

Abstract
There is an important role for in vitro assays to better understand the initial steps of malaria infection. In this section, we describe both microscopy-based and flow cytometry-based sporozoite invasion, migration and development assays with the rodent malaria parasites, Plasmodium berghei and Plasmodium yoelii, and the human malaria parasite, Plasmodium falciparum.
Keywords: Invasion, Attachment, Cell traversal, Exoerythrocytic stages, Sporozoites, Malaria, Plasmodium, Flow cytometry
1. Introduction
Sporozoite invasion of hepatocytes and subsequent development into exoerythrocytic forms (EEF) is an essential step in the establishment of malaria infection. A better understanding of the molecular interactions that underlie these processes could lead to novel vaccine strategies and new drug targets.
The information in this chapter builds on previous studies in which these techniques were first described (1–7). The assays described have several applications: they can be performed with genetically modified parasites to characterize proteins critical for invasion and preerythrocytic stage development (8–11) and with inhibitors to validate drug targets and identify metabolic or signaling pathways required for infectivity (12, 13). Additionally, they can be used to study the role of host genes in Plasmodium invasion and development, either by employing genetically modified cell lines or using RNA interference to down-modulate the expression of the gene(s) of interest (14–16). Importantly, one can pinpoint the timeframe at which a protein or pathway functions by distinguishing between an effect on attachment, invasion, early or late EEF development (9, 17). Development assays can be adapted to a moderate throughput format for screening compound libraries by using fluorescent parasites and high content imaging or flow cytometry (18) and they can be used to quantify the effect of immune sera on sporozoite infectivity and to document the developmental arrest of genetically attenuated sporozoites (19, 20). Lastly, using flow cytometry, one can sort cells infected with fluorescent parasites from a majority of noninfected cells in order to determine the transcriptional profile of host cells or parasites at different time points (14).
In this chapter, we outline both microscopy and flow cytometry assays for the quantification of sporozoite infectivity. In many cases the methodology used will depend upon the available facilities and parasites. Transgenic parasites expressing a fluorophore under the control of a housekeeping promoter are required for flow cytometry assays and can be used in microscopy-based assays where they will facilitate the workflow by eliminating some steps. Several fluorescent lines of Plasmodium berghei and Plasmodium yoelii are available (21, 22) and recently, the first transgenic fluorescent Plasmodium falciparum line has been generated (23). Flow cytometry assays offer faster data acquisition, which has clear advantages for high throughput screens. The main disadvantage with flow cytometry is host cell autofluorescence when using primary hepatocytes; this can make it difficult to differentiate infected from noninfected cells at early time points after infection, before the parasites have grown and their fluorescence intensity has increased. Microscopy has the advantage that one can visualize the parasite, increasing the amount of information that the investigator can obtain from an assay. For example, distinguishing attachment from invasion is not possible by flow cytometry.
It is important to note that infectivity of sporozoites in vivo involves more than the ability to attach to, invade, and develop in hepatocytes. Thus, there are a few instances in which one can observe normal infectivity in vitro and significantly decreased infectivity in vivo. This is seen in mutants that cannot traverse cells, which makes them unable to exit the dermal inoculation site and traverse the liver sinusoid in vivo, but does not significantly affect their infectivity in vitro where they are placed directly on top of hepatocytes (24–26). Assays to quantify cell traversal are described in this chapter. Additionally, a discrepancy between in vivo and in vitro infectivity can be observed when parasites arrest late in EEF development such that they appear normal by microscopy and of normal size by flow cytometry but cannot establish a blood stage infection in vivo due to a defect in some final maturation step or egress from the hepatocyte (27, 28). In vitro assays to better characterize fully mature EEF are described in Chapter 29.
The rodent malaria parasites, P. berghei and P. yoelii, are relatively easy to manipulate in vitro and should be used for all initial studies. P. berghei is somewhat promiscuous and will develop in many cell types in vitro (29). Nonetheless, infection is always more robust in hepatocyte cell lines and these should be used for the assays described herein. P. yoelii will only infect hepatocytes expressing CD81 and HepG2 cells expressing high levels of CD81 are the most robust system for this purpose (30). Experiments with the human malaria parasite, P. falciparum, are more difficult. Although HepG2 cells can be used for invasion assays, P. falciparum sporozoites will not develop in these cells. Recently, a new hepatoma cell line, HC04, was described that can support P. falciparum development to mature EEF (31) and a protocol using this cell line is described.
2. Materials
2.1. Media and Solutions
Complete medium for P. berghei (Pb) and P. yoelii (Py): Dulbecco’s Modified Eagle Medium (DMEM), 2 mM L-glutamine, 10% fetal bovine serum (FBS), 2% penicillin/streptomycin. Filter through 0.22-μm filter, label, date and store at 4°C for no longer than 1 month.
