

Abstract
Although it is widely assumed that species orthologs of hormone-responsive G protein-coupled receptors will be activated by the same endogenously produced ligand(s), variation in potency, particularly in cases in which more than 1 receptor responds to the same hormone, can result in challenges in defining the contribution of individual receptors in different species. This can create considerably greater issues when using synthetic chemical ligands and, in some cases, may result in a complete lack of efficacy of such a ligand when used in animal models of pathophysiology. In man, the concept that distinct responses of individuals to medicines may reflect differences in the ability of such drugs to bind to or activate single nucleotide polymorphism variants of receptors is more established as a concept but, in many cases, clear links between such variants that are associated with disease phenotypes and substantial differences in receptor ligand pharmacology have been more difficult to obtain. Herein we consider each of these issues for the group of free fatty acid receptors, FFA1-FFA4, defined to be activated by free fatty acids of varying chain length, which, based on their production by 1 tissue or location and action in distinct locations, have been suggested to possess characteristics of hormones.
In recent years it has become apparent that a wide range of biomolecules previously considered simply as intermediates of metabolic activity also act as key sensors of metabolic state and do so, at least in part, by activating members of the family of G protein-coupled receptors (GPCRs) (1, 2). This is true for molecules derived from each of dietary carbohydrate, lipid, and fiber, and on this basis, food had been described as a hormone (3). In many ways the capacity of simple biomolecules derived from the catabolism of food to signal nutritional status is both obvious and provides the most immediate means to initiate feedback loops. Moreover, the production of, for example, short-chain fatty acids (SCFAs) via fermentation of dietary fiber by the intestinal microbiota fulfills a key criterion for definition as a hormone because, as well as acting directly in the gut in which they are generated, these molecules function on cell types ranging from adipocytes to immune cells after transport throughout the systemic circulation.
The low potency of a number of these biomolecules at the GPCRs at which they are now considered to be the true endogenous regulators meant that their initial pairing with previously orphan GPCRs was met with a degree of skepticism because the potencies of the metabolic ligands to induce GPCR activation and therefore produce the modulation of secondary messenger levels and physiological function were much lower than is the case for many traditional circulating hormones and neuropeptides. For example, in initial efforts to identify ligands that activate the receptors GPR41 and GPR43 (now redesignated FFA3 and FFA2, respectively) (4), it was the recognition that the common feature of a number of apparently active ligands that shared little or no chemical similarity was that they were all present as acetate salts (5). This subsequently resulted in an appreciation that acetate, and other SCFAs including propionate and butyrate, activated these 2 GPCRs directly and were indeed the likely endogenously generated ligands of these receptors. Although the potency of these molecules at both FFA2 and FFA3 is indeed modest, this is only to be expected, given their small size and resultantly low binding energy (6), and, of key importance, high concentrations of the SCFAs, particularly acetate, are present in both the blood (7) and particularly in the gut lumen (8). This is consistent with steady-state levels of these ligands being sufficient to cause a degree of receptor activation and with changes in SCFA levels being able to modify cellular responsiveness.
The free fatty acid receptors
The human genome encodes more than 800 GPCRs and approximately 350 of these have either been shown or are believed to be activated by endogenously produced ligands (9). Given the key roles of many of these in controlling major physiological responses, the development of synthetic chemical ligands that act as either agonists or antagonists of specific GPCRs has been integral to understanding the functions of individual receptors. Furthermore, as an extension of this, ligands at many GPCRs are major components of our armorium of medicines to mask or mitigate symptoms of disease.
Among the large number of GPCRs that respond to metabolic intermediates, 4 distinct receptors are currently accepted and defined by their ability to bind and to be activated by free fatty acids of varying chain length (4). Free fatty acid receptor (FFA)-1, FFA2, and FFA3 form a highly related family group. The genes encoding them are closely linked at chromosome 19q13.1 in man (4) and therefore presumably developed from a single common ancestor via gene duplication. Alignment of the protein sequences of these receptors, linked to an appreciation that each is activated by the relevant fatty acids but not the corresponding amides, suggested that conserved, positively charged residues within the 7-transmembrane helical domains that provide the common architectural feature of GPCRs, and located close to the extracellular surface, would be central to binding of the fatty acid ligands. Mutagenesis studies have defined the central role of a pair of arginine residues, 1 in transmembrane domain V and 1 in transmembrane domain VII, in coordinating the carboxylate moiety of the fatty acids (10, 11). Moreover, a conserved histidine residue in transmembrane domain IV and a second histidine (in FFA2 and FFA3, but an asparagine in FFA1) in transmembrane domain VI, also contribute to recognition of the carboxylate (10, 11). By contrast, FFA4 (previously designated GPR120), despite binding and being activated by an overlapping group of medium- and longer-chain fatty acids as FFA1, is not closely related in sequence to the other fatty acid receptors. However, as with FFA1, an arginine residue, in this case close to the extracellular face of transmembrane domain II, provides the interaction point for the carboxylate of the fatty acids (12, 13).
The expression pattern of free fatty acid receptors
Even for poorly characterized GPCRs, expression patterns, at least as defined by the presence of appropriate mRNA transcripts, have provided suggestions of possible functional roles and hinted at potential disease associations. Indeed, studies based on transcript expression or promoter activity have been informative for the FFA family members as, for example, the high expression of FFA1 mRNA in pancreatic islets and in various enteroendocrine cells of the gut has been integral in identifying this receptor as a target for the treatment of type 2 diabetes (14, 15). Similarly, expression of FFA4 transcript in islets, gut, adipocytes, and macrophages (16–18) has suggested that this might also be an equally appropriate target for the control of diabetes and hyperglycemia. Furthermore, the identification of the expression of the FFA2 transcript in the lower gut and colon as well as in a range of immune cells (5) has resulted in this receptor being considered as a possible target for inflammatory diseases of the bowel. By contrast, although FFA3 transcript is expressed in a number of immune cell types, it is present in many of these at relatively low and similar levels. Perhaps because of this, the therapeutic potential of targeting FFA3 is much less clear cut or discussed. Although expression studies based on the receptor transcript have been important, a need still remains for the development of immunological reagents that will allow for direct identification of FFA receptor protein expression in native cells and tissues. At the current time, such antibodies are generally of both limited availability and questionable specificity.
