-α) may cause apoptosis and inflammation in amyotrophic lateral sclerosis (ALS) and spinal cord injury (SCI). Recent studies suggest that estrogen (EST) provides neuroprotection against SCI. We tested whether 1,3,5-tris (4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT) (EST receptor alpha (ERα) agonist), 2,3-bis (4-hydroxyphenyl) propionitrile (DPN) (EST receptor beta (ERβ) agonist), or EST itself would prevent apoptosis in VSC4.1 motoneurons following exposure to TNF-α. Cells were exposed to TNF-α and 15 min later treated with PPT, DPN, or EST. Posttreatment with 50 nM PPT, 50 nM DPN, or 150 nM EST prevented cell death in VSC4.1 motoneurons. Treatment of VSC4.1 motoneurons with PPT, DPN, or EST induced overexpression of ERα, ERβ, or both, which contributed to neuroprotection by upregulating expression of anti-apoptotic proteins (p-AKT, p-CREB, Bcl-2, and p-Src). Our analyses also revealed that EST agonists and EST increased phosphorylation of extracellular signal-regulated kinase (ERK). The L-type Ca2+ channel inhibitor, nifedipine (10 μM), partially inhibited EST agonist and EST-induced increase in phosphorylated ERK expression. The mitogen-activated protein kinase inhibitor, PD98059 (5 μM), partially prevented ER agonists and EST from providing neuroprotection to TNF-α toxicity. Presence of the nuclear ER antagonist, ICI 182 780 (10 μM), blocked the neuroprotection provided by all three ER agonists tested. Taken together, our data indicate that both ERα and ERβ contribute to PPT, DPN, or EST-mediated neuroprotection with similar signaling profiles. Our data strongly imply that PPT, DPN, or EST can be used as effective neuroprotective agents to attenuate motoneuron death in ALS and SCI.
Introduction
Estrogen (EST) provides neuroprotection in traumatic brain injury, spinal cord injury (SCI), and ischemic injury and also in neurodegenerative diseases (Sribnick et al. 2003, Nilsen & Brinton 2004, Soustiel et al. 2005, Gerstner et al. 2009). The neuroprotective effects of EST may be attributable to a number of characteristics of this steroid hormone. In particular, EST acts as a powerful antioxidant and also an anti-inflammatory agent (Sribnick et al. 2005). Studies from our laboratory indicated that EST could prevent increases in intracellular free [Ca2+], activation of calpain, and apoptosis (Sur et al. 2003, Sribnick et al. 2004). The protective effects of EST are mediated via the EST receptor alpha (ERα, also called ESR1) and EST receptor beta (ERβ, also called ESR2). The effects of these ER subtypes are tissue and compartment specific. Previous reports showed that the relative contribution of either ER-dependent or ER-independent mechanism to EST-mediated neuroprotection could be due to EST concentration administered. At a physiological concentration, EST may tend to exert neuroprotection through an ER-mediated signaling pathway. Along with the increase in EST concentration used, an ER-independent (e.g. antioxidant mechanism) may impose a greater impact on the neuroprotective outcomes. EST has been shown to improve outcomes in experimental trauma, hemorrhage, shock, sepsis, myocardial ischemia-reperfusion, and acute lung injury (Lahm et al. 2008). ERα decreases pro-inflammatory cytokine production and ERβ decreases inflammatory markers (Lahm et al. 2008). Only recently have selective EST agonists become available, allowing the detailed analysis of the role of ERα and ERβ in central nervous system (CNS) injuries and other neurodegenerative diseases. In particular, the effects of selective ER agonists on VSC4.1 motoneurons have not yet been investigated. It also remains unknown whether the contribution of ERα and ERβ differs in responses to particular cytokines.
A better mechanistic understanding of the effects of ER agonists or EST may help future targeted therapeutic interventions in CNS injuries and other neurodegenerative diseases. As to the mechanisms underlying the ER-mediated neuroprotective action of EST, recent studies showed that both ERα and ERβ are expressed in nonnuclear regions. As demonstrated in a number of studies, EST and its structural analogs acted as significant neuroprotectants through their antioxidant properties in a number of model systems (Behl et al. 1997, Green & Simpkins 2000). Previous studies also demonstrated that EST or ER agonists protected hippocampal neurons (Wu et al. 2005, Zhao et al. 2005) and primary cortical neurons (Cordey & Pike 2005) following activation of mitogen-activated protein kinase (MAPK) signaling cascades.
In this study, we examined the effects of EST in prevention of apoptosis in VSC4.1 motoneurons exposed to tumor necrosis factor-alpha (TNF-α). In order to clarify the role of ER, we also examined whether the ERα agonist 1,3,5-tris (4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT)) or the ERβ agonist 2,3-bis (4-hydroxyphenyl) propionitrile (DPN) could attenuate cell death. Our results demonstrated that posttreatment with PPT, DPN, or EST was significantly protective against supraphysiological TNF-α toxicity in VSC4.1 motoneurons. Furthermore, we found that PPT, DPN, or EST significantly increased expression of anti-apoptotic proteins (e.g. AKT, Bcl-2), which paralleled their neuroprotective efficacy, indicating that both ERα and ERβ could contribute to EST-mediated neuroprotection. We also evaluated the impact of the ER subtype agonists PPT and DPN on EST-inducible signaling cascades.