Complete medium for P. falciparum (Pf): DMEM/F-12 (Invitrogen), insulin 10 μg/ml (Sigma), 10% FBS, 2% penicillin/streptomycin. Filter and store as above.
Dissection medium: DMEM alone or with 2% penicillin/streptomycin. In some cases we add 5 μg/ml fungizone or 100 U/ ml nystatin to wash mosquitoes and cells (see Notes 1 and 2).
70% Ethanol in deionized water.
Phosphate buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4, 10 mM Na2HPO4, pH 7.4, filter-sterilize. Bovine serum albumin (BSA) is added to PBS in varying amounts (either 1% or 0.1%) for use as a blocking buffer. This solution should be filter-sterilized and stored at 4°C.
4% Paraformaldehyde in PBS, pH 7.4 (P. berghei and P. yoelii invasion assay only) (see Note 3).
100% Cold methanol (store at −20°C).
Collagen type I from rat tail (for HepG2 and HepG2-CD81 cells; BD Biosciences #354236).
Entactin-collagen-laminin (for HC04 cells; ECL matrix; Upstate Biotechnology).
TOTO-1 dimeric cyanine nucleic acid dye, 1,303 MW or Dextran 10,000 MW conjugated to a fluorophore (Invitrogen; for cell traversal assays). TOTO-1 is stored as outlined by the manufacturer. High MW dextran should be resuspended in PBS at 10 mg/ml, aliquoted and stored at −20°C. Repeated freeze–thaws should be avoided.
Vectashield mounting media with DAPI (Vector Labs).
Permount (Fisher; P. falciparum invasion assays only).
Trypsin–EDTA or TrypLE (Invitrogen).
Cytochalasin D (CD, Sigma; required for cell traversal, P. falciparum invasion and flow cytometry assays).
2.2. Cells (See Note 4)
Hepa1–6, mouse liver hepatoma cell line (ATCC Number: CRL-1830).
HepG2, human hepatocellular carcinoma cell line (ATCC Number: HB-8065).
HepG2-CD81, HepG2 cells expressing high levels of the tetraspanin CD81 (30).
Huh7 cells (Japanese Collection of Research Bioresources, Cell Bank JCRB0403).
2.3. Antibodies (See Notes 6 and 7)
Monoclonal antibodies (mAb) specific for the repeat region of the circumsporozoite protein (CSP): mAb 3D11 (P. berghei), mAb 2F6 (P. yoelii), mAb 2A10 (P. falciparum).
mAb 2E6, which recognizes HSP70 of all Plasmodium species.
Secondary antibodies (see Note 8): anti-mouse Ig conjugated to rhodamine, anti-mouse Ig conjugated to fluorescein, anti-mouse Ig conjugated to horseradish peroxidase (HRP) and the HRP substrate, diaminobenzadine reagent (DAB; KPL).
2.4. Plastic and Glassware
Permanox or glass lab-tek chamber slides (NalgeNunc). For P. falciparum invasion assay, glass must be used; for the other assays, we prefer permanox, but either can be used. If using glass with HepG2 or HepG2-CD81 cells, it is best to coat with collagen or ECL for better adherence.
24-Well plates, tissue-culture treated (NalgeNunc): use for flow cytometry assays and if using glass coverslips for microscopy-based assays (see Note 9).
3. Methods
3.1. Plating Cells
Precoat chamber slides 2 days prior to the experiment. HepG2, HepG2-CD81, and HC04 cells all tend to clump, so chamber slides are precoated with collagen or ECL matrix, which results in better monolayer formation. For HepG2 or HepG2-CD81 cells, coat with collagen by diluting collagen stock 1:10 in sterile H2O and adding 10 μg of collagen per cm2 of surface (see Note 10). Incubate at 37°C for 3–5 h, aspirate and dry under a UV light overnight to sterilize. Store at 4°C in ziplock bags if not using immediately. Rinse with sterile PBS before use. If using HC04 cells, coat chamber slides with ECL matrix diluted 1:200 in PBS adding at least 0.2 ml/well for 1 h at 37°C or overnight at 4°C.
Prepare cells 1 day prior to the experiment. Resuspend the appropriate cell line in complete medium at the following concentrations for microscopy assays: Hepa1–6 and Huh7, 3–4×105/ml; HepG2 and HepG2-CD81, 4.5 × 105/ml; HCO4, 1.5–3×105/ml. For flow cytometry assays, resuspend cells as follows: Hepa1–6 and Huh7, 8 × 104/ml; HepG2 and HepG2-CD81, 105/ml (see Notes 11–13).
For plating onto lab-tek chamber slides, aliquot 400 μl (Hepa1–6, HepG2 and HepG2-CD81) or 300 μl (for HC04) cells per well. If using 24-well plates, plate 1 ml of cell solution per well. For flow cytometry, we normally use 24-well plates. For high-throughput applications, 96-well plates can be used; just reduce the cell number/well by approximately fivefold.