Synthetic ligands for free fatty acid receptors
Knockout mouse models have been described for each of FFA1-FFA4 (19–22), and these have both provided insight into the function of each receptor and suggested disease areas that might be treated by small molecule pharmacological medicines. However, the overlap of function of fatty acids that activate FFA2 and FFA3 (23) and, similarly, of those that activate FFA1 and FFA4 (24) means that selective synthetic ligands are required to fully understand the roles of each receptor in human cells and tissues and in animal models other than mouse.
At the current time, a surprisingly limited set of such tool compounds are available to the research community. Partially because FFA1 has attracted the most attention as a therapeutic target, there is the greatest number of ligands available from commercial sources for this receptor. These include the agonists GW9508 (25) and TUG-424 (26) as well as the antagonist GW1100 (25). By contrast, markedly selective orthosteric ligands described in the primary literature for FFA2, including the agonist compound 1 [3-benzyl-4-(cyclopropyl-[4-(2,5-dichlorophenyl)thiazol-2-yl]amino)-4-oxobutanoic acid] (27) and the antagonist CATPB [(S)-3-(2-(3-chlorophenyl)acetamido)-4-[4-(trifluoromethyl)phenyl]butanoic acid] (27, 28) are not currently available from commercial sources but were synthesized for the published studies from details provided in the patent literature. No selective ligands for FFA3 have yet been published, whereas the only high-potency and highly selective ligand for FFA4, TUG-891, has, so far, been described in a single publication (13) and very recently has become available through commercial sources. Despite these current limitations, the developing interest in the potential therapeutic targeting of these receptors is likely to result in the wider availability of ligands in the future.
Species ortholog and single-nucleotide polymorphism variation in receptor pharmacology
Although species orthologs of many GPCRs are highly similar in their ability to bind and respond to the endogenously produced ligands because there has been evolutionary pressure to maintain function, there are a number of examples in which physiological adaptation has resulted in variation in potency. An interesting example of this can be seen for FFA2. In comparisons of the responsiveness of human and bovine FFA2 with fatty acids of varying chain length, Hudson et al (29) noted the response of the bovine ortholog to longer chain-length fatty acids than the human receptor and the lower potency of the bovine receptor for acetate. Detailed analysis of these characteristics identified a single cysteine-glycine amino acid variation that was central to these differences (29). Moreover, the alignment of FFA2 from a wide range of species now available from genome sequencing projects indicated that this variation is seen in all suborder Ruminantia species including the goat, sheep, yak, and zebu. This suggests that FFA2 from these species will likely show the same, distinct pharmacology as recorded for the cow, although it may be some time before this prediction is tested directly. Interestingly, this may reflect a physiological adaptation to diet as the rumen of cow and, particularly, sheep contains very high concentrations of SCFAs (30). Moreover, as discussed later, there is marked variation in potency and/or affinity at each of FFA1 and FFA2 from different species of various synthetic ligands.
In addition to variations in ligand potency between species orthologs, the possibility also exists that species orthologs of a receptor may couple with differing efficiencies to their downstream effectors, thus adding another level of complexity that should be considered. For example, we find that although both the human and bovine orthologs of FFA2 efficiently recruit β-arrestin-2 as measured in a bioluminescence resonance energy transfer-based assay, we have been unable to detect β-arrestin-2 recruitment to either the mouse or rat orthologs of FFA2 in this same assay (unpublished observation, B Hudson, and G Milligan). Although it is unclear whether this represents a fundamental difference in the ability of the species orthologs to recruit β-arrestin-2 or instead simply a limitation of the assay format, this clearly highlights that verifying the activity of ligands across multiple assay end points at species orthologs of these receptors is critical. An appreciation and an understanding of the basis of ligand selectivity between species are central to defining the issues that might limit interpretation of the effectiveness or otherwise of various ligands in animal models of disease or in cells and tissues isolated from different species. A number of other GPCRs display marked variation between species in response to various synthetic ligands, and recent reviews of this issue on both the receptors for histamine (31) and GPR35 (32) provide useful insight.
Even within species, and of particular relevance to the development and prescription of effective medicines in man, variation in the sequence of the same GPCR between individuals, resulting from nonsynonomous single nucleotide polymorphisms (SNPs), can result in substantial differences in pharmacology and function of the receptor. The contribution of this to stratified medicine and the successful use of therapeutics has been dominated to date by variation in the function and capacity of enzymes involved in drug metabolism (33). However, variations in pharmacology can also contribute, and GPCRs are often linked to disease via such SNPs in genome-wide association studies (34).
Free fatty acid receptor 1
Of the members of the free fatty acid receptor family, it is FFA1 that has received by far the most attention to date. This is due in large part to its high level expression in pancreatic islet β-cells and its ability to enhance glucose stimulated insulin secretion (GSIS) (14). In addition to this well-established direct effect on insulin secretion in β-cells, FFA1 agonists have also been found to stimulate glucagon-like peptide-1 (GLP-1) release from enteroendocrine cells (15, 35), and it has been demonstrated that at least part of the beneficial effect of FFA1 agonists on glycemic control involves indirect actions mediated by GLP-1 (36). Not surprisingly, given its effects on GSIS and GLP-1, there has been significant interest in FFA1 as a novel target for the treatment of type 2 diabetes, and indeed, at least 1 FFA1 agonist, TAK-875, has now progressed through phase II clinical trials with generally favorable results (37, 38). However, despite its promise as a therapeutic target, a number of factors, including SNPs in human FFA1, conflicting results from knockout studies, differences in species ortholog pharmacology, and differential biological outcomes of FFA1 agonists between species have presented challenges and complicate the use of animal models to study this receptor.
