

Abstract
The developing fetus must actively learn to tolerate benign antigens, or suffer the consequences of broken tolerance. Tolerance of self-antigens prevents development of autoimmune diseases, and is achieved by both deletion of autoreactive T cell clones in the thymus (central tolerance) and by the suppressive influence of CD4+CD25+FoxP3+ regulatory T cells (Tregs) in the periphery. Fetal CD4+ T cells have a strong predisposition to differentiate into tolerogenic Tregs that actively promote self-tolerance, as well as tolerance to non-inherited antigens on chimeric maternal cells that reside in most fetal tissues. As the fetus nears birth, a crucial transition must occur between the tolerogenic fetal immune system and a more defensive adult-type immune system that is able to combat pathogens. This paper will review the unique tolerogenic nature of the human fetal immune system and will examine evidence for a novel model of fetal immune development: the layered immune system hypothesis.
Keywords: Fetal T Cells, FoxP3, Human Developmental Immunology, Layered Immune System, Microchimerism, Regulatory T Cells (Treg), Tolerance
Introduction
A developing mammalian fetus expresses a set of polymorphic major histocompatibility complex (MHC) molecules inherited from both its mother and father, meaning that up to half of those MHC molecules can potentially be recognized by the maternal immune system as allogeneic foreign tissue. A great deal of attention and thought has been given to the quandary of how the immune system of the mother deals with this antigen mismatch to avoid immune rejection of her developing fetus [recently reviewed by Chaouat et. al.]1–10. Less scrutiny has been devoted to the reciprocal problem: how does the immune system of the fetus deal with the equally monumental challenge of developing in a semi-allogeneic host? One potential answer to this question is that the fetal adaptive immune system avoids rejection of the mother because it is inert, functionally impaired, and/or compromised due to limited antigen experience and a resultant insufficient cache of immunological memory. A historic and growing body of evidence that is discussed in this review argues that this hypothesis is not sufficient to explain fetal non-rejection of the maternal host. It is now clear that the human fetal immune system is highly active11–20 but its activity cannot be defined entirely by traditional metrics of sterilizing immunity. Rather, we must consider the developmental state of the fetus to understand that its primary goal of its immune system is quite different: to achieve tolerance. The developing mammalian fetus exists in a relatively pathogen-free environment compared to the diverse microbial environment it will encounter after birth. This is not to say, however, that it is not exposed to or responsive against antigens. The fetal immune system must develop in such a way that it learns to tolerate benign and/or necessary antigens, including self-antigens, as well as environmental and food antigens that are transferred across the placenta. It is also now clear that hematopoietic cells of maternal origin are commonly found in tissues of the developing fetus, and several studies have shown persistence of maternal microchimerism even into adulthood19, 21–25. Given the genetic diversity of the polymorphic MHC locus, this means that up to half of the MHC antigens borne by maternal cells are different than those inherited by the fetus. If the fetus were to respond to these non-inherited maternal alloantigens (NIMAs) by generating a cytotoxic response, the result could theoretically be disastrous in that it could lead to fetal-anti-maternal alloimmune rejection and loss of pregnancy. To the contrary, however, there is now evidence that the fetus actively generates tolerance to these antigens, thereby avoiding a rejection response19. This review will focus on the best-defined mediator of this response, the fetal CD4+CD25+FoxP3+ regulatory T cell (Treg), how it contributes to the fetal tolerance of NIMA, and what has been learned about fetal immune development from the study of fetal-maternal tolerance.
Mechanisms of fetal immune tolerance: central and peripheral
Throughout life, the acquired immune system must operate in balance between aggressive defense against potentially harmful invading pathogens and the tolerance of self-antigens and non-harmful commensal organisms. Furthermore, once a defensive immune response is initiated, mechanisms must come into effect to temper and eventually terminate that response lest it cause damage to host tissues resulting from ongoing and uncontrolled activation. A large body of literature demonstrates the unique nature of the developing fetal immune system and the importance of its unique ability to generate tolerance. In 1945, Ray Owen’s studies in dizygotic freemartin twin cattle (which exchange blood during development in utero via placental anastomoses) provided early evidence that fetal exposure to non-self antigens results in enduring tolerance to those antigens. Though these twins are genetically non-identical, both autologous and non-self (twin-derived) erythrocytes can be persistently detected throughout the life of the animal, indicating an established immunological tolerance for the foreign antigens specific to the twin26. This observation was then extended to demonstrate that these animals are tolerant to post-natal transplantation of the twin’s skin27. Experiments described by Billingham, Brent and Medawar in their seminal 1953 work, “Actively Acquired Tolerance of Foreign Cells” then went on to demonstrate that fetal exposure to antigenically dissimilar cells in fetal chicks and mice resulted in acquisition of enduring post-natal immune tolerance to tissues of the mismatched antigenic background. They showed that fetal exposure to foreign antigen lead to the absence of cells that would mount such an immune response, suggesting that elimination of such cells during development was the major mechanisms for establishing immune tolerance27.
Central immune tolerance is orchestrated by the culling of autoreactive T cell precursors during differentiation in the thymus. Stochastic somatic rearrangement of T-cell receptor (TCR) genes in developing immature thymocytes results in a staggering diversity of potential T cell antigen specificities, many of which have potential to react against self-MHC or crucial structural, developmental, and/or functional proteins (e.g. insulin or collagen). To prevent these autoreactive cells from developing to maturity and causing damage, immature thymocytes traffic through the thymic medulla where they encounter self-antigen and other cross-presented antigens in the context of self-MHC. Those cells whose TCRs bind avidly to self-antigen/MHC and transmit a strong signal through their T cell receptor then undergo apoptosis, leading to deletion of potentially autoreactive T cells. This process, known as clonal deletion, is also complemented by other mechanisms of eliminating autoreactive clones, including clonal diversion, TCR editing, and induction of anergy28. These first-line mechanisms to prevent anti-self immune reactions and autoimmune disease are crucial but not perfect, and autoreactive T cells can and do escape the thymus, as is evident in the case of autoimmune diseases that are driven by these cells. It is now clear, however, that periodic escape of autoreactive clones from thymic negative selection is likely the rule rather than the exception, even in the absence of clinical autoimmune disease. This is clearly demonstrated by experiments in which tissue-specific transgenic expression of co-stimulatory molecules and cytokines can result in antigen-specific immune attack of those tissues, meaning that mature lymphocytes escape clonal deletion and are capable of mediating reactions against self-antigen29. To deal with these rogue cells, a secondary line of defense exists in the form of active peripheral tolerance that is enforced by populations of regulatory cells with the ability to suppress potentially harmful autoimmune responses. The most extensively studied, and best understood, of these is the CD4+CD25+FoxP3+ Treg. In the following section, their role in establishing and maintaining tolerance will be explored, including evidence that they contribute to prevention of autoimmune diseases.