Cells tend to accumulate in the corners and edges of wells and so should be redistributed for a better distribution. For this, allow cells to settle for ~5 min at room temperature (RT) and then pipet the corners/edges of each well.
Check the cells for contamination and confluency the day after they are plated by looking at the wells with an inverted microscope (see Note 13). The cells should be sub-confluent and there should be no bacteria in the wells with the medium being red-orange, and not yellow.
3.2. Dissecting and Plating of Sporozoites
Anesthetize infected Anopheles stephensi mosquitoes on ice so they are no longer active. Transfer to a small Petri dish containing 70% ethanol for several sec and then transfer to a small Petri dish containing DMEM (see Note 1).
Dissect salivary glands from the washed mosquitoes (see Note 14).
Grind the salivary glands to release sporozoites. If there is a lot of mosquito debris, remove it by centrifuging at 100 × g for 4 min at 4°C. Transfer sporozoite-containing supernatant to a new tube, add 200–400 μl fresh dissection medium to the pellet, mix, spin again and add the wash supernatant to the tube containing the sporozoites.
Determine the number of sporozoites using a hemocytometer. Spin sporozoites at 16,000 × g for 4 min at 4°C to pellet and resuspend in the desired volume of complete medium (see Notes 15 and 16).
Carefully remove the medium from the cells and add 1.0–7.5×104 sporozoites per well (see Note 17), taking care not to damage the adherent cell monolayer. For assays with P. berghei and P. yoelii, use 150 μl of medium per lab-tek well; for P. falciparum assays, plate 50 μl per well. Each test condition and controls should be plated in triplicate (see Note 18). Cell traversal activity can be assayed by plating the sporozoites in medium containing 1 μM TOTO-1 or 1 mg/ml high MW dextran conjugated to a fluorophore (see Note 19).
Invasion efficiency of the rodent malaria sporozoites is somewhat enhanced if sporozoites are spun onto the cells in a refrigerated centrifuge set at 15°C for 3 min at 314 × g. Seal the lid of the lab-tek with parafilm and tape the entire lab-tek slide onto the top of a 24-well plate, which can be placed in a plate spinner. Since this procedure does not enhance the efficiency of P. falciparum invasion, it is not necessary in this case.
Incubate at 37°C with 5% CO2 for 3 h (see Note 20).
After incubation, invasion and cell traversal assays are processed for either microscopy or flow cytometry readouts, whereas development assays are carefully washed three times with complete medium to remove excess sporozoites and then returned to the incubator.
Development assays should have their medium changed daily thereafter. P. falciparum cultures can be kept for 3–7 days and are usually developed between 3 and 6 days post-invasion. P. berghei and P. yoelii cultures are usually developed 2 days post-invasion, although early development can be quantified at 24 h post-invasion and very late events can be observed at 60–70 h post-invasion (see Notes 2 and 21).
3.3. Quantification of EEF by Microscopy
Carefully remove medium from cells and fix sporozoites by gently adding 100% cold methanol and let sit at RT for 10–15 min (see Note 22).
Carefully wash three times with PBS.
Remove last wash and block with 200 μl 1% BSA/PBS and incubate for 30–60 min at 37°C.
Remove blocking solution and add 150 μl of 10 μg/ml of primary antibody in 1% BSA/PBS and incubate 1 h at 37°C (see Note 7).
Wash four times with PBS.
Remove PBS, add appropriate dilution of secondary antibody in 1%BSA/PBS and incubate 1 h at 37°C (see Note 8).
Wash four times with PBS.
Remove last wash and then remove plastic upper structure and silicone gasket of lab-tek chamber slide. Allow to dry briefly at RT, add approximately three drops (~20 μl) of Vectashield mounting medium, and place coverslip on slide. Mounted slides will not dry out and can be reviewed for 2 weeks afterwards. For prolonged storage, coverslips can be permanently sealed around the perimeter with nail polish and stored at 4°C.
View and count using an epi-fluorescence microscope (if using fluorophore-conjugated secondary antibodies) or under brightfield (if using a visual readout, i.e., an enzyme-linked secondary with a colored reaction product). For rodent malaria EEF, there are sufficient numbers such that 50 representative fields per well can be counted (see Note 23). For P. falciparum EEF, count the entire well. EEF will be round fluorescent structures inside of the hepatocytes frequently near the host cell nucleus (see Fig. 1).
Fig. 1.