Materials and Methods
Cell culture
The VSC4.1 motoneuron cell line was formed by fusion of dissociated embryonic rat ventral spinal cord neuron with mouse N18TG2 neuroblastoma cell (Crawford et al. 1992, Smith et al. 1994). The motoneuron–neuroblastoma hybrid cell line (VSC4.1) was a generous gift from Dr S H Appel (Baylor College of Medicine, TX, USA). The VSC4.1 motoneurons were grown in monolayer to subconfluency in 75 cm2 flasks containing 10 ml of DMEM/F12 medium with 15 mM HEPES, pyridoxine, and NaHCO3 (Sigma), supplemented with 2% Sato’s components, 1% penicillin, and streptomycin (GIBCO-Invitrogen), and 2% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA). Optimal doses of TNF-α (Sigma), EST (Sigma), PPT, DPN, and ICI 182 780 (Tocris Cookson, Ellisville, MO, USA) for VSC4.1 cells were determined by the Trypan blue dye exclusion test. Cells were treated with 50 ng/ml TNF-α alone for 24 h to examine cell death. Moreover, cells were treated first with 50 ng/ml TNF-α for 15 min and then with 150 nM EST or 50 nM PPT or 50 nM DPN for 24 h. Cells from all treatment groups were used for determination of morphological and biochemical features of apoptosis, analysis of mRNA expression of the genes regulating apoptosis, and analysis of specific protein expression and activity by western blotting. Neuronal viability was determined by the Trypan blue dye exclusion test, which could indicate the metabolically active cells in the cultures (Das et al. 2005, 2010a,b). All of the remaining experiments were repeated in triplicate with n≥3 for each treatment group.
Detection of apoptosis
The VSC4.1 motoneurons (1×105) from each treatment group were washed with PBS, pH 7·4, sedimented onto the microscopic slide using Cytobucket in Centra CL2 centrifuge (IEC, Needham Heights, MA, USA) at 150 g for 5 min and fixed in methanol. Cells from each treatment were washed with PBS, pH 7·4, sedimented onto the microscopic slide, and fixed. The morphological (Wright staining) and biochemical (ApopTag assay) features of apoptosis were examined, as described previously (Das et al. 2005, 2010a,b). After Wright staining and ApopTag assay, cells were counted under the light microscope to determine the percentage of apoptosis. At least 500 cells were counted from four randomly selected fields in each treatment and the percentage of apoptotic cells was calculated from three separate experiments.
Determination of intracellular free [Ca2+] using Fura-2 assay
The level of intracellular free [Ca2+] was measured in VSC4.1 motoneurons using the fluorescence Ca2+ indicator Fura-2/AM, as described previously (Das et al. 2005, 2010a,b). The value of Kd, a cell-specific constant, was determined experimentally to be 0·169 M for the VSC4.1 cells using standards of the Calcium Calibration Buffer kit with magnesium (Molecular Probes, Eugene, OR, USA).
Analysis of mRNA expression
The levels of mRNA expression of ERα, ERβ, bax, bcl-2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were measured using the reverse transcription-PCR (RT-PCR). Total RNA was extracted from each of the 12 treatment groups. Primers for ERα, ERβ, bax, bcl-2, and GAPDH genes were designed using Oligo software (National Biosciences, Plymouth, MN, USA). The RT-PCR products were resolved by agarose gel electrophoresis. The levels of mRNA expression of the targeted genes were determined by calculating the optical density (OD) of the bands using Quantity One software (Bio-Rad; Table 1).
Table 1.
Primers used in RT-PCR for amplification of mRNA of specific genes
| Gene
|
Primer sequences
|
Product size (bp)
|
|---|---|---|
| GAPDH | Forward: 5′-TTC ACC ACC ATG GAG AAG GC-3′ Reverse: 5′-GGC ATG GAC TGT GGT CAT GA-3′ |
237 |
| ERβ | Forward: 5′-TCC CTC TTT GCG TTT GGA CTA-3′ Reverse: 5′-TTC CCG GCA GCA CCA GTA ACC-3′ |
262 |
| ERα | Forward: 5′-AAT TCT GAC AAT CGA CGC CAG-3′ Reverse: 5′-GTG CTT CAA CAT TCT CCC TCC TC-3′ |
344 |
| bax | Forward: 5′-GCA GAG AGG ATG GCT GGG GAG A-3′ Reverse: 5′-TCC AGA CAA GCA GCC GCT CAC G-3′ |
352 |
| bcl-2 | Forward: 5′-GGA TGA CTT CTC TCG TCG CTA C-3′ Reverse: 5′-TGC AGA TGC CGG TTC AG-3′ |
255 |
Antibodies
Monoclonal antibodies against β-actin (1:2000 dilution; Sigma) and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used to standardize cytosolic protein loading on the SDS-PAGE. Anti-cytochrome c oxidase subunit IV (COX4) antibody (Molecular Probes) was used to standardize the mitochondrial protein levels. COX4 is a membrane protein in the inner mitochondrial membrane and it remains in the mitochondria regardless of activation of apoptosis. Antibodies against ERα and ERβ agonists were purchased from Santa Cruz Biotechnology. All other primary IgG antibodies were purchased from Santa Cruz Biotechnology or Calbiochem (Gibbstown, NJ, USA). All primary antibodies were diluted at a concentration of 1:200, unless otherwise stated. Secondary antibodies were HRP-conjugated goat anti-mouse IgG (ICN Biomedicals, Aurora, OH, USA) and HRP-conjugated goat anti-rabbit IgG (ICN Biomedicals, Solon, OH, USA) and diluted at a concentration of 1:2000.
Western blotting
Western blotting was performed, as described previously (Das et al. 2005, 2010a,b). The isolation of cytosolic, mitochondrial, and nuclear fractions was performed by standard procedures. Cytochrome c in the supernatants and pellets and also caspase-3-activated DNase (CAD) in the nuclear fractions were analyzed by western blotting. The autoradiograms were scanned using Photoshop software (Adobe Systems) and OD of each band was determined using Quantity One software (Bio-Rad).