FFA1 polymorphisms
An examination of the 1000 genomes project database (39) identifies 29 missense variants of human FFA1. Of these, only Arg211His has a minor allele frequency (MAF) greater than 1%, in which the arginine (Arg) variant is the minor allele with an MAF of 18.2% (Table 1). One early study examining the effect of the Arg211His polymorphism in Japanese men indicated that individuals homozygous for the less common Arg variant had reduced serum insulin and β-cell function, suggesting that this polymorphism may contribute to insulin secretion capacity (40). However, a second study found that this polymorphism did not contribute to insulin release assessed through oral glucose tolerance tests (41), and both studies found no differences in allelic frequency between healthy and diabetic individuals. Furthermore, at least 2 in vitro studies have now examined the pharmacology of the Arg211His variant and found no measurable effect on FFA1 function (41, 42).
Table 1.
Open Reading Frame SNPs of Human FFA1-FFA4 That Have Been Linked With Disease or Show Minor Allele Frequency Greater Than 1%
| Variant | Minor Allele | MAF, % |
|---|---|---|
| FFA1 | ||
| Asp175Glu | Glu | 0.2 |
| Gly180Ser | Ser | 0.2 |
| Arg211His | Arg | 18.2 |
| FFA2 | ||
| Ile46Val | Val | 1.0 |
| Leu211His | His | 3.6 |
| FFA3 | ||
| Gln7His | His | 5.6 |
| Arg45His | His | 18.5 |
| FFA4 | ||
| Arg67Cys | Cys | 14.9 |
| Arg254His/Arg270Hisa | His | 0.7 |
Polymorphism is at position 254 in short isoform of FFA4 and 270 in long isoform.
In addition to the relatively common Arg211His polymorphism, several rare human polymorphisms of FFA1 have also been examined. For example, 1 study reported that the MAF of a Gly180Ser polymorphism continuously increased from nonobese (0.42%) to moderately obese (1.07%) to severely obese (2.60%) individuals (43). Oral glucose tolerance testing of individuals with this polymorphism indicated that insulin secretion was significantly reduced in individuals with the minor Ser allele, whereas in vitro experiments suggested that this FFA1 variant resulted in a receptor with greatly reduced function in a Ca2+ mobilization assay (43). However, a subsequent study failed to reproduce this reduced function in vitro (42). A second rare polymorphism, Asp175Asn, has also been described to reduce the efficacy of FFA1 in vitro (41), although as with the other FFA1 polymorphisms, contradictory results found that the pharmacology of this variant is not different from the wild type (42). These studies have not, however, examined any potential variation in function of the synthetic ligands that are currently undergoing clinical trials, eg, TAK-875, and it would certainly be of considerable interest to examine this directly.
In addition to the missense polymorphisms, there are also 2 SNPs upstream of the FFA1 gene (rs1978013 and rs1978014) that have been associated with β-cell function (44). Although this study did not find a statistical link between either of these SNPs and type 2 diabetes alone, the haplotype consisting of the T and G alleles did confer protection (44). Taken together, although several studies have examined polymorphisms of FFA1, there is at present no clear evidence indicating that any of these significantly alter FFA1 function or increase risk of obesity and type 2 diabetes.
Species ortholog variation in pharmacology at FFA1
Although the most studied of the free fatty acid GPCRs, little work has examined potential differences in the pharmacology between human and rodent orthologs of FFA1 in detail and the possible implications of this for preclinical assessment of FFA1 ligands. In cases in which agonists have been compared between the human and rodent, perhaps not surprisingly, given that drug development programs are typically designed to optimize ligands for the human receptor, most synthetic agonists of FFA1 have been found to be between 2- and 10-fold more potent in functional assays at human FFA1 compared with either the mouse or rat orthologs (45–47). One exception is a series of thiazolidinedione ligands developed by Merck, which were found to be 4- to 6-fold more potent at the mouse than the human receptor (48). Although binding assays to obtain more direct measures of ligand affinity have been challenging with the free fatty acid receptors, at least in part due to the marked lipophilicity of many of the available ligands, such studies have been carried out with the clinical candidate TAK-875, and indeed, the affinity of this compound was found to be approximately 3.5-fold higher at the human (dissociation constant = 0.038 μM) than at the rat (0.14 μM) FFA1 (49).
At least 2 chemical series of FFA1 antagonists have also now been described; one by GlaxoSmithKline (GW1100) (25) and the other by Pfizer (50). Although both series have been successfully used to block FFA1 responses in rodent insulinoma cell lines (25, 51), we find that, at least in a Ca2+ mobilization assay, although GW1100 has similar activity at human and rodent orthologs, the Pfizer compounds are significantly human selective (Figure 1), suggesting that there are likely to be significant species ortholog issues to be considered when using these FFA1 antagonists in nonhuman systems.
Figure 1.
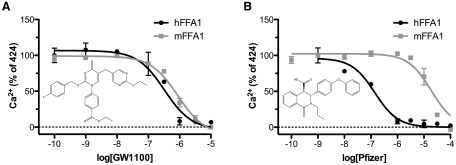
Species ortholog selectivity of FFA1 antagonists. Concentration-response inhibition curves were generated for either GW1100 (A) (25), or an antagonist from the Pfizer chemical series (B) (50) against a 300-μM concentration of TUG-424 (26) using a Ca2+ mobilization assay in Flp-In T-REx 293 cells designed to express either human (black lines) or mouse (gray lines) FFA1. Compound structures for the antagonists are shown as inserts. For GW1100, similar pIC50 values are observed for human (6.48 ± 0.17) and mouse (6.05 ± 0.07) orthologs, whereas, in contrast, the Pfizer compound is approximately 100-fold selective for the human ortholog, with pIC50 values of 6.85 ± 0.12 at human compared with 4.73 ± 0.09 at mouse. hFFA1, human FFA1; mFFA1, mouse FFA1.