Tregs and peripheral tolerance
Tregs play a crucial role in immunity as a ‘rheostat’ of the immune response: both preventing autoimmunity by inhibiting anti-self responses and also acting to suppress defensive immune responses at an appropriate stage to prevent host tissue damage. Basic molecular mechanisms governing the differentiation and function of Tregs have been recently reviewed by other authors and will not be discussed at length30–36. Is is important, however, to consider the role of Tregs in establishing and maintaining peripheral tolerance in order to understand their participation in fetal immunity. The existence of regulatory cell populations was suspected and pursued for many years, and the concept of the T suppressor cells was originally proposed and championed by Gershon and colleagues37–40. It is only in the last 10–15 years, however, that CD4+CD25+FoxP3+ Tregs have been accepted as a crucial controller of the immune response. The initial discovery of Tregs came from experiments in which a specific population of CD4+ cells that also highly express the high-affinity interleukin (IL)-2 receptor α-chain, CD25, were found to be protective in mouse models of autoimmunity41–44. It was subsequently shown that the transcription factor forkhead box P3 (FoxP3) was not only a specific marker for CD4+CD25+ Tregs, but was also crucial for their development, maintenance of phenotype, and function45–51. The importance of this factor came to light from both human clinical observations and genetic mutant mouse models which together demonstrated that disruption of the FoxP3 gene results in an absence or paucity of regulatory cells leading to autoimmunity45–58. In 1982, a human syndrome was described that was defined by early neonatal onset in males of autoimmune disease in multiple organs, including thyroid, pancreas, gut, and skin52. The manifestations of the disease included type I diabetes, thyroiditis, inflammatory enteropathy, atopic dermatitis, and death from overwhelming infection, and the syndrome was named IPEX (for Immune Dysregulation, Polyendocrinopathy, Enteropathy, and X-linked). The syndrome was initially described as universally fatal, with decreased fetal viability or death within the first year of life52. Meanwhile, a mouse strain called scrufy was identified as a spontaneously arising mutant with a strikingly similar phenotype to patients with IPEX54–56, 58. Hemizygous scurfy males die within the first three weeks after birth with disease characterized by T cell over-proliferation and extensive multi-organ leukocyte infiltration and autoimmunity53–56. The gene defective in the scurfy mouse was mapped to the FoxP3 locus, and genetic complementation with FoxP3 rescued the scurfy phenotype46. It was subsequently demonstrated that induced disruption of FoxP3 resulted in the absence of Tregs and reproduced the characteristics of the scurfy phenotype48. FoxP3 is strongly conserved between mice and humans, and subsequent studies confirmed that FoxP3 disruption and the consequent absence of Tregs is the primary immune lesion in IPEX45, 57, 58.
The mechanisms by which Tregs function to suppress immune responses have been intensely studied, and there seem to be diverse Treg responses that come into play depending on the context of activation and the environment in which they are operating59, 60. Specifically, the mechanisms of Treg-mediated suppression seem to be determined at least in part by whether they are maintaining immune quiescence to prevent immune activation in the physiological homeostatic steady state or are responding to dampen an active inflammatory response61. The mechanisms used by Tregs to suppress immune responses include: transmission of inhibitory signals via cell-cell surface interactions or secreted cytokines, diminishing conventional T cell activation or fitness by limiting growth factors like IL-2 or essential amino acids, direct target-cell cytotoxicity, and/or modulation of antigen presenting cell function36, 59–61. Like other αβ TCR-utilizing T cells, Tregs have a diverse TCR repertoire and can respond to a wide range of antigens. Though they do not seem to have an absolute requirement for recognition of specific self-antigens to mediate suppression, clonal Treg pools responding against a specific antigen recognized by their TCRs seem to be more effective suppressors than polyclonal populations mediating non-specific suppression62–65.
In the years since their discovery and acceptance as being functional regulatory cells, it has become clear that Tregs play a crucial role in maintaining peripheral tolerance and immune homeostasis. Insufficient or dysfunctional Treg responses are thought to contribute to the pathogenesis of several disease states resulting from broken self-tolerance, including Type I diabetes63, 65–67. Not only are Tregs a dominant mediator of peripheral self-tolerance, they also appear to be important in modulating the innate and acquired immune responses to foreign antigen68–71. Most circulating Tregs differentiate from T cell precursors in the thymus, and are thereafter phenotypically and functionally distinctive compared to conventional FoxP3−CD4+ T cells36, 72. These ‘thymic’ or ‘natural’ Tregs (nTreg) likely play a crucial role in maintenance of tolerance to self-antigen, and to other antigens presented in the thymus. Tregs can also, however, be generated under specific circumstances from FoxP3−CD4+ conventional T cells after thymic egress73–82. These cells have been called ‘peripheral’ or ‘induced’ Treg (iTreg), and may play a role more in the tempering of responses to antigens not encountered in the thymus, including pathogen-related antigens. One of the best-studied iTreg populations resides in the colon, develops in response to commensal bacteria, and is thought to be important for maintenance of tolerance to these commensals68, 70. Differentiation or ‘conversion’ of conventional Tregs into iTregs is dependent on several factors, including the strength of antigen signal, specific signals from antigen presenting cells, and the nature of the local cytokine milieu. In particular, activation of T cells under the influence of transforming growth factor-β (TGF-β) and IL-2 can induce naïve CD4+ T cells to induce FoxP3 expression and adopt a regulatory phenotype83–85. The role of Tregs in promoting both solid organ and hematopoietic transplantation tolerance has also been of great interest71. Infusion of Tregs that are polyclonally expanded ex vivo is now being studied in phase I and II clinical trials of hematopoietic stem cell transplantation, and demonstrates great promise as a strategy to reduced graft-versus host disease69, 71, 86, 87. This begs the important question whether Tregs might play a role in establishing and maintaining tolerance to the only naturally occurring allograft encountered by the immune system: the fetus.
Tregs contribute to maternal tolerance of the fetus
First clearly demonstrated by Alexander Betz, and colleagues a significant body of evidence from mouse models now demonstrates that maternal Tregs that are induced by, and are reactive against, paternal alloantigen contribute to successful implantation and maintenance of pregnancy7, 88–98. Though challenging to demonstrate definitively, it also now seems that maternal Tregs also play a similar role in protecting the fetus from rejection during human pregnancy93, 95, 99–102. During normal early (5–9 gestational weeks) human pregnancy, Sasaki et al. observed that suppressive CD4+CD25high T cells are enriched in the decidua, and that this enrichment was not found in cases of spontaneous abortion100. Similarly, the frequency of CD4+CD25high T cells was found to increase in the peripheral blood of pregnant women at increasing frequencies that peaked in the second trimester and declined after birth99. More recent evidence suggests that maternal failure to increase circulating Treg frequency is correlated with pregnancy loss102. It has also been shown that in preeclampsia there is also a relative failure to increase circulating Tregs, leading to a relative imbalance of regulatory and inflammatory cells compared to normal pregnancy101. Together, these observations suggest that maternal Treg responses in humans also contribute to maternal tolerance of fetus. The mother may also be primed toward tolerance by exposure to paternal antigens in the context of immunomodulatory factors in semen or at the utero-placental interface1–6, 103–108. It is also clear, though, that cells from the fetus transit into the mother, establishing residence in maternal tissues and resulting in microchimerism that can last for many years22, 24, 25. These microchimeric cells likely provide a stimulus for initiation and generation of the tolerogenic Treg response demonstrated in pregnancy. Immune chimerism also occurs in the opposite direction, and maternal cells have been found to routinely (if not universally) reside in fetal tissues19, 21–25. This begs the question: why does the fetus not reject the mother? Work by many investigators has demonstrated that the human fetal peripheral immune system is highly active, and that fetal T cells are intrinsically capable of becoming activated in response to foreign antigen11–20. Could this then suggest that active peripheral tolerogenic mechanisms might contribute to the physiological absence of fetal anti-maternal rejection?