Caspase-3, caspase-8, and capase-9 colorimetric assays
Measurements of caspase-3, caspase-8 (Sigma), and caspase-9 (Invitrogen) activities were performed using the commercially available assay kits. Concentration of pNA released from the substrate was calculated on the basis of absorbance values at 405 nm. Experiments were performed in triplicate.
Statistical analysis
All results obtained from different treatments of VSC4.1 cells were analyzed using StatView software (Abacus Concepts, Berkeley, CA, USA). Statistically significant differences were determined by one-way ANOVA followed by the Newman–Keuls post hoc analysis. Data were expressed as mean±S.E.M. of separate experiments (n≥3). Significant difference between control (CON) and TNF-α, or TNF-α+ER agonist (PPT or DPN), or EST+ER antagonist (ICI) treatment was indicated by ★P≤0·05 or ★★P≤0·01. Significant difference between TNF-α and TNF-α+ER agonist (PPT or DPN) or EST posttreatment (15 min) was indicated by #P≤0·05 or ##P≤0·01.
Results
PPT, DPN, and EST prevented apoptotic death in VSC4.1 motoneurons exposed to TNF-α
We examined whether PPT, DPN, and EST could protect VSC4.1 motoneurons from TNF-α toxicity (Fig. 1). Cells were treated first with 50 ng/ml TNF-α for 15 min and then posttreated with PPT, DPN, or EST in the absence or presence of ICI for a total of 24 h (Fig. 1). Cell death was examined by Wright staining to assess morphological features (Fig. 1A) and ApopTag assay to detect DNA fragmentation as a biochemical feature of apoptosis (Fig. 1B). Cell survival was determined by Trypan blue dye exclusion test. Our dose–response analyses indicated that 50 nM PPT, 50 nM DPN, and 150 nM EST provided maximal neuroprotection (data not shown). The results obtained from Wright staining (Fig. 1A) were further confirmed by the ApopTag assay (Fig. 1B). We calculated the percentages of apoptosis based on Wright staining. The posttreatment with PPT, DPN, and EST prevented TNF-α toxicity and significantly decreased apoptotic death, compared with control (CON), in VSC4.1 motoneurons (Fig. 1C). Treatments with ER agonists and EST caused inhibition of TNF-α-mediated DNA fragmentation, showing little or no brown color cells. Compared with CON cells, treatment with 50 ng/ml TNF-α induced more than 50% increase (P<0·01) in apoptotic cells (Fig. 1C). The posttreatment of cells with PPT, DPN, or EST decreased TNF-α-induced apoptosis by three- to four-fold. Our data also suggested that two ER subtypes could contribute to neuroprotection and thus the ER antagonist ICI prevented PPT, DPN, or EST-mediated protection of cells.
Figure 1.

Posttreatment with PPT, DPN, or EST prevented TNF-α-induced apoptosis in VSC4.1 motoneurons. Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). (A) Wright staining showing representative cells from each treatment group. Arrows indicate apoptotic cells. (B) ApopTag assay showing representative cells from each treatment group. (C) Bar graphs indicating the percentage of apoptotic cells counted from each group (based on Wright staining). **P<0·01 compared to control; #P<0·05 compared to TNF-α; ##P<0·01 compared to TNF-α. Full colour version of this figure available via http://dx.doi.org/10.1677/JOE-10-0338.
Involvement of ERα and ERβ in neuroprotection
To ascertain whether both ERα and ERβ can contribute to protection of VSC4.1 motoneurons against TNF-α toxicity, we measured the expression of ERα and ERβ at the mRNA and protein levels after the treatments (Fig. 2). Treatment of cells with PPT, DPN, or EST induced overexpression of ERα and ERβ at mRNA (Fig. 2A) and protein (Fig. 2B) levels. The level of expression of ERβ was less than that of ERα in PPT-treated cells, while the level of expression of ERα was less than that of ERβ in DPN or EST-treated cells (Fig. 2C). Cells treated with TNF-α alone showed significantly (P<0·05) reduced levels of ERβ and ERα at mRNA and protein levels when compared with PPT, DPN, or EST-treated cells. The posttreatment with PPT, DPN, or EST restored ER expression at protein level in cells exposed to TNF-α, while ER expression was decreased in the presence of ER antagonist. No significant changes (P=0·234) were seen in ERα and ERβ expression at the mRNA or protein level in the presence of ER antagonist, compared with CON cells. Thus, our results demonstrated that PPT, DPN, and EST activated overexpression of ERα and ERβ to block the events required for initiation of downstream apoptotic signaling cascade.
Figure 2.

RT-PCR and western blotting for expression of estrogen receptor beta (ERβ) and estrogen receptor alpha (ERα). Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). Representative pictures to show levels of ERβ, ERα, and GAPDH at mRNA (RT-PCR) (A) and protein levels (western blotting) (B). (C) Bar graphs indicating the changes in expression of ERβ and ERα over CON. *P<0·05 compared to control.