In addition to differences in basic ligand pharmacology between species, several other factors have complicated the study of FFA1 function in rodent systems. For example, although TAK-875 has been found to suppress glucagon release from isolated pancreatic rat islets, in the same study, it was found to have no such effect on glucagon secretion from human islets (52). Another factor that has greatly complicated the understanding of FFA1 function in rodent systems has been the fact that knockout mouse models of this receptor have often given confusing or conflicting results. In particular, although FFA1 knockout lines have generally confirmed that fatty acids mediate acute enhancement of GSIS through FFA1 (53), knockout models have been less clear on whether the chronic inhibitory effect of fatty acids on GSIS also reflects activation of FFA1. In one study FFA1 knockout mice were protected from the negative effects of chronic exposure to fatty acids (53), whereas other studies have found this not to be the case (19, 48, 54, 55). These conflicting results led to speculation that either FFA1 agonists or antagonists might be useful therapeutically. This controversy highlights the need for selective pharmacological tool compounds with good activity at rodent receptor orthologs suitable for in vivo work to complement and confirm the results of knockout studies.
Free fatty acid receptor 2
Both FFA2 and FFA3 are activated by binding of SCFAs, although there are distinct differences in potency of the SCFAs at these receptors (5, 29). FFA2 is activated by high micromolar or millimolar SCFA concentrations, most notably acetate (C2) or propionate (C3), and substantially less so by caproate (C6) and formate (C1). A rank order of SCFA potency at FFA2 has been described as C2 = C3 > C4 > C6 > C5 > C1 (5, 57).
FFA2 is most highly expressed in immune cells, including neutrophils, monocytes, and polymorphonuclear cells (5), stimulating immune cell recruitment in inflammatory responses (57, 58). It is also present in adipose tissue and primary adipocytes, in which it promotes adipogenesis by increasing lipid accumulation (59) and inhibiting lipolysis (60). FFA2 expression in the enteroendocrine L cells mediates SCFA-induced GLP-1 release (61). FFA2 is also present in pancreatic islets, specifically the β-cells, although its role in β-cell function remains unclear (62). Studies with FFA2 knockout mice have shown that these animals have improved glucose control, reduced body fat mass, and increased insulin sensitivity (63). However, in an acute model of colitis, 2 separate studies have reported conflicting results with FFA2 knockouts, in that one showed reduced (20) and the other showed heightened inflammatory responses (58). Altogether these observations indicate an importance of FFA2 function in the immune system and metabolism. Selective FFA2 ligands could therefore be of therapeutic benefit.
Free fatty acid receptor 3
FFA3 is activated preferentially by the SCFAs propionate (C3), butyrate (C4), and valerate (C5), with a rank order potency of C3 = C4 = C5 > C6 > C2 > C1 (5). Although FFA2 and FFA3 share the same endogenous ligands, ligand activation of these receptors initiates different signaling pathways. FFA3 couples solely to Gαi/o signaling, whereas FFA2 signals via Gαi/o and Gαq, pathways (4). FFA3 expression is widespread, but its function in many of these tissues is unknown. A recent study has uncovered a role for FFA3 expressed in the sympathetic ganglion in controlling energy expenditure in the fed state and during fasting (64). Sympathetic outflow is dysregulated in metabolic disorders such as obesity, thus making FFA3 a potential drug target for the treatment of such conditions.
Early FFA3 studies reported that the receptor is highly expressed in adipose tissue and in adipocyte cell lines (5). Furthermore, Xiong et al (65) demonstrated that propionate-induced leptin production from adipocytes is mediated by FFA3. These observations have since proved to be controversial, with many conflicting reports as to whether FFA3 and/or FFA2 is expressed in adipose tissue and adipocytes (21, 59, 66). A more recent study has now determined that leptin production in response to SCFAs is mediated by FFA2, not FFA3 (66). Data from FFA3−/− mouse studies have also generated conflicting observations in terms of the animals' glucose tolerance levels and body fat mass (21). It is evident that FFA3 knockdown or knockout can be associated with concomitant down-regulation of FFA2 expression (66), thus making data interpretation difficult. FFA3-selective ligands would therefore be useful tools in such investigations to dissect FFA2 vs FFA3 responses in tissues in which these receptors are coexpressed.
FFA3 has been shown to be expressed in pancreatic β-cells and the insulin-producing β-cell lines, MIN6, NIT-1, and βTC-6 (62, 66). A patent detailing a group of FFA3 agonists and antagonists/inverse agonists cites that these FFA3 ligands can modulate β-cell function, eliciting either inhibitory (FFA3 agonists) or stimulatory (FFA3 antagonists/inverse agonists) effects on insulin secretion (67). The therapeutic potential of FFA3-selective ligands in metabolic disorders may therefore be an encouraging prospect.
Species ortholog variation in pharmacology at FFA2 and FFA3
The development of novel selective ligands for FFA2 and FFA3 has been complicated further by recent observations describing pharmacological variation between the human and rodent orthologs of these receptors (28). Rodents are routinely used in GPCR drug development to assess the function and efficacy of potential therapeutic drugs, which means that variation in pharmacology between species is likely to confound interpretation of potential drug efficacy. Mammalian species GPCR orthologs are assumed to respond to the same ligands. However, for modulatory GPCRs such as FFA2 and FFA3, which respond to a number of ligands, the potencies and rank order of function may differ between species, which is certainly true for the bovine FFA2 ortholog, as described earlier (29).