Part of the challenge in studying fetal immunity has to do with the diversity and variability in immune development between species, and especially the difference between humans and laboratory mice. Inbred mouse strains commonly used in the laboratory begin to populate the thymus with T cell precursors at about 12 days of gestation109. The developing fetal mouse does not start to populate secondary lymphoid structures until near the end of gestation, making the newborn mouse more closely resemble a human at a much early fetal developmental stage110, 111. T cells are absent in the newborn mouse spleen and are very sparse in the lymph nodes, where they have a primitive, simplified spatial organization110. During the first week of life, lymph node T cells become more abundant and adopt a more mature architecture, while the spleen becomes populated with T cells110. Tregs typically do not exit the thymus in mice until at least three days after birth, and increase to adult levels over three weeks112. This developmental schedule would lend support to the assumption that the fetus was not significantly challenged by maternal antigen in the immune periphery, and that tolerance of self-antigen in the developing fetus was entirely due to central deletion of autoreactive cells. The human fetal schedule of immune development is highly distinct from the mouse however, and suggests the need for active peripheral tolerance mechanisms. In the human fetus, T cell precursors transit to the thymus by 9 weeks of gestation14. Mature naïve and memory αβ T cells are readily found by 12–14 weeks in spleen and lymph nodes, and are abundant by the end of the second trimester11–14, 18, 19. Impaired fetal survival and early demise in IPEX syndrome clearly demonstrates the clinical importance of a peripheral tolerogenic mechanism in human immune development. Another compelling hint that the human fetal immune system may actively generate tolerance came from the studies of Burlingham and colleagues, who demonstrated that long-term survival of kidney grafts was enhanced when the donor carried mismatched human leukocyte antigens (HLA) that were the same as non-inherited alleles from the recipient’s mother (i.e. NIMAs)113. Although the recipient had no opportunity to generate post-natal tolerance to these unshared maternal antigens, there was nevertheless some factor preventing their rejection that was likely to have arisen during the period of exposure to NIMAs in utero. This finding suggested a fundamental shift in the understanding of how the fetal immune system develops, and implied that humans have the ability to generate active, long-lasting, post-natal tolerance to antigens experienced in utero.
Tregs are abundant in the developing human fetus
The developing human fetal immune system in the second trimester is distinct from that of the newborn, or the mature adult. One of the most striking differences is that fetal tissues have an increased frequency of Tregs compared to any other time in development16, 18, 19, 114. The earliest observation of this phenomenon may have been in the work of Cooper and colleagues, who demonstrated that fetal spleen and premature cord blood were enriched for a CD45RO+ memory population that also expressed CD25, was highly proliferative in response to IL-2, but not to standard mitogenic stimuli (e.g. anti-CD3)15. After the description of Tregs, and FoxP3 as their definitive marker, it was confirmed that secondary fetal lymphoid tissues in the second trimester contain a surprisingly high abundance of CD4+CD25+FoxP3+ Tregs, on average 15–20% of CD4+ cells16, 18. This is in contrast to full-term umbilical cord blood, adult peripheral blood, or lymph nodes from adults, in which Tregs typically represent less than 5% of total CD4+ cells18. The abundance of Tregs in secondary fetal lymphoid organs was not reflected in the thymus at equivalent gestational ages, where the frequency of CD25+FoxP3+ single CD4 (sp4) thymocytes was comparable to the infant thymus (about 10–12%)18. This suggested that a significant proportion of the Treg-enriched fetal T cell population consisted of Tregs that expanded from nTregs, or were generated from conventional CD4+FoxP3− T cells, in response to antigen. Upon depletion of CD25+ T cells from fetal lymph node-derived T cells, a significant proportion of the remaining conventional T cells both (1) proliferate spontaneously; and (2) produce interferon-γ (IFN-γ) in response to SEB stimulation (which they did not in the presence of Tregs)18. These findings demonstrated the suppressive influence of Tregs in human fetal lymph nodes, and implied that the fetal immune system may have the ability to generate active peripheral tolerance via these cells
Fetal Tregs generate specific tolerance toward maternal alloantigen
Given that a significant frequency of human T cells have the capacity to recognize alloantigen, the fetal immune response against NIMA was used as an in vivo model system to test the hypothesis that fetal Tregs are generated from conventional fetal T cells in response to antigen stimulation.19 Maternal cells were confirmed to be in relatively high abundance (up to 0.8%) in second-trimester fetal lymph nodes, and cells were from each major hematopoietic lineage were found to be present in full-term umbilical cord blood (including T, B, NK, and monocytes)19. In mixed lymphocyte reaction (MLRs), fetal immune responses against maternal antigen presenting cells (APCs) bearing NIMAs were dampened compared to responses against unrelated alloantigen from third-party donors19. This demonstrated that fetuses are more tolerant toward NIMAs, and knowing that they are exposed to those antigens on resident maternal cells, it was proposed that this tolerance was mediated not only by central clonal deletion, but also by peripherally generated Tregs. Fetal T cells that were depleted of CD25+ Tregs prior to MLR had significantly enhanced immune reactions against self and maternal APC, but not against unrelated donors, confirming the presence of NIMA-specific Tregs19. It was unknown whether these were nTregs generated against maternal antigen presented in the thymus, or if iTregs generated in the periphery in response to stimulation by maternal cells in secondary lymphoid organs may contribute to anti-NIMA tolerance. To determine if these cells could be generated from non-Tregs, conventional FoxP3− naïve (CD45RA+CCR7+) T cells were stimulated with foreign APCs. Surprisingly, alloantigen-stimulated fetal T cells overwhelmingly differentiated into CD25+FoxP3+ Tregs that were confirmed to mediate alloantigen-specific suppression19. This effect was dependent on TGFβ signaling, and fetal lymph nodes were found to express significantly higher levels of TGF-β family members compared to adult lymph nodes. These findings do not rule out the possibility that fetal Tregs represent a mixed population of nTregs and iTregs, but do clearly demonstrate that fetal T cells have a strong propensity to become functional tolerogenic Tregs upon antigen stimulation, and that the fetal peripheral immune niche is tuned to support such a response.