Both ER agonists and EST rapidly increased extracellular signal-regulated kinase 1/2 phosphorylation with involvement of L-type Ca2+ channels
We investigated whether ER agonists (PPT and DPN) and EST would cause activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in VSC4.1 motoneurons and whether this activation was partially mediated by an increase in intracellular free [Ca2+] (Fig. 3). Our western blotting results demonstrated that both ER agonists (PPT and DPN) and EST induced a rapid and dramatic increase in activation or phosphorylation of ERK1/2 (p-ERK1/2), which quickly peaked at 0·5 h (fourfold increase compared with CON cells) and remained elevated above the baseline level for 24 h (Fig. 3A). We monitored the expression of β-actin to demonstrate equal loading of protein samples in all lanes of the western blot. We also observed slight upregulation of intracellular free [Ca2+] for ~0·5 h before returning to the baseline level in PPT, DPN, or EST-treated VSC4.1 cells (Fig. 3B). Thus, our results showed an apparent correlation between ERK1/2 activation and intracellular free [Ca2+] rise. Cells treated with TNF-α and subsequently (after 15 min) exposed to PPT, DPN, or EST significantly attenuated the [Ca2+] rise, compared with CON cells. Cells treated with TNF-α for 24 h demonstrated a significant increase (P=0·009) of more than 2- to 2·5-fold in intracellular free [Ca2+], compared with CON cells. This increase was significantly attenuated (P=0·005) by posttreatment with both ER agonists and EST. Furthermore, there was no significant difference (P=0·437) in intracellular free [Ca2+] between CON cells and those treated with TNF-α plus PPT, DPN, or EST (Fig. 3B). In our studies, we examined whether L-type Ca2+ channel was involved in increasing intracellular free [Ca2+] for ERK1/2 activation in the presence of PPT, DPN, or EST (Fig. 3C). Our results indicated that 5 min pretreatment with nifedipine (an L-type Ca2+ channel blocker) partially blocked the increase in ERK1/2 activation induced by the ER agonists and EST (Fig. 3C). To investigate whether ERK1/2 activation was required for ER agonists or EST to maintain neuronal survival, we measured ERK1/2 activation in cells treated with PPT, DPN, or EST alone and also in the presence of MAPK inhibitor PD98059 (Fig. 3D). Our results demonstrated that PD98059 partially blocked the expression of p-ERK1/2 and increased neuronal viability, compared with cells treated with TNF-α alone (Fig. 3E). We also examined the effects of posttreatment with ICI 182 780 (ICI), a well-known nuclear ER antagonist (Dauvois et al. 1993), in VSC4.1 cells. Treatment of cells with TNF-α followed by PPT, DPN, or EST along with ICI for 24 h exhibited significant decrease in cell viability (Fig. 3E). Our results also demonstrated that treatment with nifedipine, PD98059, and ICI alone did not affect survival of VSC4.1 motoneurons.
Figure 3.

Determination of ERK phosphorylation, intracellular free [Ca2+], and involvement of L-type Ca2+ channels in VSC4.1 cells. Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). (A) Posttreatment with PPT, DPN, or EST rapidly increased ERK phosphorylation. Western blot analysis to show levels of phosphorylation of p44/42 ERK1/2 at 0·5 and 24 h. (B) Determination of intracellular free [Ca2+] at 0·5 and 24 h. Western blot analysis to show levels of phosphorylation of p44/42 ERK1/2 after posttreatment with nifedipine (C) and PD98059 (D). (E) Treatment with nifedipine and PD98059 increased cell viability. Trypan blue dye exclusion assay was used to assess cell viability. **P<0·01 compared to control.
Impact of PPT, DPN, or EST on the expression of cell survival markers
A number of anti-apoptotic (survival) proteins play a central role in preventing programed cell death and decrease in their expression has been associated with various neurodegenerative disorders. In order to investigate the impact of ER agonists and EST on the activation of some survival proteins or inactivation of pro-apoptotic proteins, we measured their levels in VSC4.1 cells after the treatments (Fig. 4). Our western blotting demonstrated that cells treated with TNF-α and subsequently posttreated with PPT, DPN, or EST increase phosphorylation of AKT (p-AKT, an activated survival protein) and also phosphorylation of Bad (p-Bad, an inactivated pro-apoptotic protein), thus preventing its binding and inhibition of anti-apoptotic Bcl-2 protein (Fig. 4A). Furthermore, both ER agonists and EST induced phosphorylation of cAMP response element-binding protein (p-CREB) and upregulated Bcl-2 (Fig. 4A). Because CREB is known to be a transcription factor for Bcl-2, our results suggest that PPT, DPN, or EST exert their anti-apoptotic effects significantly on CREB phosphorylation and Bcl-2 upregulation via nongenomic activation of the PI3K/AKT pathway in VSC4.1 cells (Fig. 4B). Since Src is a critical upstream regulator of the EST-stimulated PI3K/AKT/eNOS pathway, we determined the expression of Src. There was a significant increase (P<0·05) in phosphorylation of Src (p-Src) in PPT, DPN, or EST-treated cells (Fig. 4B). Posttreatment with the ER antagonist ICI inhibited expression of survival proteins, which were initially upregulated by PPT, DPN, or EST alone (Fig. 4B). These results indicated the involvement of ERα and ERβ in protection of cells.
Figure 4.
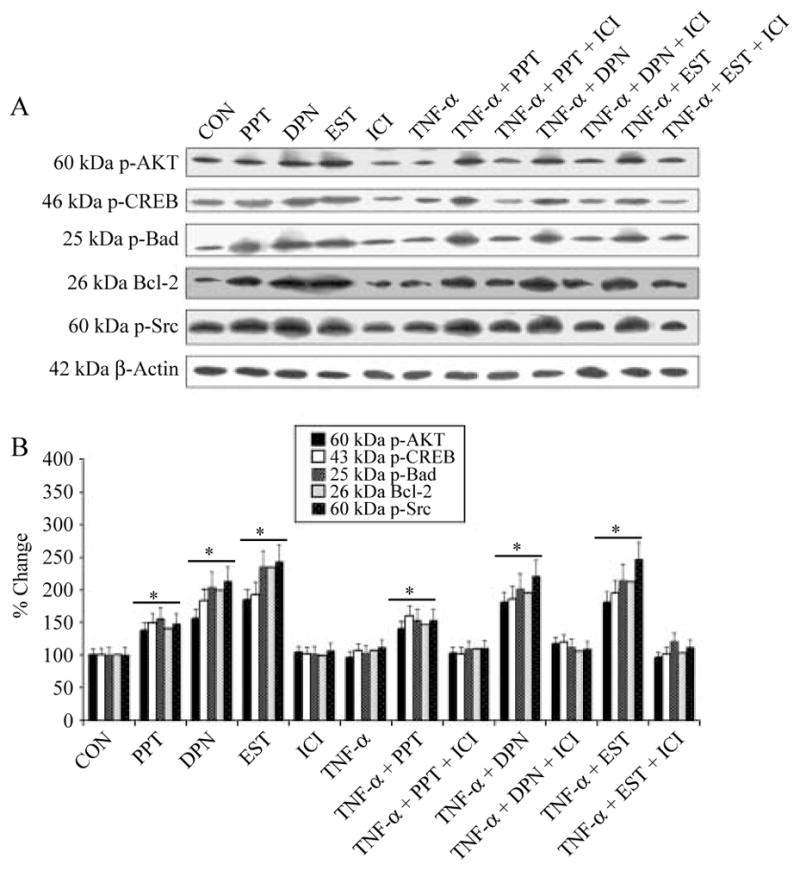
Determination of survival proteins. Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). (A) Western blotting to show levels of p-AKT, p-CREB, p-Bad, Bcl-2, p-Src, and β-actin. (B) Densitometric analysis showing percent change in OD of the p-AKT, p-CREB, p-Bad, Bcl-2, and p-Src bands. *P<0·05 compared to control.