For the human orthologs, acetate (C2) is reported to be approximately 20-fold more potent at human FFA2 than human FFA3, whereas propionate (C3) is equally potent at the 2 receptors. On this basis, C2 has been described as FFA2 selective, whereas C3 is considered nonselective. In a recent pharmacological analysis of the mouse orthologs, we found significant differences in C2/C3 selectivity and ligand potencies from those of the human orthologs (28). In [35S]GTPγS binding assays, C2 was found to be nonselective at mouse FFA2 and mouse FFA3, whereas C3 was approximately 12-fold more selective for mouse FFA3 over mouse FFA2. This altered selectivity to C2 and C3 was as a direct result of changes in SCFA potency at the mouse ortholog. The orthologs at which the SCFAs were most potent, human FFA2 and mouse FFA3, displayed high levels of constitutive activity, whereas the orthologs with lower SCFA potency, mouse FFA2 and human FFA3, showed limited constitutive activity. Constitutive activity and associated SCFA potency of the species orthologs was shown to be regulated by the absence (human FFA2 and mouse FFA3) or presence (mouse FFA2 and human FFA3) of an extracellular ionic lock interaction between a nonconserved acidic residue of the second extracellular loop and arginine residues within the orthosteric binding pocket (28). As proof of this, the ligand-independent activity of the species orthologs could be reversed by performing reciprocal mutations of the nonconserved acidic residue, which tended to yield corresponding changes in SCFA potency.
Acetate is often used in studies as a FFA2-selective agonist to differentiate between human FFA2 and human FFA3 signaling in cells that coexpress these receptors (57). It has also been used in rodent studies as an indicator of FFA2 activity (66). As we have now shown, C2 is actually nonselective between mouse FFA2 and mouse FFA3, so it is extremely difficult to assess the functional contributions of FFA2 vs FFA3 using endogenous ligands. Studies with knockout mice have proved more useful in defining FFA2 and FFA3 function, although it is clear that C2 cannot be used in these mouse models to imply FFA2-specific function. Furthermore, as described earlier, there have been some conflicting observations from knockout mouse models of FFA2 and FFA3, thus making it difficult to determine whether a positive or negative modulator of receptor function would be of therapeutic value. To address such issues, selective potent ligands that can effectively discriminate between FFA2 and FFA3 in both human and rodent experimental systems are required.
FFA2 polymorphisms
There are 40 FFA2 missense variants detailed in the 1000 genomes project database (39). The Leu211His allele is the most common verified variant with a MAF value of 3.6% (Table 1). At present there are no reports of this polymorphism being of functional significance or being associated with any disease/clinical phenotype. However, such a deleterious change in amino acid composition could possibly impact receptor function. Given its position within the receptor's third intracellular loop, the Leu211His variation would appear most likely to alter G protein coupling. Also of note is a rarer missense variant, Arg255Gln. The Arg residue at this position is a conserved polar residue at the upper face of transmembrane domain VII, which tethers SCFAs to their binding site. Expression of this minor allelic variant would therefore be anticipated to be unresponsive to endogenous ligands and hence behave as a knockout.
FFA3 polymorphisms
In addition to FFA1, FFA2, and FFA3, an additional, structurally related, GPCR sequence, GPR42, is encoded in tandem at the same position on human chromosome 19q13.1 (68). GPR42 is thought to have arisen by tandem duplication of FFA3 (GPR41). FFA3 and GPR42 share 98% sequence identity, differing at only 6 amino acid positions (Figure 2), but GPR42 has been reported to be inactive and a potential pseudogene (5). Amino acid position 174 is responsible for determining functionality of FFA3 and GPR42 because mutation of Arg174 (found in FFA3) to Trp174 (found in GPR42) makes the receptor nonresponsive to SCFAs (5). The reciprocal mutation in GPR42 is sufficient to restore partial functional responsiveness. Although originally thought to be an inactive pseudogene, the 6 amino acid differences between FFA3 and GPR42 are now known to be polymorphic variants (69).
Figure 2.

Comparisons of the sequence variation of FFA3 and GPR42. Amino acid sequences corresponding to human FFA3 and human GPR42 were aligned using the ClustalW algorithm. The 6 polymorphic variants are highlighted in boxes (solid). Predicted transmembrane regions are boxed (dashed). Accession numbers are as follows: FFA3, O14843; and GPR42, O15529.
A genotyping study of the GPR41 FFA3 and GPR42 alleles from a population of more than 100 subjects revealed that the functional Arg174 allele was detected in 61% of those subjects at the GPR42 locus (69). Based on this study, it is likely that a substantial proportion of the population encode an active GPR42 variant. Coexpression of FFA3 with an active variant of GPR42 could therefore influence an individual's response to FFA3 ligands. Furthermore, the discovery that GPR42 may encode a functional gene has important implications for previous FFA3 expression studies. Techniques for determining mRNA expression, including Northern blot analysis, TaqMan, and RT-PCR, would fail to discriminate between FFA3 and GPR42 mRNA transcripts. Thus, previously reported FFA3 mRNA expression data could represent composites of GPR41 and GPR42 expression.
The 1000 genomes project database (39) also details 51 missense variants of FFA3. The most common allelic variant is Arg45His, with a MAF score of 18.5% (Table 1). Amino acid position 45 is one of the nonconsensus residues between FFA3 and GPR42. The His45 minor allele of FFA3 was reported in 1 subject in the original FFA3/GPR42 genetic study (69). This allele is also found in a number of expressed sequence tag clones (GenBank accession number CF147780). It is likely that the His45 allele is a benign variant because interchange with the GPR42 allele at this site, Arg45Cys, did not alter FFA3 function as determined previously (5).
Two relatively rare described missense variants, Asp158Asn and Arg185Gln, would be expected to have significant consequential effects on FFA3 function. We have previously shown that FFA3 residue Asp158 is involved in forming an ionic lock interaction between arginine residues in the SCFA binding pocket that limit ligand-independent activation of the receptor (28). The mouse ortholog, which displays marked constitutive activity, has an Asn residue at this position. Expression of the Asp158Asn allele in humans would therefore yield a FFA3 receptor with increased constitutive activity and potential ligand potency for the endogenous agonist ligands. Arg185 of FFA3 is conserved between FFA1-FFA3 and is critical for ligand recognition and anchoring of the carboxylate moiety of the SCFAs to the binding pocket (10, 11). Mutation at this site has been shown to ablate SCFA interactions. On this basis, the Arg185Gln variant would be anticipated to be nonresponsive to SCFAs.