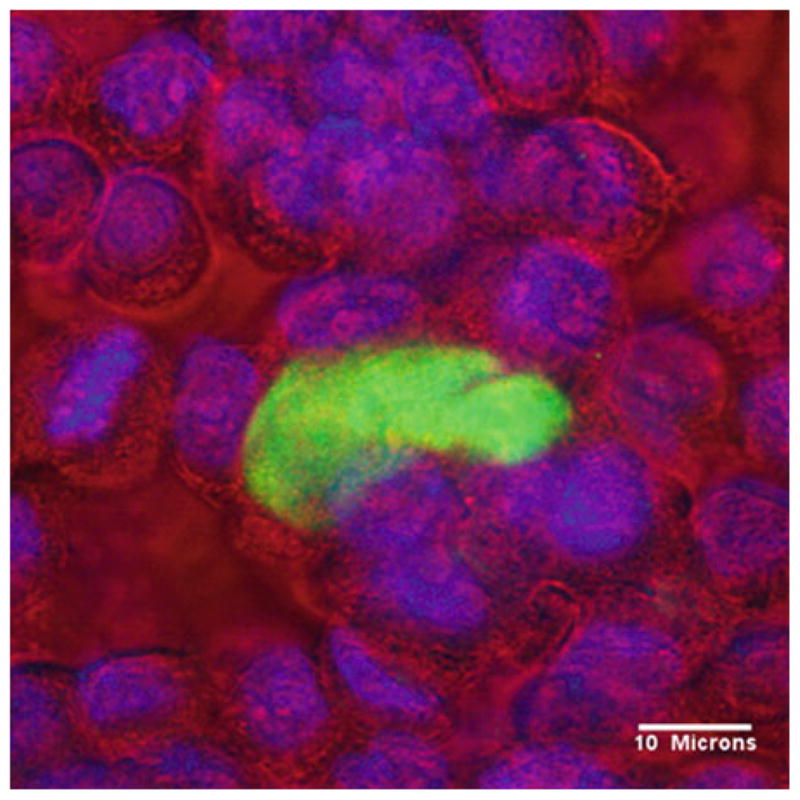
Day 6 Plasmodium falciparum EEF (green) growing in HC04 cells.
3.4. Quantification of P. berghei and P. yoelii Invasion and Cell Traversal by Microscopy
Carefully remove medium, fix sporozoites with 400 μl 4% PFA and incubate at 37°C for 1 h (see Note 24).
Remove PFA and wash two times with PBS. For cell traversal assays, proceed to step 15 of this section after fixation and washing.
Remove last wash and block with 200 μl 1% BSA/PBS per well for 30–60 min at 37°C.
Remove blocking solution, add 150 μl of 1–5 μg/ml of mAb 3D11 (for P. berghei) or mAb 2F6 (for P. yoelii) in 1% BSA/ PBS and incubate 45 min to 1 h at 37°C.
Wash four times with PBS.
Remove last wash and add 200 μl of appropriate dilution of anti-mouse rhodamine in 1% BSA/PBS and incubate 45 min to 1 h at 37°C (see Note 25).
Wash four times with PBS.
Remove last wash and add 400 μl cold 100% methanol and incubate at RT for 15 min.
Wash three times with PBS.
Remove last wash and block with 200 μl 1% BSA/PBS per well for 30 min at 37°C.
Remove blocking solution and add 150 μl of 1–5 μg/ml of mAb 3D11 (for P. berghei) or mAb 2F6 (for P. yoelii) in 1% BSA/PBS and incubate 45 min to 1 h at 37°C.
Wash four times with PBS.
Remove last wash and add 200 μl of the appropriate dilution of anti-mouse FITC in 1% BSA/PBS and incubate 45 min to 1 h at 37°C.
Wash four times with PBS and once with ddH2O.
Mount slide (see Subheading 3.3, step 8).
Count 50–100 fields per well using a 40× objective on an epi-fluorescence microscope (see Notes 23 and 26). For invasion assays, each field should be counted using both red and green filters. Attached sporozoites that have not invaded will appear red using the rhodamine filter (e.g., 528–553 nm), whereas all sporozoites (those on the outside and those that have invaded) will appear green using the FITC filter (e.g., 450–490 nm). The percent invasion equals: [(Total spz - Attached spz)/Total spz] × 100. Slides can be saved in the dark at RT for later viewing. For best results, however, view within a week since attached but not invaded sporozoites can become detached and float in the mounting medium, skewing the results. For cell traversal assays, count the number of fluorescent host cells in 50–100 fields per well. Controls using CD-treated sporozoites will enable you to gauge background fluorescence.
3.5. Quantification of P. falciparum Invasion by Microscopy
Carefully remove medium and wash cells three times with PBS.
Remove last wash and fix with cold methanol for 10 min at RT.
Wash three times with PBS.
Remove last wash and add 100 μl of 10 μg/ml mAb 2A10 in PBS and incubate 30 min at RT.
Wash two times with PBS.
Remove last wash and block twice, 10 min each time, with 300 μl 0.1% BSA/PBS.
Wash two times with PBS.
Remove last wash and add 100 μl of the appropriate dilution of anti-mouse Ig conjugated to HRP in PBS for 30 min at RT.
Wash three times with PBS.