Posttreatment with ER agonists and EST blocked extrinsic and intrinsic pathways of apoptosis
We examined the inhibitory effects of the ER agonists and EST on apoptotic cascade in VSC4.1 cells exposed to TNF-α (Fig. 5). Caspase-8 is a key enzyme in the death receptor of extrinsic pathway of apoptosis and it is also linked to the mitochondrial or intrinsic pathway of apoptosis. We performed western blotting to determine the inhibitory effects of PPT, DPN, or EST on caspase-8 activation and caspase-8-mediated cleavage of Bid to tBid for its translocation to the mitochondria (Fig. 5A). Our results showed significant increases (P=0·001) in 18 kDa active caspase-8 and 15 kDa tBid (mitochondrial fraction) in VSC4.1 cells treated with TNF-α (Fig. 5A). We monitored the levels of β-actin and COX4 expression to ensure that equal amounts of cytosolic and mitochondrial proteins respectively were loaded in all lanes. We also observed that posttreatment with ER agonists and EST completely blocked caspase-8 activation (western blotting) and activity (colorimetric assay) and proteolytic cleavage of Bid to tBid in cells exposed to TNF-α (Fig. 5B). In contrast, posttreatment with ICI significantly decreased inhibitory effects of PPT, DPN, or EST on caspase-8 activation and activity (Fig. 5B).
Figure 5.

Examination of components involved in extrinsic and intrinsic apoptotic pathways. Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). (A) Western blotting to show levels of caspase-8, β-actin, tBid, and COX4. (B) Densitometric analysis showing percent change in OD of the caspase-8 and tBid bands and determination of caspase-8 activity (colorimetrically). (C) Alteration in Bax and Bcl-2 expression at mRNA and protein levels. Representative pictures to show Bax, Bcl-2, and GAPDH at mRNA (RT-PCR) and protein (western blotting) levels after different treatments. (D) Densitometric analysis showing the Bax:Bcl-2 ratio. (E) Western blotting to show levels of cytochrome c, COX4, caspase-9, and β-actin. (F) Densitometric analysis showing percent change in OD of the mitochondrial and cytosolic 15 kDa cytochrome c and 39 kDa active caspase-9, and determination of caspase-9 activity (colorimetrically). **P<0·01 compared to control; ##P<0·01 compared to TNF-α.
An increase in the Bax:Bcl-2 ratio is very important in initiation of mitochondrial pathway of apoptosis. We examined Bax and Bcl-2 expression at both mRNA and protein levels and found that the cells treated with TNF-α increased Bax but decreased Bcl-2 expression at both mRNA and protein levels (Fig. 5C), leading to a significant increase (P<0·01) in the Bax:Bcl-2 ratio in the cells (Fig. 5D). In this study, it should be noted that we used a monoclonal antibody in the western blotting to detect 21 kDa Baxα and 24 kDa Baxβ isoforms, both of which were considered in determining total amount of Bax expression. The increase in the Bax:Bcl-2 ratio (Fig. 5D) could promote the mitochondrial release of the pro-apoptotic factors. Treatment of cells with the ER agonists or EST significantly attenuated (P=0·003) the increase in the Bax:Bcl-2 ratio, compared with cells treated with TNF-α. In addition, there was no significant difference (P=0·687) between CON cells and cells treated with TNF-α and posttreated with PPT, DPN, or EST. Notably, treatment of cells with PPT, DPN, or EST alone did not significantly (P=0·435) alter the Bax:Bcl-2 ratio.
We also examined the cytochrome c levels in both cytosolic and mitochondrial fractions after all treatments of the cells (Fig. 5E). After TNF-α treatment, there was a significant (P<0·01) disappearance of 15 kDa cytochrome c from the mitochondrial fraction and its subsequent appearance in the cytosolic fraction (Fig. 5F). The mitochondrial release of cytochrome c into the cytosol was responsible for caspase-9 activation. We detected significant increases in 37 kDa active caspase-9 fragment and caspase-9 activity (colorimetrically) in TNF-α-treated cells (Fig. 5F). Posttreatment with PPT, DPN, or EST significantly blocked the mitochondrial release of cytochrome c into the cytosol and thereby inhibited caspase-9 activation in TNF-α-treated cells. Treatment of cells with the ER antagonist ICI significantly decreased the effect of PPT, DPN, or EST.