Implications for fetal and neonatal infection
The strong predisposition of fetal T cells to differentiate into Tregs has many potential implications for the overall function of the fetal immune response and the nature of its interactions with both benign (self, maternal, environmental, commensal microbial, and food) antigens as well as antigens associated with potentially harmful pathogens. Given that the in utero environment is relatively protected against microbial infection, it makes teleological and evolutionary sense that T cells in the developing fetus may be predisposed to mount tolerogenic responses, and that the niche in which they develop may support such responses. As seen in the example of IPEX, the absence of such tolerance is disastrous52. In the face of microbial or viral infection, however, a dominant tolerogenic response might theoretically be detrimental. This raises an interesting possibility that the fetal predisposition toward tolerance could contribute to the enhanced susceptibility to serious infection that is well recognized in fetuses and newborns, and particularly in premature newborns.
Infection is a leading cause of death and morbidity in newborns. Not only are neonates susceptible to more severe forms of disease caused by typical human pathogens, they are also subject to serious infection by microbes that are considered commensal flora in adults. For example, even after implementation of intensive screening and prevention practices, the estimated rate of Group B Streptococal sepsis in the first week of life is 0.34 per 1000 live births, resulting in 60–70 deaths per year in the United States alone115. Premature infants are especially predisposed to more severe infections from all pathogens and can also succumb to fatal infection by microbes that infrequently cause severe disease in adults116. This increased susceptibility to infection is accompanied by a relatively ineffective response to neonatal vaccination117, 118. Compared to adults and older children, even full-term newborns produce less, and generally less effective, antibody in response to most immunizations. They are also less able to generate effector T cells that mediate effective antimicrobial responses117, 119–122. Together, these deficiencies render the fetus and neonate a vulnerable target for a host of invading pathogens. Many mechanisms of classical host defense are compromised in the fetus and neonate, but this has often been attributed to the immature developmental state of the immune system, or to the absence of antigen exposure. The work discussed here demonstrates that the fetal immune system is most certainly not inert, but rather is extremely active and capable of responding to antigenic stimulation. The nature of its response, however, while appropriately developmentally tuned to create tolerance, may predispose the fetus toward tolerance of harmful microbes in the face of infection. The negative implications of this tolerance-promoting mandate are clear: when faced with an invading microbe, the human fetal tendency to generate tolerance to antigens associated with that microbe may be detrimental if it allows infection to proceed unchecked. This hypothetical explanation for fetal and neonatal susceptibility to infection remains to be tested, but does provide a novel model with which to frame such pressing questions. It also raises the important question of how a tolerogenic immune program that is crucial for survival and development in utero is ultimately converted into an immune response that can functionally meet the outside world.
A (not-so) new model for fetal immune development: the layered immune system
As the fetus nears birth, the tolerogenic fetal T cell response must be converted into a response dominated by T cells capable of supporting defensive antimicrobial immunity. To begin to understand this process, and the mechanisms by which it might be governed, it was necessary to delve further into the nature of the fetal immune system, and how it might be different from that of the adult. Global gene expression profiling on human fetal and adult CD4+ naïve T cells and Tregs followed by comparative bioinformatic analysis revealed that phenotypically similar T cells in the fetus and adult had strikingly different gene expression profiles123. This was true of both Tregs and naïve T cells, and many of the genes that were differentially expressed in fetal naïve T cells were also similarly differentially expressed in fetal regulatory T cells. The commonality of fetal naïve and Treg gene expression profiles suggested that fetal T cells could represent a unique hematopoietic lineage. There is, in fact, strong historical evidence in both avian and mouse models that fetal hematopoietic stem-progenitor cells (HSPCs) can give rise to unique subsets of lymphocytes in the fetus that cannot be generated from adult HSPCs124–132. In the mouse, the first wave of T cell progenitors that colonize the fetal thymus differentiate into a unique fetal subset of γδ TCR-utilizing T cells with a restricted Vγ3/Vδ1 TCR125. These cells are eventually replaced by a wave of more diverse γδ T cells and ultimately, prior to birth, by a wave of T cells utilizing the αβ TCR125, 129. This phenomenon echoes earlier reports in quail/chick chimeras in which three waves of thymocytes populate the thymus, and replace one another in sequence133, 134. A similar phenomenon has been described and well characterized in mouse B lymphocyte development. Two distinct lineages can be defined based on surface phenotype staining, the B-1 and B-2 lineage, initially defined by CD5 (Ly-1) expression135, 136. The B-1 lineage seems to be a more primitive one that is found in newborn mice and cannot be efficiently generated by adoptive transfer of adult bone marrow128. Together, these findings suggest a model whereby maturation of progeny cells derived from distinct hematopoietic progenitors results in waves of mature immune effector cells that are tuned to the specific developmental needs of the organism. As these progressive, distinct waves accumulate, they co-exist for a period of time and result in ‘layers’ of immune cell lineages. This model, first formally proposed by Lee and Len Herzenberg, has become known as the layered model of immune development (Figure 1)137. Given the highly unique gene-expression profile of fetal T cells (including fetal Tregs), it stood to reason that these cells could represent their own wave of T cell progeny arising from a distinct fetal hematopoietic progenitor.
Figure 1.
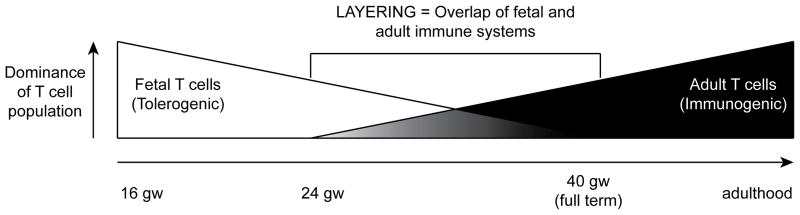
Representation of the layered immune system in the human fetus. Tolerogenic fetal T cells derived from fetal hematopoietic stem–progenitor cells (HSPC) dominate fetal immune responses until the third trimester, when a population of adult HSPC-derived immunogenic T cells come to dominance. During the transition period, a layered immune system occurs, and the prevailing nature of the immune response may be governed, in part, by the degree of layering of fetal and adult T cells and the relative influence they exert.
To test the hypothesis that fetal T cells represent a unique cell lineage compared to adult T cells, a series of experiments were carried out in which fetal HSPCs from human fetal liver and bone marrow (18–22 gestational weeks) and adult bone marrow were injected directly into the human Thy/Liv organ of the SCID-hu Thy/Liv mouse123. This humanized mouse model allows for reproducible, multi-lineage hematopoiesis (including thymopoiesis) from human HSPCs138, 139. Global gene expression analysis was carried out on mature CD3+CD4+CD8−CD25− sp4 thymocytes that were differentiated from fetal liver-, fetal bone marrow-, or adult bone marrow-derived HSPCs using this model. Surprisingly, HSPCs from both fetal liver and bone marrow gave rise to identical populations of sp4 thymocytes on the basis of gene expression. By contrast, adult bone marrow-derived HSPCs had a radically different gene expression profile compared to each population of fetal HSPC-derived thymocytes. There was significant overlap between genes expressed in primary peripheral CD4+ fetal T cells and CD4+ thymocytes derived from fetal HSPC, and the same was true of adult peripheral CD4+ T cells and CD4+ thymocytes derived from adult HSPC. This strongly suggested that fetal and adult HSPCs give rise to developmentally unique lineages of fetal T cells, thus, providing strong evidence that the layered immune system hypothesis can be extended to describe human T cell development. Considered in context, this hypothesis proposes that fetal immune development proceeds in such a manner that a dominant tolerogenic fetal immune system is progressively replaced by an immunogenic adult-type immune system, resulting in co-existing populations (or layers) of tolerogenic and immunogenic T cells. These two immune systems serve different roles based on developmental state of the organism, and the sum immune response that is generated toward antigenic stimulation is the result of the relative contribution of these two opposing T cell layers. Future perspectives for peripheral fetal tolerance and the layered immune system hypothesis.