Prepare DAB reagent just before use following manufacturer’s instructions. Discard PBS from chamber slide and add 100 μl DAB reagent per well and allow to develop until color begins to appear, approximately 2 min. It can help to watch this development using an inverted microscope under low power. Stop the reaction with the addition of water, wash twice with water and remove plastic upper structure and silicone gasket of lab-tek chamber slide.
Dry slide for 1 h and mount with Permount by applying 3–4 drops (~20 μl) per slide and gently placing the coverslip on top, applying a small amount of pressure to eliminate any air bubbles. Allow mounting medium to dry overnight. Count the number of intracellular sporozoites per well using a phase contrast microscope and a 40× objective (see Note 27). Positive controls should yield at least 200 intracellular sporozoites per well.
3.6. Quantification of P. berghei, P. yoelii, and P. falciparum Invasion, Cell Traversal, and EEF Development by Flow Cytometry
At the time point appropriate for your assay, (3 h for invasion and cell traversal assays, 24–40 h for EEF development assay), carefully remove medium from cells and wash once with 1 ml of PBS (less if using 96-well plates).
Add 150 μl of trypsin and incubate for 5 min at 37°C.
Add 400 μl of PBS with 10% FCS to each well to inactivate the trypsin.
Place the contents of each well in a separate microfuge tube, centrifuge at 134 × g for 3 min at 4°C and resuspend in 150 μl of PBS with 2% FCS.
Analyze cells on a flow cytometer with the appropriate settings for the fluorophores used.
Analyze data with the appropriate software packages for the flow cytometer. For each condition tested and respective controls, triplicate wells should be collected and analyzed independently. Infected cells can be distinguished from their noninfected counterparts on the basis of the fluorescence they emit, which, in turn, is proportional to the number of parasites in the cell. Thus, invasion rates can be measured by determining the percentage of fluorescence-positive cells at early time points after sporozoite addition. Cell traversal and invasion can be simultaneously assessed if fluorescent dextran with a fluorophore distinct from the one expressed by the parasite, was included in the assay. By carefully gating the different cell populations, one can distinguish between (1) cells whose membrane has been disrupted, (2) those containing a parasite, (3) double positive cells, i.e., containing parasites “in transit” or that were traversed and subsequently invaded. At later time points, the percentage of cells harboring developing parasites can be determined and the fluorescence intensity of these cells is an effective measure of EEF development. Note that fluorescence intensity will only be proportional to parasite development if the fluorophore expressed by the parasite is under the control of a promoter of a housekeepking gene. Addition of a defined number of fluorescent beads to a sample of known volume can be used to determine absolute numbers of infected cells.
Footnotes
In EEF development assays, contamination can be a problem because mosquitoes are not sterile and sporozoites can be contaminated with bacteria and yeast during dissection. All mosquitoes are washed in 70% ethanol; however, supplemental washes may be necessary. For EEF development assays with rodent malaria parasites, we wash mosquitoes twice in DMEM containing 5 μg/ml fungizone and 2% penicillin–streptomycin. P. falciparum sporozoite infecticity can be inhibited by fungizone so washes are performed with DMEM containing 100 U/ml of nystatin and 2% penicillin–streptomycin. These washes should be followed by a wash in DMEM with no additives.
Development of mature EEF takes 7 days for P. falciparum and 2 days for the rodent malaria parasites. The length of time that you keep the EEF cultures going will depend upon the goal of your experiment.
Work in the fume hood when making the paraformaldehyde solution. In the hood, weigh out and dissolve the PFA in PBS pH 7.4 using low to medium heat and continuous stirring (keep covered). It is important that the temperature does not rise above 65–70°C. When the solution becomes clear, cool to RT, adjust volume with PBS and check that pH is still between 7.2 and 7.4. Filter-sterilize, aliquot, and store at −20°C. PFA can be stored at 4°C for up to a week.
Your choice of which cell line to use will depend on the Plasmodium species you are working with. P. berghei sporozoites invade and develop well in all cell lines. P. yoelii invades and develops well in HepG2-CD81 but only moderately well in Hepa1–6 cells and not at all in HepG2 cells. P. falciparum invades HepG2 cells but only develops in HC04 cells. Huh7 cells can be used for P. berghei and P. yoelii; although the efficiency of invasion is lower, the cells adhere in nice monolayers without the need for an adhesion matrix. Do not use host cells that have been passaged more than 15 times as invasion and development rates will be lower. For any of these assays primary hepatocytes can also be used, however, their isolation and maintenance is beyond the scope of this protocol. Note that P. falciparum will only grow and develop in primary human hepatocytes whereas the rodent malaria parasites grow well in both mouse and rat primary hepatocytes.