Posttreatment with PPT, DPN, or EST blocked downstream events of apoptosis
Our finding of increased intracellular free [Ca2+] in VSC4.1 cells treated with TNF-α for 24 h (Fig. 3B) suggested activation of the Ca2+-dependent protease calpain and its involvement in cell death. Therefore, we examined the levels of calpain and caspase-3 expression and their activation for mediation of apoptosis (Fig. 6). Western blotting showed an increase in m-calpain expression and a decrease in calpastatin (endogenous calpain inhibitor) expression (Fig. 6A) resulting in a significant increase in the m-calpain:calpastatin ratio (Fig. 6B) in TNF-α-treated cells. Activation of caspase-3 caused cleavage of the inhibitor of caspase-activated DNase (ICAD) to release and translocate CAD to the nucleus (Fig. 6A) leading to an increase in the CAD:ICAD ratio (Fig. 6B) so as to promote degradation of nuclear DNA in TNF-α-treated cells. Posttreatment with PPT, DPN, or EST significantly blocked the ICAD degradation and thereby translocation of CAD to the nucleus (Fig. 6B). Furthermore, treatment with the ER antagonist ICI significantly decreased the action of PPT, DPN, or EST. A uniform expression of β-actin served as a loading control for cytosolic proteins. We stained one set of gel with Coomassie blue to ensure the loading of equal amounts of nuclear proteins in all lanes. A colorimetric assay showed very significant increase in total caspase-3 activity in VSC4.1 cells after treatment with TNF-α (Fig. 6C). Posttreatment with PPT, DPN, or EST significantly attenuated caspase-3 activity but treatment with the ER antagonist ICI reversed the action of PPT, DPN, or EST (Fig. 6C).
Figure 6.

Examination of downstream events of apoptosis in VSC4.1 cells. Treatment groups: control (CON); 50 nM PPT (24 h); 50 nM DPN (24 h); 150 nM EST (24 h); 10 μM ICI (24 h); 50 ng/ml TNF-α (24 h); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+PPT (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+DPN (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure); 50 ng/ml TNF-α (24 h)+EST (treatment at 15 min post TNF-α exposure)+ICI (treatment at 20 min post TNF-α exposure). (A) Western blotting to show levels of m-calpain, calpastatin, active caspase-3, ICAD (cytosolic), CAD (nuclear), and β-actin. (B) Densitometric analysis showing the m-calpain:calpastatin ratio and the ICAD (cytosolic):CAD (nuclear) ratio. (C) Determination of caspase-3 activation by western blotting and total caspase-3 activity by colorimetric assay. **P<0·01 compared to control; ##P<0·01 compared to TNF-α.
Discussion
One central finding of this study is that posttreatment with the ER agonists such as PPT (ERα agonist) and DPN (ERβ agonist), and also EST rapidly prevent Ca2+ influx through the L-type Ca2+ channel and ERK phosphorylation, both of which are required for EST-mediated neuroprotection of VSC4.1 motoneurons. Our results suggest that ER-mediated cytoprotection is due to inhibition of both extrinsic and intrinsic pathways of apoptosis. The present results support a direct relationship between cytoprotection and overexpression of survival proteins and inhibition of calpain and caspase-3 activities, as described in earlier reports (Nilsen et al. 2002, 2003, Sur et al. 2003). Both ER agonists and EST also prevent Ca2+ influx through the L-type Ca2+ channel and cell death induced by TNF-α, strongly indicating the involvement of multiple mechanisms for cell death. Posttreatment with the ER antagonist ICI significantly decreased the ability of ER-mediated cell protection, confirming the involvement of PPT, DPN, or EST in neuroprotective mechanism. Thus, these results showed that PPT, DPN, or EST worked via ERα, ERβ, or both receptors to prevent apoptosis in VSC4.1 motoneurons. The underlying mechanisms of neuroprotection rendered by both ERα and ERβ are in agreement with previous studies (Zhao et al. 2004) but differ from another study showing that low-dose ICI protects hippocampus neurons (Zhao & Brinton 2007). Different results suggest that the effect of ICI treatment may be either dose-dependent or tissue/cell-specific.
The current investigation indicates that both ERα and ERβ are substantially increased at mRNA and protein levels after the treatment of cells with PPT, DPN, or EST. Our results suggest that both ER agonists and EST are responsible for protecting VSC4.1 motoneurons from TNF-α insult via the ER receptor-mediated pathway. This contention is also supported by the fact that ICI, an ER antagonist, abrogated the neuroprotective effects of PPT, DPN, or EST. It is noteworthy that threefold lower concentration of PPT and DPN than EST exerted their neuroprotective effects, making the ER agonists (PPT and DPN) more efficacious than EST for therapeutic use. A possible explanation for these findings is that only a small amount of EST may interact with ERα and ERβ, suggesting nongenomic effects. In contrast, a substantial amount of EST may bind to cytosolic ERα and ERβ and diffuse into the nucleus to exert slower, genomic effects. Alternatively, it is possible that PPT and DPN have higher affinities for their respective receptors than EST itself. In this context, it is of interest to note that PPT has a 410-fold higher binding affinity for ERα than ERβ, while DPN has a 70-fold higher affinity for ERβ over ERα (Weiser et al. 2009).
It has been previously demonstrated that EST decreases Ca2+ influx by modulating voltage-gated L-type Ca2+ channels and initiates the downstream Src/ERK signaling pathways leading to EST-mediated activation of the neuroprotection and neurotropism-associated genes in hippocampal neurons (Wu et al. 2005, Zhao et al. 2005). This study partially confirmed this unique characteristic of ER agonists (PPT and DPN) and EST for motoneuron protection via ERK activation. ERK kinases have dual function in MAPK cascade by controlling cell growth and activation of p44 and p42 MAPK (Hershberger et al. 2009) by phosphorylating both threonine and tyrosine residues. Activation of the Ca2+ signaling as a second messenger sets into motion a biochemical cascade of signaling events leading to gene expression required for neurite outgrowth and neuronal plasticity (West et al. 2001). This study further confirmed this unique regulation of intracellular Ca2+ dynamics in ER agonist and EST-treated motoneurons. Comparable magnitude of attenuation of TNF-α-mediated intracellular Ca2+ rise was exerted by selective ERα agonist (PPT), selective ERβ agonist (DPN), and EST, suggesting that ERα and ERβ may play an equivalently important role in the promotion of EST-mediated neuronal defense mechanisms leading to increased neuronal survival when exposed to neurodegenerative insults.