Free fatty acid receptor 4
Although, as noted earlier, lacking substantial homology with the other members of the FFA receptor family, FFA4 is activated by similar long-chain fatty acid ligands as FFA1. Although FFA4 expression has been reported in pancreatic islets and β-cell lines (18, 62), unlike FFA1, there is currently no evidence that FFA4 directly stimulates insulin secretion in these cells. Instead, FFA4 activation appears to affect islet function and insulin secretion through 2 independent mechanisms: 1) stimulation of GLP-1 release from gut enteroendocrine cells (16) and 2) protecting against cell death in pancreatic islet β-cells (18). In addition to these indirect effects on islet function and insulin secretion, FFA4 is also expressed in immune cells, shows antiinflammatory properties, and appears to protect against insulin resistance (17). Considering each of these factors, FFA4 has received increasing interest in recent years as a possible therapeutic target for type 2 diabetes and obesity.
FFA4 polymorphisms and isoforms
At least 31 missense polymorphisms are described for FFA4 in publically available databases (39). However, of these, only the Arg67Cys polymorphism shows a MAF greater than 1% (14.9%) (Table 1). One study examining this polymorphism found no difference in pharmacology between it and wild-type FFA4 and only a weak trend toward an association between the polymorphism and obesity in humans [odds ratio 1.16] (22). In contrast, the same study also identified a second, less common, Arg254His (Arg270His in the long isoform of FFA4; see later text) polymorphism (MAF = 0.7%–3%) that appeared to be strongly associated with obesity (odds ratio odds ratio 1.62). Examination of the in vitro properties of this polymorphism indicated that the less common His allele greatly reduced receptor function (22). Findings that individuals with a reduced functional form of FFA4 are prone to obesity are further supported by animal work demonstrating that mice lacking FFA4 develop more severe obesity and insulin resistance than their wild-type counterparts after being fed a high-fat diet (22).
In addition to the polymorphisms, 2 splice variant isoforms of FFA4 have also been described, varying by the presence or absence of a 16-amino acid insertion in the third intracellular loop (70). Interestingly, the longer isoform is present only in humans, making the study of its function in rodent or other nonhuman systems challenging. Functionally, it has been found that although the short isoform of FFA4 couples in vitro to both G protein-dependent and β-arrestin pathways, the long isoform couples only to the β-arrestin response (12), indicating that the function of FFA4 may be different in humans from other species in tissues in which the long isoform is present. At present, although expression of FFA4-long has been shown in some tissues, particularly in the colon (71), the broader importance of this isoform and its full expression pattern in human tissues remain largely unknown.
Species ortholog variation at FFA4
At present there are very few described potent and selective synthetic ligands for FFA4. Of these, the only one currently described in the academic literature, TUG-891 [compound 43 (13)], was found to have relatively similar activities at human and mouse orthologs of FFA4. In addition to compounds reported in the academic literature, there are also several patents disclosing potent FFA4 agonists (56, 72, 73); however, few data are disclosed, and no information on the activity of these compounds at nonhuman orthologs of FFA4 is currently available. In our hands, testing a range of both endogenous fatty acids, FFA4 synthetic, and FFA1 synthetic ligands that also show activity at FFA4, we found the pharmacology of most tested ligands at the human and mouse orthologs of FFA4 to be quite similar in a β-arrestin-2 recruitment assay (Figure 3). Although this perhaps suggests species ortholog variation may be less of an issue for FFA4 than the other FFA receptors, the fact that many FFA1 selective ligands are less potent at mouse than human FFA1, but not at mouse than human FFA4, indicates that the FFA1 selectivity of these compounds will be reduced in mouse systems. Furthermore, the specific assay end point measured may influence the observed selectivity of these compounds, as for example at the human ortholog TUG-891 was found to be substantially more FFA4 selective when assessed by β-arrestin-2 recruitment than when assessed using Ca2+ mobilization assays (13). This highlights the critical importance of assessing these factors at the appropriate species orthologs across multiple assay end points before selecting FFA1 or FFA4 ligands for use in nonhuman cells and tissues to ensure that the studies actually target the receptor effectively and selectively.
Figure 3.
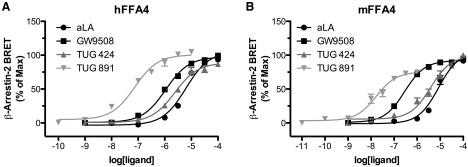
Comparison of FFA4 agonists at human and mouse orthologs. Concentration-response curves were generated for FFA4 agonists α-linolenic acid (aLA), GW9508 (25), TUG-424 (26), and TUG-891 (13) using a bioluminescence resonance energy transfer-based β-arrestin-2 recruitment assay in human embryonic kidney 293 cells transfected with either the human (A) or mouse (B) orthologs of FFA4. Although compounds displayed somewhat higher potency at the mouse ortholog, the rank order of potency of TUG-891 > GW9508 > TUG-424 > aLA was maintained between species. hFFA4, human FFA4; mFFA4, mouse FFA4.
Conclusions
Most currently studied SNP variants within the open reading frame of members of the FFA receptor group of human GPCRs generally appear to have rather modest effects on the pharmacology and response of these receptors. The potential exceptions to this are the Arg254His polymorphism of FFA4 and the functionality or otherwise of GPR42. However, to date, these issues have not been explored in detail for clinical candidate or even synthetic proof-of-concept ligands, instead centering largely on the function of the endogenously produced ligands. These topics will require significantly more attention as the FFA receptors become better appreciated and more fully validated therapeutic targets. Considerably wider variations in ligand pharmacology have been observed between species orthologs of both FFA1 and, particularly, FFA2, and considerable attention therefore needs to be given to assessing this issue before reaching conclusions on the contribution and role of these receptors in studies performed in nonhuman species. This is of particular relevance, given the importance of rodent models of both metabolic diseases and inflammatory conditions.