Although HC04 cells have near genetic identity with HepG2 cells (U. Krzych, unpublished data), they are functionally different from HepG2 cells since they support the full development of P. falciparum EEF. These cells have been deposited in MR4 and should be available in the near future.
mAb 3D11, mAb 2A10, and mAb 2E6 hybridomas are available from MR4 (http://www.mr4.org/; catalog numbers MRA-100, MRA-183, and MRA-662, respectively). mAb 2F6 hybridoma is available from P. Sinnis upon request.
mAbs specific for the CSP repeats should be used for invasion assays and can be used for the development assays if one is looking at early EEF (24 h P. berghei/P. yoelii EEF and up to 3 days P. falciparum EEF). Since CSP is not robustly expressed in mature EEF, mAb specific for Hsp70 should be used for staining this stage. For cell traversal and flow cytometry assays no antibodies are needed.
For the EEF development assays use the secondary antibody and visualization system that you are most comfortable with. For invasion assays with the rodent malaria parasites, you need two different secondary antibodies conjugated to different fluorophores so that intracellular and extracellular parasites can be distinguished. For P. falciparum invasion assays, the antibodies conjugated to HRP followed by the substrate DAB works best. Most secondary antibodies come with recommended concentrations for use in immunofluorescence assays. We test each secondary at this as well as higher and lower dilutions and use the one that gives the best signal to noise ratio. For cell traversal and flow cytometry assays, no antibodies are needed.
Invasion, cell traversal and development assays with rodent malaria parasites can also be performed using round glass coverslips (No. 1, 12 mm diameter, Fisher). Since coverslips are larger they don’t work as well for the P. falciparum assays where there are fewer events to be scored. Sterilize coverslips by autoclaving or flaming and place one in each well of a sterile 24-well plate. Plate 1 ml of cell solution per coverslip rather than 0.4 ml used per lab-tek well. Use the same total number of sporozoites per coverslip as you would use for a lab-tek well but plate them in 300 μl of solution rather than 150 μl. Follow protocol as outlined for lab-tek, except volume needed to cover the coverslip is 300 μl minimum so add 300–400 μl volume of blocking solution, antibodies, etc.. Remove coverslips from plate after last wash, let dry briefly cells-side up, invert and mount on glass slide with 3 μl mounting media and seal with nail polish.
Store collagen solution at 4°C as it polymerizes at RT. Since the concentration of the collagen varies from batch to batch, the amount of the 1:10 solution required will vary. Nonetheless, make sure it evenly covers the surface of the lab-tek wells.
All cell lines can be removed from flasks using trypsin/EDTA or TrypLE. If you have trouble with clumping of HepG2 or HepG2-CD81 cells, these can partially be broken up with vigorous pipetting when the cells are still in trypsin. Cells can also be grown in collagen-coated flasks to minimize clumping.
For development assays, which are 2–6-day assays, plate cells at a slightly lower density than you would for an invasion assay so they are not completely overgrown when you stain for EEF.
Cell density on the day of the experiment will affect the invasion efficiency. Sporozoites do not invade well if cells are too closely packed together or if they are too sparse. Ideally the cells should be subconfluent, with some but not a lot of space in between individual cells. We find that even when everyone is following the same protocol, there is variation among investigators in the cell number that is plated. For beginners, it is best to plate cells at the suggested density and at densities 25% above and 25% below this. On the day of the experiment look at your wells under a microscope and chose the wells that look best for the experiment.
It can take some time to gain experience in the proper handling of sporozoites. Although sporozoites maintain their infectivity for days, possibly weeks, while resident in mosquito salivary glands, they loose infectivity once dissected away from this environment: 1 h at 37°C will result in nonviable sporozoites, however, if kept at lower temperatures (between 4 and 20°C) infectivity can be preserved for several h. Therefore, the goal is to dissect so as to optimize sporozoite infectivity and the most important factors are speed and not rupturing the salivary glands during dissection. We find that the infectivity of the sporozoites is better preserved if we perform “dirty dissections,” i.e., the salivary glands are not completely clean of surrounding tissue. This maintains the integrity of the glands so the sporozoites are released from the glands during grinding, just before they are plated on cells. During dissection, every 10–15 min, place the salivary glands in an Eppendorf tube so that they do not heat up under the light of the microscope. If you are dissecting P. berghei, this tube should be kept on ice, if you are dissecting P. yoelii or P. falciparum, it should be kept at RT.
At this point sporozoites can be pretreated with experimental compounds or conditions.
Pelleting sporozoites is usually required to resuspend in complete medium ± inhibitors to be tested. However, if the sporozoites are sufficiently concentrated such that you can just aliquot them into complete medium, sporozoite infectivity will be enhanced. The less sporozoites are vortexed, pipetted and centrifuged, the better their infectivity.
Use 104 sporozoites per well for cell traversal assays, 2–4 × 104 per well for invasion assays and the higher end of the range for development assays. In addition, if performing invasion assays with cells on glass coverslips, you may also want to use the higher end of this range since the surface area is somewhat larger.