The layered immune system hypothesis may have potential to enhance our understanding of normal fetal immune development, and also represents a new way to model the pathophysiology of many diseases. For example, if the type and magnitude of response that is generated in utero or at birth is determined in part by the opposing fetal and adult influences, the relative contribution of each may determine whether the ultimate response is tolerogenic, immunogenic, or an intermediate of the two. Within a human population, if there is variability in the degree to which layering occurs at birth, this could theoretically also lead to equal variability in the nature of immune responses that can be generated. As an example, consider the scenario in which the transition from fetal to adult T cell predominance is delayed compared to the population norm, resulting in a newborn infant with an over-representation of fetal T cells. When faced with colonizing microbes, including GBS, this infant might be more predisposed to mount a tolerogenic response to those microbes. In that scenario, an organism that would normally become a commensal symbiotic microbe in most infants could potentially cause invasive infection and serious disease. Conversely, an infant who had precocious maturation of the adult compartment may then have over-representation of adult T cells at birth. Such an infant may then be at risk of overly zealous immune responses against normally tolerated antigens such as self, environmental, or food antigens, potentially leading to auto-immune disease, allergy, or food-antigen intolerance. To address these questions, it will be necessary to better understand the timing of, and mechanisms governing, the transition from tolerogenic fetal responses to immunogenic adult responses.
Another question to be answered is whether hematopoiesis arising from fetal HSPCs persists throughout post-natal life, and whether T cells arising from these progenitors play a role in homeostasis, health, and disease. Tregs capable of mediating specific tolerance to NIMA persist in children and young adults19. It is not clear whether these Tregs represent long-lived cells acquired during fetal development, the progeny of such cells, or Tregs arising more recently due to persistence of chimeric maternal cells in the offspring. It is intriguing to consider that fetal stem-progenitor cells (or their long-lived progeny) may persist in a normally non-dominant state throughout life, and could potentially be re-activated in situations where re-acquisition of tolerance is needed (e.g. after non-ablative chemotherapy, or during immune reconstitution after initiation of antiretroviral therapy for HIV disease). This possibility also begs the question whether, in some cases, broken or incomplete tolerance (e.g. autoimmune diseases) represents the failure of a tolerogenic fetal T cell population. Though these considerations remain hypothetical, ongoing investigations will hopefully lead to further insights into the process of normal human fetal immune development and its influence of on how our immune systems learn to interact with ourselves, and the environment in which we exist.
Summary
In conclusion, the immune environment of the developing human fetus is especially well suited to generate peripheral tolerance. Fetal T cells, which are derived from a developmentally restricted hematopoietic lineage, appear to be a major player in this tolerogenic disposition because they are relatively enriched in Tregs, and there is a strong tendency for naïve CD4+ fetal T cells to differentiate into Tregs upon antigen stimulation. These fetal Tregs are capable of quelling immune responses against maternal alloantigen, and may therefore provide a means by which the fetus prevents maternal rejection to allow for maintenance of pregnancy. A deeper understanding of the nature of the developing human fetal immune system has profound potential to help us understand health and disease in the fetus, newborn, and likely every stage of life thereafter.
Acknowledgments
Thanks to Dr. Mike McCune for helpful discussion and reading of this manuscript. The author is supported by a grant from the NIH/NICHD (K08-HD067295).
References
- 1.Rouas-Freiss N, Goncalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci U S A. 1997;94:11520–11525. doi: 10.1073/pnas.94.21.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 3.Mellor AL, Munn DH. Immunology at the maternal-fetal interface: lessons for T cell tolerance and suppression. Annu Rev Immunol. 2000;18:367–391. doi: 10.1146/annurev.immunol.18.1.367. [DOI] [PubMed] [Google Scholar]
- 4.Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 5.Mellor AL, Munn DH. Tryptophan catabolism prevents maternal T cells from activating lethal anti-fetal immune responses. J Reprod Immunol. 2001;52:5–13. doi: 10.1016/s0165-0378(01)00118-8. [DOI] [PubMed] [Google Scholar]
- 6.Makrigiannakis A, Zoumakis E, Kalantaridou S, Coutifaris C, Margioris AN, Coukos G, Rice KC, Gravanis A, Chrousos GP. Corticotropin-releasing hormone promotes blastocyst implantation and early maternal tolerance. Nat Immunol. 2001;2:1018–1024. doi: 10.1038/ni719. [DOI] [PubMed] [Google Scholar]
- 7.Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004;5:266–271. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 8.Zenclussen AC, Schumacher A, Zenclussen ML, Wafula P, Volk HD. Immunology of pregnancy: cellular mechanisms allowing fetal survival within the maternal uterus. Expert reviews in molecular medicine. 2007;9:1–14. doi: 10.1017/S1462399407000294. [DOI] [PubMed] [Google Scholar]
- 9.Chaouat G, Petitbarat M, Dubanchet S, Rahmati M, Ledee N. Tolerance to the foetal allograft? Am J Reprod Immunol. 2010;63:624–636. doi: 10.1111/j.1600-0897.2010.00832.x. [DOI] [PubMed] [Google Scholar]
- 10.Leber A, Zenclussen ML, Teles A, Brachwitz N, Casalis P, El-Mousleh T, Jensen F, Woidacki K, Zenclussen AC. Pregnancy: tolerance and suppression of immune responses. Methods Mol Biol. 2011;677:397–417. doi: 10.1007/978-1-60761-869-0_25. [DOI] [PubMed] [Google Scholar]
- 11.Kay HE, Doe J, Hockley A. Response of human foetal thymocytes to phytohaemagglutinin (PHA) Immunology. 1970;18:393–396. [PMC free article] [PubMed] [Google Scholar]
- 12.Ceppellini R, Bonnard GD, Coppo F, Miggiano VC, Pospisil M, Curtoni ES, Pellegrino M. Transplantation antigens: introductory symposium. Mixed leukocyte cultures and HL-A antigens. I. Reactivity of young fetuses, newborns and mothers at delivery. Transplant Proc. 1971;3:58–63. [PubMed] [Google Scholar]
- 13.Hayward AR, Ezer G. Development of lymphocyte populations in the human foetal thymus and spleen. Clin Exp Immunol. 1974;17:169–178. [PMC free article] [PubMed] [Google Scholar]