EST is known to modulate voltage-dependent L-type Ca2+ channels and both ERα and ERβ may be involved in this modulatory mechanism. In this study, we used nifedipine, an L-type Ca2+ channel inhibitor, which moderately blocked the ER agonist and EST-induced increase in ERK phosphorylation, suggesting that L-type Ca2+ channels partially regulate ERα- and ERβ-mediated ERK activation. These results suggest that in addition to L-type channels, ER agonist and EST may also interact with other channels, which may modulate downstream signaling events. Furthermore, we observed that the inhibition of the MAPK signaling by PD98059 partially blocked ER agonist and EST-mediated neuroprotection. These results suggest that activation of the MAPK signaling is essential for EST-mediated neuroprotection.
Signaling via AKT activation participates in neuronal survival and regulating other downstream neuronal survival markers such as CREB and Bcl-2 (Yune et al. 2008). Our data indicate that ER agonists and EST induced AKT activation that precedes and potentially modulates CREB activation for regulating transcription of Bcl-2. It also activates Src and induces P85 (regulatory subunit of PI3K) complex formation. This complex formation results in the successive activation of PI3K, AKT, and eNOS with consequent enhanced NO release, implicating Src as a critical upstream regulator of the EST-stimulated PI3K/AKT/eNOS pathway (Haynes et al. 2003). Our results also supported the general hypothesis that AKT phosphorylates Bad and thus preventing its binding and inhibition to Bcl-2 (Zoubeidi et al. 2010). Furthermore, our results demonstrated that both ER agonists and EST promote neuronal survival mainly through ER-mediated MAPK signaling pathway, ER-dependent mechanism, and also ER-independent mechanism.
The effects of PPT, DPN, and EST are clearly not limited to its MAPK signaling or upregulation of survival protein ability. Both ER agonists and EST may signal through a variety of regulated pathways. Our results show that ER agonists and EST block the extrinsic pathway of apoptosis involving the caspase-8 activation and Bid clevage, and also the intrinsic pathway of apoptosis that engages the mitochondrial permeabilization to release of cytochrome c (Das et al. 2010a,b). Since the mitochondria play a key role both in maintaining cellular homeostasis and triggering the activation of cell death pathways, we evaluated the effect of PPT, DPN, and EST on expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 and release of cytochrome c in TNF-α-treated cells, as reported previously (Das et al. 2010a,b). Our current results showed that both ER agonists and EST can protect apoptotic cells by alterating Bax and Bcl-2 expression at mRNA and protein levels with a decrease in the Bax:Bcl-2 ratio. Our results further demonstrated that PPT, DPN, and EST could prevent the release of mitochondrial pro-apoptotic factors and activation of caspase-9, leading to protection of cells.
Calpain and caspases play a crucial role in the execution of apoptosis (Das et al. 2010a,b). Since cell death can be classified to follow a classical apoptotic mode if execution of cell death is dependent on calpain and caspase activity, we analyzed the downstream events in VSC4.1 cells after different treatments. The processing of pro-caspase-3 to its active form is considered to be a point of no return in the death-signaling cascade. Our observation that treatment with TNF-α alone leads to an increase in calpain and caspase-3 activity and downregulation of calpastatin is in agreement with the results of previous studies (Das et al. 2010a,b). We also found that TNF-α increased the activation of caspase-3 activity and the levels of cleavage of ICAD (DFF45) in VSC4.1 cells. Notably, ICAD is a caspase-3 substrate that has to be cleaved before beginning apoptotic DNA fragmentation. CAD (DFF40) remains inactive while bound to ICAD; however, caspase-3 cleaves ICAD at two sites, thereby releasing the endonuclease, which then cleaves DNA. From our own observations, a reduction in calpain and caspase-3 activity correlates well with the reduction in the Bax:Bcl-2 ratio in PPT, DPN, and EST-treated cells. An increase in the Bax:Bcl-2 ratio is thought to be upstream of the caspases in the mitochondria-mediated apoptotic pathway. Taken together, our current data support earlier evidence that ER agonists and EST mediate cytoprotection, in part, due to inhibition of the protease activity (Sur et al. 2003, Sribnick et al. 2004).
Our results indicate that posttreatment with PPT, DPN, or EST inhibits TNF-α-mediated cell death in VSC4.1 motoneurons. Posttreatment with PPT, DPN, or EST is associated with increases in expression of ERα and ERβ. The neuroprotective effect of PPT, DPN, or EST on VSC4.1 cells was blocked by the ER antagonist ICI, favoring the idea of involvement of ERα and ERβ for rendering neuroprotection. In summary, this study establishes the importance of ER agonists and EST in neuroprotection. Most importantly, our investigation implies that ER agonists at a very low dose can be used as potential therapeutic agents for the treatment of CNS injuries and various neurodegenerative diseases.
Acknowledgments
Funding
This investigation was supported in part by the R01 grants from the NINDS (NS-31622, NS-41088, NS-45967, and NS-57811) and the NIH (C06 RRO15455).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that may be perceived as prejudicing the impartiality of the research reported.