Controls are essential: sporozoite infectivity varies among different batches of infected mosquitoes and even with the same batch of mosquitoes different investigators can obtain different results. The positive control is untreated, wild-type sporozoites to assess overall sporozoite infectivity and this is required for all assays. For the rodent malaria parasites, positive controls should have an invasion rate of ≥50%, which means that 50% of sporozoites that are left adherent to cells after washing should be intracellular. Lower invasion rates suggest problems with dissection or handling. If you are performing a development assay, positive controls should yield 1–2 EEF per field when using a 40× objective. Numbers for P. falciparum are lower: for invasion assays one should count ≥200 intracellular sporozoites per well in positive control wells, and for development assays there should be at least 50–100 EEF per control well. When analyzing by flow cytometry, invasion rates should be above 1%, meaning ≥1% of total cells should have intracellular sporozoites, and for development assays approximately 0.8% of cells should be infected with a developing parasite. Furthermore, fluorophore intensity of cells with mature parasites should be at least 10- to 50-fold (and ideally 102- to 103-fold) greater than that of cells with newly invaded sporozoites. A negative control is necessary for cell traversal assays, P. falciparum invasion assays and all flow cytometry assays. This can be best achieved with cytochalasin-D (CD) which immobilizes sporozoites and prevents invasion but not attachment (17). Preincubate sporozoites with 1 μM CD for 10 min at RT and add them to cells in the continued presence of CD since this is a reversible inhibitor. For cell traversal and flow cytometry assays, negative controls enable you to gauge background fluorescence. For the P. falciparum invasion assay it helps to see the difference between intracellular and extracellular sporozoites.
TOTO-1 and high MW dextran are cell-impermeable and will only enter cells whose plasma membranes have been breached by sporozoites; thus after washing the cell monolayer of excess TOTO or high MW dextran, only cells that have been traversed by sporozoites will fluoresce. TOTO-1 is a nucleic acid stain that only fluoresces after it is bound to nucleic acid and gives somewhat cleaner results than the dextran. If you want to visualize sporozoites in traversed cells, do not permeabilize with methanol as this will lead to leakage of the fluorescent tracer, use 0.1% saponin in PBS.
After 1 h at 37°C in vitro, sporozoites are no longer infectious and both invasion efficiency and cell traversal activity reach a plateau. Invasion and cell traversal assays that will be scored by microscopy can therefore be processed at this time if it is more convenient. However, invasion assays that will be scored by flow cytometry should not be processed until 3 h as the additional fluorescence in partially developed parasites is helpful for the analysis.
Contamination is a concern for EEF development assays. To minimize contamination, EEF cultures can be washed with antibiotic-containing media once or twice a day. Rodent malaria EEF cultures are washed twice with DMEM containing 2% penicillin/streptomycin and 5 μg/ml fungizone followed by a third wash without fungizone. Each wash should be 2 min. Since even low exposures to fungizone decreases development of P. falciparum EEF, we wash twice with DMEM containing 2% penicillin/streptomycin and if you suspect fungal growth, 100 U/ml of nystatin can be added to these washes. A third wash without antibiotics is then performed. In all cases, add complete medium to the cultures following the washes.
If using GFP parasites do not fix with methanol but with 4% paraformaldehyde, block with 1% BSA/PBS for 1 h at 37°C, wash with PBS and proceed to step 8, Subheading 3.3.
If you are not counting the entire well, you must sample the well in a systematic way, beginning at one corner and moving in a methodical fashion through the well.
Fixing too long with paraformaldehyde can permeabilize membranes so if you prefer to fix overnight, use 2% paraform-aldehyde and leave at 4°C.
If using GFP parasites you can skip the second staining (steps 7–13, Subheading 3.4) and go to step 14, Subheading 3.4.
P. berghei and P. yoelii sporozoite attachment to cells can be assessed with this assay and is a reflection of the number of intracellular plus extracellular sporozoites counted per field. An inhibitor of attachment will reduce the total number of sporozoites counted when compared with controls. Some inhibitors of attachment do not inhibit invasion so the percent of total sporozoites that are intracellular remains the same as the control although total number of sporozoites is lower. Other inhibitors inhibit both attachment and invasion.
Intracellular sporozoites are thick, bright, refringent and delineated by a dark line. They can be clearly distinguished from extracellular sporozoites, which are dimmer and fade into the background.