- 14.Haynes BF, Martin ME, Kay HH, Kurtzberg J. Early events in human T cell ontogeny. Phenotypic characterization and immunohistologic localization of T cell precursors in early human fetal tissues. J Exp Med. 1988;168:1061–1080. doi: 10.1084/jem.168.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrne JA, Stankovic AK, Cooper MD. A novel subpopulation of primed T cells in the human fetus. J Immunol. 1994;152:3098–3106. [PubMed] [Google Scholar]
- 16.Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T? cells in the human fetus. Eur J Immunol. 2005;35:383–390. doi: 10.1002/eji.200425763. [DOI] [PubMed] [Google Scholar]
- 17.Lewis D, Wilson CB. Developmental Immunology and Role of Host Defenses in Susceptibility to Infection. In: Remington JS, Klein JO, Wilson CB, Baker CJ, editors. Infectious Diseases of the Fetus and Newborn Infant. 6. Philadelphia, PA: Elsevier Saunders; 2006. pp. 87–210. [Google Scholar]
- 18.Michaelsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol. 2006;176:5741–5748. doi: 10.4049/jimmunol.176.10.5741. [DOI] [PubMed] [Google Scholar]
- 19.Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, Lee T-H, Nixon DF, McCune JM. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–1565. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mold JE, McCune JM. Immunological tolerance during fetal development: from mouse to man. Adv Immunol. 2012;115:73–111. doi: 10.1016/B978-0-12-394299-9.00003-5. [DOI] [PubMed] [Google Scholar]
- 21.Maloney S, Smith A, Furst DE, Myerson D, Rupert K, Evans PC, Nelson JL. Microchimerism of maternal origin persists into adult life. J Clin Invest. 1999;104:41–47. doi: 10.1172/JCI6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo YM, Lau TK, Chan LY, Leung TN, Chang AM. Quantitative analysis of the bidirectional fetomaternal transfer of nucleated cells and plasma DNA. Clinical chemistry. 2000;46:1301–1309. [PubMed] [Google Scholar]
- 23.Loubiere LS, Lambert NC, Flinn LJ, Erickson TD, Yan Z, Guthrie KA, Vickers KT, Nelson JL. Maternal microchimerism in healthy adults in lymphocytes, monocyte/macrophages and NK cells. Lab Invest. 2006;86:1185–1192. doi: 10.1038/labinvest.3700471. [DOI] [PubMed] [Google Scholar]
- 24.Gammill HS, Nelson JL. Naturally acquired microchimerism. The International journal of developmental biology. 2010;54:531–543. doi: 10.1387/ijdb.082767hg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol. 2012;33:421–427. doi: 10.1016/j.it.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen RD. Immunogenetic Consequences of Vascular Anastomoses between Bovine Twins. Science. 1945;102:400–401. doi: 10.1126/science.102.2651.400. [DOI] [PubMed] [Google Scholar]
- 27.Billingham RE, Lampkin GH, Medawar PB, Williams HLL. Tolerance to homografts, twin diagnosis and the freemartin condition in cattle. Heredity. 1952;6:201–212. [Google Scholar]
- 28.Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harbor perspectives in biology. 2012;4:1–15. doi: 10.1101/cshperspect.a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci U S A. 1994;91:5138–5142. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 33.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 34.Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol. 2009;10:689–695. doi: 10.1038/ni.1760. [DOI] [PubMed] [Google Scholar]
- 35.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 36.Benoist C, Mathis D. Treg cells, life history, and diversity. Cold Spring Harbor perspectives in biology. 2012;4:a007021. doi: 10.1101/cshperspect.a007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18:723–737. [PMC free article] [PubMed] [Google Scholar]
- 38.Gershon RK, Cohen P, Hencin R, Liebhaber SA. Suppressor T cells. J Immunol. 1972;108:586–590. [PubMed] [Google Scholar]
- 39.Eardley DD, Hugenberger J, McVay-Boudreau L, Shen FW, Gershon RK, Cantor H. Immunoregulatory circuits among T-cell sets. I. T-helper cells induce other T-cell sets to exert feedback inhibition. J Exp Med. 1978;147:1106–1115. doi: 10.1084/jem.147.4.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green DR, Flood PM, Gershon RK. Immunoregulatory T-cell pathways. Annu Rev Immunol. 1983;1:439–463. doi: 10.1146/annurev.iy.01.040183.002255. [DOI] [PubMed] [Google Scholar]
- 41.Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J Exp Med. 1982;156:1577–1586. doi: 10.1084/jem.156.6.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 43.Smith H, Sakamoto Y, Kasai K, Tung KS. Effector and regulatory cells in autoimmune oophoritis elicited by neonatal thymectomy. J Immunol. 1991;147:2928–2933. [PubMed] [Google Scholar]
- 44.Suri-Payer E, Amar AZ, Thornton AM, Shevach EM. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol. 1998;160:1212–1218. [PubMed] [Google Scholar]
- 45.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 47.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 48.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 50.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 51.Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–737. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 53.Lyon MF, Peters J, Glenister PH, Ball S, Wright E. The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc Natl Acad Sci U S A. 1990;87:2433–2437. doi: 10.1073/pnas.87.7.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blair PJ, Bultman SJ, Haas JC, Rouse BT, Wilkinson JE, Godfrey VL. CD4+CD8− T cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J Immunol. 1994;153:3764–3774. [PubMed] [Google Scholar]
- 55.Kanangat S, Blair P, Reddy R, Daheshia M, Godfrey V, Rouse BT, Wilkinson E. Disease in the scurfy (sf) mouse is associated with overexpression of cytokine genes. Eur J Immunol. 1996;26:161–165. doi: 10.1002/eji.1830260125. [DOI] [PubMed] [Google Scholar]
- 56.Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–2554. [PubMed] [Google Scholar]
- 57.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 58.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 59.Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Frontiers in immunology. 2012;3:178. doi: 10.3389/fimmu.2012.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 61.Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 2005;202:1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bluestone JA, Bour-Jordan H. Current and future immunomodulation strategies to restore tolerance in autoimmune diseases. Cold Spring Harbor perspectives in biology. 2012;4:1–23. doi: 10.1101/cshperspect.a007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harbor perspectives in medicine. 2012;2:a007807. doi: 10.1101/cshperspect.a007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D, Aloisi T, Perruccio K, Ruggeri L, Balucani C, Pierini A, Sportoletti P, Aristei C, Falini B, Reisner Y, Velardi A, Aversa F, Martelli MF. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 70.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sagoo P, Lombardi G, Lechler RI. Relevance of regulatory T cell promotion of donor-specific tolerance in solid organ transplantation. Frontiers in immunology. 2012;3:184. doi: 10.3389/fimmu.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geiger TL, Tauro S. Nature and nurture in Foxp3(+) regulatory T cell development, stability, and function. Hum Immunol. 2012;73:232–239. doi: 10.1016/j.humimm.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 75.Finney CA, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4(+)CD25(+) regulatory T cells in Heligmosomoides polygyrus infection. Eur J Immunol. 2007;37:1874–1886. doi: 10.1002/eji.200636751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 77.McSorley HJ, Harcus YM, Murray J, Taylor MD, Maizels RM. Expansion of Foxp3+ regulatory T cells in mice infected with the filarial parasite Brugia malayi. J Immunol. 2008;181:6456–6466. doi: 10.4049/jimmunol.181.9.6456. [DOI] [PubMed] [Google Scholar]