Acknowledgments
This work was supported by Grant 11-116196 from the Danish Council for Strategic Research, Grant 089600/Z/09 from the Wellcome Trust, Grant UGW854/11 from the Strategic Partnership on Food and Drink Science, Scottish Government Programme of Research (to G.M.), and the Canadian Institutes of Health Research (fellowship to B.D.H.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Arg
- arginine
- FFA
- free fatty acid receptor
- GLP-1
- glucagon-like peptide-1
- GPCR
- G protein-coupled receptor
- GSIS
- glucose stimulated insulin secretion
- MAF
- minor allele frequency
- SCFA
- short-chain fatty acid
- SNP
- single-nucleotide polymorphism.
References
- 1. Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov. 2012;11:603–619 [DOI] [PubMed] [Google Scholar]
- 2. Smith NJ. Low affinity GPCRs for metabolic intermediates: challenges for pharmacologists. Front Endocrinol (Lausanne) 2012;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ryan KK, Seeley RJ. Food as a hormone. Science. 2013;339:918–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008;60:405–417 [DOI] [PubMed] [Google Scholar]
- 5. Brown AJ, Goldsworthy SM, Barnes AA, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319 [DOI] [PubMed] [Google Scholar]
- 6. Schmidt J, Smith NJ, Christiansen E, et al. Selective orthosteric free fatty acid receptor 2 (FFA2) agonists: identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J Biol Chem. 2011;286:10628–10640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolever TM, Josse RG, Leiter LA, Chiasson JL. Time of day and glucose tolerance status affect serum short-chain fatty acid concentrations in humans. Metabolism. 1997;46:805–811 [DOI] [PubMed] [Google Scholar]
- 8. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272 [DOI] [PubMed] [Google Scholar]
- 10. Sum CS, Tikhonova IG, Neumann S, et al. Identification of residues important for agonist recognition and activation in GPR40. J Biol Chem. 2007;282:29248–29255 [DOI] [PubMed] [Google Scholar]
- 11. Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008;283:32913–32924 [DOI] [PubMed] [Google Scholar]
- 12. Watson SJ, Brown AJ, Holliday ND. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol. 2012;81:631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimpukade B, Hudson BD, Hovgaard CK, Milligan G, Ulven T. Discovery of a potent and selective GPR120 agonist. J Med Chem. 2012;55:4511–4515 [DOI] [PubMed] [Google Scholar]
- 14. Itoh Y, Kawamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176 [DOI] [PubMed] [Google Scholar]
- 15. Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57:2280–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirasawa A, Tsumaya K, Awaji T, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94 [DOI] [PubMed] [Google Scholar]
- 17. Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taneera J, Lang S, Sharma A, et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2011;16:122–134 [DOI] [PubMed] [Google Scholar]
- 19. Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes. 2008;57:2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bellahcene M, O'Dowd JF, Wargent ET, et al. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br J Nutr. 2012;31:1–10 [DOI] [PubMed] [Google Scholar]
- 22. Ichimura A, Hirasawa A, Poulain-Godefroy O, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483:350–354 [DOI] [PubMed] [Google Scholar]
- 23. Milligan G, Stoddart LA, Smith NJ. Agonism and allosterism: the pharmacology of the free fatty acid receptors FFA2 and FFA3. Br J Pharmacol. 2009;158:146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hudson BD, Smith NJ, Milligan G. Experimental challenges to targeting poorly characterized GPCRs: uncovering the therapeutic potential for free fatty acid receptors. Adv Pharmacol. 2011;62:175–218 [DOI] [PubMed] [Google Scholar]
- 25. Briscoe CP, Peat AJ, McKeown SC, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christiansen E, Urban C, Merten N, et al. Discovery of potent and selective agonists for the free fatty acid receptor 1 [FFA(1)/GPR40], a potential target for the treatment of type II diabetes. J Med Chem. 2008;51:7061–7064 [DOI] [PubMed] [Google Scholar]
- 27. Hudson BD, Due-Hansen ME, Christiansen E, et al. Defining the molecular basis for the first potent and selective orthosteric agonists of the FFA2 free fatty acid receptor. J Biol Chem. 2013;288:17296–17312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hudson BD, Tikhonova IG, Pandey SK, Ulven T, Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J Biol Chem. 2012;287:41195–41209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hudson BD, Christiansen E, Tikhonova IG, et al. Chemically engineering ligand selectivity at the free fatty acid receptor 2 based on pharmacological variation between species orthologs. FASEB J. 2012;26:4951–4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70:567–590 [DOI] [PubMed] [Google Scholar]
- 31. Strasser A, Wittmann HJ, Buschauer A, Schneider EH, Seifert R. Species-dependent activities of G-protein-coupled receptor ligands: lessons from histamine receptor orthologs. Trends Pharmacol Sci. 2013;34:13–32 [DOI] [PubMed] [Google Scholar]
- 32. Milligan G. Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol Sci. 2011;32:317–325 [DOI] [PubMed] [Google Scholar]
- 33. Zhou SF, Liu JP, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev. 2009;41:89–295 [DOI] [PubMed] [Google Scholar]
- 34. Insel PA, Tang CM, Hahntow I, Michel MC. Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim Biophys Acta. 2007;1768:994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiong Y, Swaminath G, Cao Q, et al. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol. 2013;369:119–129 [DOI] [PubMed] [Google Scholar]
- 36. Luo J, Swaminath G, Brown SP, et al. A Potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One. 2012;7:e46300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burant CF, Viswanathan P, Marcinak J, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–1411 [DOI] [PubMed] [Google Scholar]