References
- 1.Hollingdale MR, et al. Entry of Plasmodium berghei sporozoites into cultured cells and their transformation into trophozoites. Am J Trop Med Hyg. 1983;32:685–690. doi: 10.4269/ajtmh.1983.32.685. [DOI] [PubMed] [Google Scholar]
- 2.Hollingdale MR, et al. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in a hepatoma cell line. Am J Trop Med Hyg. 1983;32:682–684. doi: 10.4269/ajtmh.1983.32.682. [DOI] [PubMed] [Google Scholar]
- 3.Mazier D, et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science. 1985;227:440–442. doi: 10.1126/science.3880923. [DOI] [PubMed] [Google Scholar]
- 4.Renia L, et al. Malaria sporozoite penetration: a new approach by double staining. J Immunol Meth. 1988;112:201–205. doi: 10.1016/0022-1759(88)90358-4. [DOI] [PubMed] [Google Scholar]
- 5.Mota M, et al. Migration of Plasmodium sporozoites through cells before infection. Science. 2001;291:141–144. doi: 10.1126/science.291.5501.141. [DOI] [PubMed] [Google Scholar]
- 6.VanBuskirk KM, et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc Natl Acad Sci USA. 2009;106:13004–13009. doi: 10.1073/pnas.0906387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prudencio M, et al. Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell Microbiol. 2008;10:218–224. doi: 10.1111/j.1462-5822.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- 8.Coppi A, et al. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011;208:341–356. doi: 10.1084/jem.20101488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu M, et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver stage malarial parasites. Cell Host Microbe. 2008;4:567–578. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haussig JM, et al. Inactivation of a Plasmodium apicoplast protein attenuates formation of liver merozoites. Mol Microbiol. 2011;81:1511–1525. doi: 10.1111/j.1365-2958.2011.07787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller AK, et al. Plasmodium liver stage developmental arrest by depletion of a protein at the parasite-host interface. Proc Natl Acad Sci USA. 2005;102:3022–3027. doi: 10.1073/pnas.0408442102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobbs CV, et al. HIV protease inhibitors inhibit the development of preerythrocytic-stage Plasmodium parasites. J Infect Dis. 2009;199:134–141. doi: 10.1086/594369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parvanova I, et al. A small molecule inhibitor of signal peptide peptidase inhibits Plasmodium development in the liver and decreases malaria severity. PLoS One. 2009;4:e5078. doi: 10.1371/journal.pone.0005078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albuquerque SS, et al. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics. 2009;10:270. doi: 10.1186/1471-2164-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues CD, et al. Host scavenger receptor SR-B1 plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe. 2008;4:271–282. doi: 10.1016/j.chom.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Prudencio M, et al. Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in Plasmodium sporozoite infection. PLoS Pathog. 2008;4:e1000201. doi: 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinzon-Ortiz C, et al. The binding of the circumsporozoite protein to cell surface heparan sulfate proteoglycans is required for Plasmodium sporozoite attachment to cells. J Biol Chem. 2001;276:26784–26791. doi: 10.1074/jbc.M104038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gego A, et al. New approach for high-throughput screening of drug activity on Plasmodium liver stages. Antimicrob Agents Chemother. 2006;50:1586–1589. doi: 10.1128/AAC.50.4.1586-1589.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller AK, et al. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature. 2005;433:164–167. doi: 10.1038/nature03188. [DOI] [PubMed] [Google Scholar]
- 20.van Dijk MR, et al. Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc Natl Acad Sci USA. 2005;102:12194–12199. doi: 10.1073/pnas.0500925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franke-Fayard B, et al. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Tarun AS, et al. Quantitative isolation and in vivo imaging of malaria parasite liver stages. Int J Parasitol. 2006;36:1283–1293. doi: 10.1016/j.ijpara.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Talman AM, et al. A Plasmodium falciparum strain expressing GFP throughout the parasite’s life-cycle. PLoS One. 2010;5:e9156. doi: 10.1371/journal.pone.0009156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishino T, et al. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol. 2004;2:77–84. doi: 10.1371/journal.pbio.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amino R, et al. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe. 2008;3:88–96. doi: 10.1016/j.chom.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Moreira CK, et al. The Plasmodium TRAP/MIC2 family member, TRAP-Like Protein (TLP), is involved in tissue traversal by sporozoites. Cell Microbiol. 2008;10:1505–1516. doi: 10.1111/j.1462-5822.2008.01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Combe A, et al. Clonal conditional mutagenesis in malaria parasites. Cell Host Microbe. 2009;5:386–396. doi: 10.1016/j.chom.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Falae A, et al. Role of Plasmodium berghei cGMP-dependent protein kinase in late liver stage development. J Biol Chem. 2010;285:3282–3288. doi: 10.1074/jbc.M109.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calvo-Calle JM, et al. In vitro development of infectious liver stages of P. yoelii and P. berghei malaria in human cell lines. Exp Parasitol. 1994;79:362–373. doi: 10.1006/expr.1994.1098. [DOI] [PubMed] [Google Scholar]
- 30.Silvie O, et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat Med. 2003;9:93–96. doi: 10.1038/nm808. [DOI] [PubMed] [Google Scholar]
- 31.Sattabongkot J, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am J Trop Med Hyg. 2006;74:708–715. [PubMed] [Google Scholar]