- 78.Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, Li SH, Simpson PM, Chatila TA, Williams CB. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. 2009;182:3461–3468. doi: 10.4049/jimmunol.0802535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Daniel C, Wennhold K, Kim HJ, von Boehmer H. Enhancement of antigen-specific Treg vaccination in vivo. Proc Natl Acad Sci U S A. 2010;107:16246–16251. doi: 10.1073/pnas.1007422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci U S A. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, Finney CA, Greenwood EJ, Knox DP, Wilson MS, Belkaid Y, Rudensky AY, Maizels RM. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med. 2010;207:2331–2341. doi: 10.1084/jem.20101074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 85.Peng Y, Laouar Y, Li MO, Green EA, Flavell RA. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci U S A. 2004;101:4572–4577. doi: 10.1073/pnas.0400810101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hippen KL, Riley JL, June CH, Blazar BR. Clinical perspectives for regulatory T cells in transplantation tolerance. Semin Immunol. 2011;23:462–468. doi: 10.1016/j.smim.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zenclussen AC, Gerlof K, Zenclussen ML, Sollwedel A, Bertoja AZ, Ritter T, Kotsch K, Leber J, Volk HD. Abnormal T-cell reactivity against paternal antigens in spontaneous abortion: adoptive transfer of pregnancy-induced CD4+CD25+ T regulatory cells prevents fetal rejection in a murine abortion model. Am J Pathol. 2005;166:811–822. doi: 10.1016/S0002-9440(10)62302-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zenclussen AC. CD4(+)CD25+ T regulatory cells in murine pregnancy. J Reprod Immunol. 2005;65:101–110. doi: 10.1016/j.jri.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 90.Zenclussen AC, Gerlof K, Zenclussen ML, Ritschel S, Zambon Bertoja A, Fest S, Hontsu S, Ueha S, Matsushima K, Leber J, Volk HD. Regulatory T cells induce a privileged tolerant microenvironment at the fetal-maternal interface. Eur J Immunol. 2006;36:82–94. doi: 10.1002/eji.200535428. [DOI] [PubMed] [Google Scholar]
- 91.Zhao JX, Zeng YY, Liu Y. Fetal alloantigen is responsible for the expansion of the CD4(+)CD25(+) regulatory T cell pool during pregnancy. J Reprod Immunol. 2007;75:71–81. doi: 10.1016/j.jri.2007.06.052. [DOI] [PubMed] [Google Scholar]
- 92.Thuere C, Zenclussen ML, Schumacher A, Langwisch S, Schulte-Wrede U, Teles A, Paeschke S, Volk HD, Zenclussen AC. Kinetics of regulatory T cells during murine pregnancy. Am J Reprod Immunol. 2007;58:514–523. doi: 10.1111/j.1600-0897.2007.00538.x. [DOI] [PubMed] [Google Scholar]
- 93.Leber A, Teles A, Zenclussen AC. Regulatory T cells and their role in pregnancy. Am J Reprod Immunol. 2010;63:445–459. doi: 10.1111/j.1600-0897.2010.00821.x. [DOI] [PubMed] [Google Scholar]
- 94.Zenclussen ML, Thuere C, Ahmad N, Wafula PO, Fest S, Teles A, Leber A, Casalis PA, Bechmann I, Priller J, Volk HD, Zenclussen AC. The persistence of paternal antigens in the maternal body is involved in regulatory T-cell expansion and fetal-maternal tolerance in murine pregnancy. Am J Reprod Immunol. 2010;63:200–208. doi: 10.1111/j.1600-0897.2009.00793.x. [DOI] [PubMed] [Google Scholar]
- 95.Clark DA, Chaouat G. Regulatory t cells and reproduction: how do they do it? J Reprod Immunol. 2012;96:1–7. doi: 10.1016/j.jri.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 96.Yin Y, Han X, Shi Q, Zhao Y, He Y. Adoptive transfer of CD4+CD25+ regulatory T cells for prevention and treatment of spontaneous abortion. Eur J Obstet Gynecol Reprod Biol. 2012;161:177–181. doi: 10.1016/j.ejogrb.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 97.Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012;150:29–38. doi: 10.1016/j.cell.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rowe JH, Ertelt JM, Xin L, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature. 2012;490:102–106. doi: 10.1038/nature11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Somerset DA, Zheng Y, Kilby MD, Sansom DM, Drayson MT. Normal human pregnancy is associated with an elevation in the immune suppressive CD25+ CD4+ regulatory T-cell subset. Immunology. 2004;112:38–43. doi: 10.1111/j.1365-2567.2004.01869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sasaki Y, Sakai M, Miyazaki S, Higuma S, Shiozaki A, Saito S. Decidual and peripheral blood CD4+CD25+ regulatory T cells in early pregnancy subjects and spontaneous abortion cases. Mol Hum Reprod. 2004;10:347–353. doi: 10.1093/molehr/gah044. [DOI] [PubMed] [Google Scholar]
- 101.Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas de St Groth B, Nanan R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol. 2009;183:7023–7030. doi: 10.4049/jimmunol.0901154. [DOI] [PubMed] [Google Scholar]
- 102.Winger EE, Reed JL. Low circulating CD4(+) CD25(+) Foxp3(+) T regulatory cell levels predict miscarriage risk in newly pregnant women with a history of failure. Am J Reprod Immunol. 2011;66:320–328. doi: 10.1111/j.1600-0897.2011.00992.x. [DOI] [PubMed] [Google Scholar]
- 103.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 104.Robertson SA, Guerin LR, Moldenhauer LM, Hayball JD. Activating T regulatory cells for tolerance in early pregnancy - the contribution of seminal fluid. J Reprod Immunol. 2009;83:109–116. doi: 10.1016/j.jri.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 105.Robertson SA, Guerin LR, Bromfield JJ, Branson KM, Ahlstrom AC, Care AS. Seminal fluid drives expansion of the CD4+CD25+ T regulatory cell pool and induces tolerance to paternal alloantigens in mice. Biol Reprod. 2009;80:1036–1045. doi: 10.1095/biolreprod.108.074658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Remes Lenicov F, Rodriguez Rodrigues C, Sabatte J, Cabrini M, Jancic C, Ostrowski M, Merlotti A, Gonzalez H, Alonso A, Pasqualini RA, Davio C, Geffner J, Ceballos A. Semen promotes the differentiation of tolerogenic dendritic cells. J Immunol. 2012;189:4777–4786. doi: 10.4049/jimmunol.1202089. [DOI] [PubMed] [Google Scholar]