- 38. Kaku K, Araki T, Yoshinaka R. Randomized, double-blind, dose-ranging study of TAK-875, a novel GPR40 agonist, in Japanese patients with inadequately controlled type 2 diabetes. Diabetes Care. 2013;36:245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogawa T, Hirose H, Miyashita K, Saito I, Saruta T. GPR40 gene Arg211His polymorphism may contribute to the variation of insulin secretory capacity in Japanese men. Metabolism. 2005;54:296–299 [DOI] [PubMed] [Google Scholar]
- 41. Hamid YH, Vissing H, Holst B, et al. Studies of relationships between variation of the human G protein-coupled receptor 40 Gene and type 2 diabetes and insulin release. Diabet Med. 2005;22:74–80 [DOI] [PubMed] [Google Scholar]
- 42. Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J Biol Chem. 2009;284:17527–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vettor R, Granzotto M, De Stefani D, et al. Loss-of-function mutation of the GPR40 gene associates with abnormal stimulated insulin secretion by acting on intracellular calcium mobilization. J Clin Endocrinol Metab. 2008;93:3541–3550 [DOI] [PubMed] [Google Scholar]
- 44. Kalis M, Levéen P, Lyssenko V, Almgren P, Groop L, Cilio CM. Variants in the FFAR1 gene are associated with beta cell function. PLoS One. 2007;2:e1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin DC, Zhang J, Zhuang R, et al. AMG 837: a novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents. PLoS One. 2011;6:e27270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Christiansen E, Due-Hansen ME, Urban C, et al. Free fatty acid receptor 1 (FFA1/GPR40) agonists: mesylpropoxy appendage lowers lipophilicity and improves ADME properties. J Med Chem. 2012;55:6624–6628 [DOI] [PubMed] [Google Scholar]
- 47. Christiansen E, Due-Hansen ME, Urban C, et al. Discovery of a potent and selective free fatty acid receptor 1 agonist with low lipophilicity and high oral bioavailability. J Med Chem. 2013;56:982–992 [DOI] [PubMed] [Google Scholar]
- 48. Tan CP, Feng Y, Zhou YP, et al. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes. 2008. 2008;57:2211–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Negoro N, Sasaki S, Mikami S, et al. Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist. ACS Med Chem Lett. 2010;1:290–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Humphries PS, Benbow JW, Bonin PD, et al. Synthesis and SAR of 1,2,3,4-tetrahydroisoquinolin-1-ones as novel G-protein-coupled receptor 40 (GPR40) antagonists. Bioorg Med Chem Lett. 2009;19:2400–2403 [DOI] [PubMed] [Google Scholar]
- 51. Christiansen E, Urban C, Grundmann M, et al. Identification of a potent and selective free fatty acid receptor 1 (FFA1/GPR40) agonist with favorable physicochemical and in vitro ADME properties. J Med Chem. 2011;54:6691–6703 [DOI] [PubMed] [Google Scholar]
- 52. Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther. 2011;340:483–489 [DOI] [PubMed] [Google Scholar]
- 53. Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258 [DOI] [PubMed] [Google Scholar]
- 54. Latour MG, Alquier T, Oseid E, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lan H, Hoos LM, Liu L, et al. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes. 2008;57:2999–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ma J, Novack A, Nashashibi I, et al. Aryl GPR120 receptor agonists and uses thereof. International patent application, WO 2010/048207 April 29, 2010
- 57. Le Poul E, Loison C, Struyf S, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–25489 [DOI] [PubMed] [Google Scholar]
- 58. Sina C, Gavrilova O, Förster M, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol. 2009;183:7514–7522 [DOI] [PubMed] [Google Scholar]
- 59. Hong YH, Nishimura Y, Hishikawa D, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146:5092–5099 [DOI] [PubMed] [Google Scholar]
- 60. Ge H, Li X, Weiszmann J, et al. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology. 2008;149:4519–4526 [DOI] [PubMed] [Google Scholar]
- 61. Tolhurst G, Heffron H, Lam YS, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61:364–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kebede MA, Alquier T, Latour MG, Poitout V. Lipid receptors and islet function: therapeutic implications? Diabetes Obes Metab 2009;11(suppl 4):10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bjursell M, Admyre T, Göransson M, et al. 2011 Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 300:E211–E220 [DOI] [PubMed] [Google Scholar]
- 64. Kimura I, Inoue D, Maeda T, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci USA. 2011;108:8030–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xiong Y, Miyamoto N, Shibata K, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc Natl Acad Sci USA. 2004;101:1045–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zaibi MS, Stocker CJ, O'Dowd J, et al. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett. 2010;584:2381–2386 [DOI] [PubMed] [Google Scholar]
- 67. Leonard JN, Chu ZL, Bruce MA, Boatman PD. GPR41 and modulators thereof for the treatment of insulin-related disorders. PCT Int Appl. 2006;WO2006052566 [Google Scholar]
- 68. Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O'Dowd BF. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun. 1997;239:543–547 [DOI] [PubMed] [Google Scholar]
- 69. Liaw CW, Connolly DT. Sequence polymorphisms provide a common consensus sequence for GPR41 and GPR42. DNA Cell Biol. 2009;28:550–560 [DOI] [PubMed] [Google Scholar]
- 70. Moore K, Zhang Q, Murgolo N, Hosted T, Duffy R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: comparison with human GPR120 splice variants. Comp Biochem Physiol B Biochem Mol Biol. 2009;154:419–426 [DOI] [PubMed] [Google Scholar]
- 71. Galindo MM, Voigt N, Stein J, et al. G protein-coupled receptors in human fat taste perception. Chem Senses. 2012;37:123–139 [DOI] [PubMed] [Google Scholar]
- 72. Arkawa K, Nishimura T, Sugimoto Y, Takahashi H. Novel isoindolin-1-one derivative. International patent application, WO 2010/104195 September 16, 2010
- 73. Hashimoto N, Sasaki Y, Nakama C, Ishikawa M. Novel phenyl-isoxayol-3-ol derative. US patent application, US 2010/0130559 May 27, 2010