References
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure–activity relationship. Molecular Pharmacology. 1997;51:535–541. [PubMed] [Google Scholar]
- Cordey M, Pike CJ. Neuroprotective properties of selective estrogen receptor agonists in cultured neurons. Brain Research. 2005;1045:217–223. doi: 10.1016/j.brainres.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Crawford GD, Le WD, Smith RG, Xie WJ, Appel SH. A novel N18TG2×mesencephalon cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. Journal of Neuroscience. 1992;12:3392–3398. doi: 10.1523/JNEUROSCI.12-09-03392.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Sribnick EA, Wingrave JM, Del Re AM, Woodward JJ, Appel SH, Banik NL, Ray SK. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. Journal of Neuroscience Research. 2005;81:551–562. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- Das A, McDowell M, Pava MJ, Smith JA, Reiter RJ, Woodward JJ, Varma AK, Ray SK, Banik NL. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-alpha toxicity involves membrane melatonin receptors. Journal of Pineal Research. 2010a;48:157–169. doi: 10.1111/j.1600-079X.2009.00739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Banik NL, Ray SK. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer. 2010b;116:164–176. doi: 10.1002/cncr.24699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. Journal of Cell Science. 1993;106:1377–1388. doi: 10.1242/jcs.106.4.1377. [DOI] [PubMed] [Google Scholar]
- Gerstner B, Lee J, DeSilva TM, Jensen FE, Volpe JJ, Rosenberg PA. 17β-Estradiol protects against hypoxic/ischemic white matter damage in the neonatal rat brain. Journal of Neuroscience Research. 2009;87:2078–2086. doi: 10.1002/jnr.22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Simpkins JW. Estrogens and estrogen-like non-feminizing compounds. Their role in the prevention and treatment of Alzheimer’s disease. Annals of the New York Academy of Sciences. 2000;924:93–98. doi: 10.1111/j.1749-6632.2000.tb05566.x. [DOI] [PubMed] [Google Scholar]
- Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. Journal of Biological Chemistry. 2003;278:2118–2123. doi: 10.1074/jbc.M210828200. [DOI] [PubMed] [Google Scholar]
- Hershberger PA, Stabile LP, Kanterewicz B, Rothstein ME, Gubish CT, Land S, Shuai Y, Siegfried JM, Nichols M. Estrogen receptor beta (ERβ) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 2009;116:102–109. doi: 10.1016/j.jsbmb.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, Meldrum DR. Selective estrogen receptor-alpha and estrogen receptor-beta agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2008;295:R1486–R1493. doi: 10.1152/ajpregu.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. PNAS. 2003;100:2842–2847. doi: 10.1073/pnas.0438041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Current Drug Targets. CNS and Neurological Disorders. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Chen S, Brinton RD. Dual action of estrogen on glutamate-induced calcium signaling: mechanisms requiring interaction between estrogen receptors and src/mitogen activated protein kinase pathway. Brain Research. 2002;930:216–234. doi: 10.1016/S0006-8993(02)02254-0. [DOI] [PubMed] [Google Scholar]
- Smith RG, Alexianu ME, Crawford G, Nyormoi O, Stefani E, Appel SH. Cytotoxicity of immunoglobulins from amyotrophic lateral sclerosis patients on a hybrid motoneuron cell line. PNAS. 1994;91:3393–3397. doi: 10.1073/pnas.91.8.3393. 10. 1073/pnas.91.8.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soustiel JF, Palzur E, Nevo O, Thaler I, Vlodavsky E. Neuroprotective anti-apoptosis effect of estrogens in traumatic brain injury. Journal of Neurotrauma. 2005;22:345–352. doi: 10.1089/neu.2005.22.345. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Wingrave JM, Matzelle DD, Ray SK, Banik NL. Estrogen as a neuroprotective agent in the treatment of spinal cord injury. Annals of the New York Academy of Sciences. 2003;993:125–133. doi: 10.1111/j.1749-6632.2003.tb07521.x. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Ray SK, Nowak MW, Li L, Banik NL. 17β-Estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. Journal of Neuroscience Research. 2004;76:688–696. doi: 10.1002/jnr.20124. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. Journal of Neuroscience Research. 2005;82:283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- Sur P, Sribnick EA, Wingrave JM, Nowak MW, Ray SK, Banik NL. Estrogen attenuates oxidative stress-induced apoptosis in C6 glial cells. Brain Research. 2003;971:178–188. doi: 10.1016/S0006-8993(03)02349-7. [DOI] [PubMed] [Google Scholar]
- Weiser MJ, Wu TJ, Handa RJ. Estrogen receptor-beta agonist diarylpropionitrile: biological activities of R- and S-enantiomers on behavior and hormonal response to stress. Endocrinology. 2009;150:1817–1825. doi: 10.1210/en.2008-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. PNAS. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T-W, Wang JM, Chen S, Brinton RD. 17β-Estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and Bcl-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Yune TY, Park HG, Lee JY, Oh TH. Estrogen-induced Bcl-2 expression after spinal cord injury is mediated through phosphoinositide-3-kinase/Akt-dependent CREB activation. Journal of Neurotrauma. 2008;25:1121–1131. doi: 10.1089/neu.2008.0544. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Estrogen receptor alpha and beta differentially regulate intracellular Ca2+ dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Research. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wu T-W, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Research. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]
- Zhao L, Chen S, Wang JM, Brinton RD. 17β-Estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism. Neuroscience. 2005;132:299–311. doi: 10.1016/j.neuroscience.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Zoubeidi A, Zardan A, Wiedmann RM, Locke J, Beraldi E, Fazli L, Gleave ME. Hsp27 promotes insulin-like growth factor-I survival signaling in prostate cancer via p90Rsk-dependent phosphorylation and inactivation of Bad. Cancer Research. 2010;70:2307–2317. doi: 10.1158/0008-5472.CAN-09-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]