- 107.Ramhorst R, Fraccaroli L, Aldo P, Alvero AB, Cardenas I, Leiros CP, Mor G. Modulation and recruitment of inducible regulatory T cells by first trimester trophoblast cells. Am J Reprod Immunol. 2012;67:17–27. doi: 10.1111/j.1600-0897.2011.01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balandya E, Wieland-Alter W, Sanders K, Lahey T. Human seminal plasma fosters CD4(+) regulatory T-cell phenotype and transforming growth factor-beta1 expression. Am J Reprod Immunol. 2012;68:322–330. doi: 10.1111/j.1600-0897.2012.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Douagi I, Andre I, Ferraz JC, Cumano A. Characterization of T cell precursor activity in the murine fetal thymus: evidence for an input of T cell precursors between days 12 and 14 of gestation. Eur J Immunol. 2000;30:2201–2210. doi: 10.1002/1521-4141(2000)30:8<2201::AID-IMMU2201>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 110.Friedberg SH, Weissman IL. Lymphoid tissue architecture. II. Ontogeny of peripheral T and B cells in mice: evidence against Peyer’s patches as the site of generation of B cells. J Immunol. 1974;113:1477–1492. [PubMed] [Google Scholar]
- 111.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 112.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burlingham WJ, Grailer AP, Heisey DM, Claas FH, Norman D, Mohanakumar T, Brennan DC, de Fijter H, van Gelder T, Pirsch JD, Sollinger HW, Bean MA. The effect of tolerance to noninherited maternal HLA antigens on the survival of renal transplants from sibling donors. N Engl J Med. 1998;339:1657–1664. doi: 10.1056/NEJM199812033392302. [DOI] [PubMed] [Google Scholar]
- 114.Darrasse-Jeze G, Marodon G, Salomon BL, Catala M, Klatzmann D. Ontogeny of CD4+CD25+ regulatory/suppressor T cells in human fetuses. Blood. 2005;105:4715–4721. doi: 10.1182/blood-2004-10-4051. [DOI] [PubMed] [Google Scholar]
- 115.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, Craig AS, Schaffner W, Zansky SM, Gershman K, Stefonek KR, Albanese BA, Zell ER, Schuchat A, Schrag SJ. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA. 2008;299:2056–2065. doi: 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 116.Khadilkar V, Tudehope D, Fraser S. A prospective study of nosocomial infection in a neonatal intensive care unit. J Paediatr Child Health. 1995;31:387–391. doi: 10.1111/j.1440-1754.1995.tb00843.x. [DOI] [PubMed] [Google Scholar]
- 117.Siegrist CA. Vaccination in the neonatal period and early infancy. Int Rev Immunol. 2000;19:195–219. doi: 10.3109/08830180009088505. [DOI] [PubMed] [Google Scholar]
- 118.Siegrist CA. Neonatal and early life vaccinology. Vaccine. 2001;19:3331–3346. doi: 10.1016/s0264-410x(01)00028-7. [DOI] [PubMed] [Google Scholar]
- 119.Garcia AM, Fadel SA, Cao S, Sarzotti M. T cell immunity in neonates. Immunol Res. 2000;22:177–190. doi: 10.1385/IR:22:2-3:177. [DOI] [PubMed] [Google Scholar]
- 120.Adkins B. Development of neonatal Th1/Th2 function. Int Rev Immunol. 2000;19:157–171. doi: 10.3109/08830180009088503. [DOI] [PubMed] [Google Scholar]
- 121.Adkins B. Neonatal T cell function. J Pediatr Gastroenterol Nutr. 2005;40 (Suppl 1):S5–7. doi: 10.1097/00005176-200504001-00004. [DOI] [PubMed] [Google Scholar]
- 122.Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 2009;30:585–591. doi: 10.1016/j.it.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, McCune JM. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330:1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Havran WL, Allison JP. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature. 1988;335:443–445. doi: 10.1038/335443a0. [DOI] [PubMed] [Google Scholar]
- 126.Coltey M, Bucy RP, Chen CH, Cihak J, Losch U, Char D, Le Douarin NM, Cooper MD. Analysis of the first two waves of thymus homing stem cells and their T cell progeny in chick-quail chimeras. J Exp Med. 1989;170:543–557. doi: 10.1084/jem.170.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Herzenberg LA. Toward a layered immune system. Cell. 1989;59:953–954. doi: 10.1016/0092-8674(89)90748-4. [DOI] [PubMed] [Google Scholar]
- 128.Lalor PA, Stall AM, Adams S, Herzenberg LA. Permanent alteration of the murine Ly-1 B repertoire due to selective depletion of Ly-1 B cells in neonatal animals. Eur J Immunol. 1989;19:501–506. doi: 10.1002/eji.1830190314. [DOI] [PubMed] [Google Scholar]
- 129.Havran WL, Allison JP. Origin of Thy-1+ dendritic epidermal cells of adult mice from fetal thymic precursors. Nature. 1990;344:68–70. doi: 10.1038/344068a0. [DOI] [PubMed] [Google Scholar]
- 130.Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, Weissman IL. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. 1990;62:863–874. doi: 10.1016/0092-8674(90)90262-d. [DOI] [PubMed] [Google Scholar]
- 131.Kantor AB, Stall AM, Adams S, Herzenberg LA, Herzenberg LA. Differential development of progenitor activity for three B-cell lineages. Proc Natl Acad Sci U S A. 1992;89:3320–3324. doi: 10.1073/pnas.89.8.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol. 2006;7:293–301. doi: 10.1038/ni1301. [DOI] [PubMed] [Google Scholar]
- 133.Le Douarin NM, Jotereau FV. Tracing of cells of the avian thymus through embryonic life in interspecific chimeras. J Exp Med. 1975;142:17–40. doi: 10.1084/jem.142.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jotereau FV, Le Douarin NM. Demonstration of a cyclic renewal of the lymphocyte precursor cells in the quail thymus during embryonic and perinatal life. J Immunol. 1982;129:1869–1877. [PubMed] [Google Scholar]
- 135.Hardy RR, Hayakawa K, Haaijman J, Herzenberg LA. B-cell subpopulations identifiable by two-color fluorescence analysis using a dual-laser FACS. Ann N Y Acad Sci. 1982;399:112–121. doi: 10.1111/j.1749-6632.1982.tb25667.x. [DOI] [PubMed] [Google Scholar]
- 136.Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly-1 B cells: genetic control, autoantibody production, increased lambda light chain expression. Eur J Immunol. 1986;16:450–456. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- 137.Herzenberg LA, Kantor AB, Herzenberg LA. Layered evolution in the immune system. A model for the ontogeny and development of multiple lymphocyte lineages. Ann N Y Acad Sci. 1992;651:1–9. doi: 10.1111/j.1749-6632.1992.tb24588.x. [DOI] [PubMed] [Google Scholar]
- 138.McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241:1632–1639. doi: 10.1126/science.241.4873.1632. [DOI] [PubMed] [Google Scholar]
- 139.Namikawa R, Weilbaecher KN, Kaneshima H, Yee EJ, McCune JM. Long-term human hematopoiesis in the SCID-hu mouse. J Exp Med. 1990;172:1055–1063. doi: 10.1084/jem.172.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]