

Abstract
With a wealth of disease-associated DNA variants being recently reported, the challenges of providing their functional characterization are mounting. Previously, as part of a large systematic resequencing of the X chromosome in 208 unrelated families with nonsyndromic X-linked intellectual disability, we identified three unique variants (two missense and one protein truncating) in USP9X. To assess the functional significance of these variants, we took advantage of the Usp9x knockout mouse we generated. Loss of Usp9x causes reduction in both axonal growth and neuronal cell migration. Although overexpression of wild-type human USP9X rescued these defects, all three USP9X variants failed to rescue axonal growth, caused reduced USP9X protein localization in axonal growth cones, and (in 2/3 variants) failed to rescue neuronal cell migration. Interestingly, in one of these families, the proband was subsequently identified to have a microdeletion encompassing ARID1B, a known ID gene. Given our findings it is plausible that loss of function of both genes contributes to the individual's phenotype. This case highlights the complexity of the interpretations of genetic findings from genome-wide investigations. We also performed proteomics analysis of neurons from both the wild-type and Usp9x knockout embryos and identified disruption of the cytoskeleton as the main underlying consequence of the loss of Usp9x. Detailed clinical assessment of all three families with USP9X variants identified hypotonia and behavioral and morphological defects as common features in addition to ID. Together our data support involvement of all three USP9X variants in ID in these families and provide likely cellular and molecular mechanisms involved.
Main Text
Intellectual disability (ID) affects ∼2%–3% of the population, and in developed countries the dominant cause is of genetic origin.1 Although ID is one of the most highly heterogeneous human disorders, there is currently an overrepresentation of causative mutations found on the X chromosome.2 Recently the reports of a large-scale X-exome resequencing effort coupled with high-resolution copy-number profiling provided plausible explanations for approximately half of a cohort of 208 families with evidence for X-linked ID (XLID).3,4 Because the cohort was previously excluded from the mutations and large-scale (i.e., 500G banding resolution) cytogenetic alterations known to cause XLID at that time, most variants discovered were located in genes that were not previously associated with XLID.3 For the vast majority of the variants identified, further genetic and functional evidence was required (e.g., Shoubridge et al.5) to support their causal involvement in ID of the respective individuals and families studied.
Variants in USP9X (MIM 300072) have been identified as being potentially involved in XLID.3 The X-exome sequencing initially identified a single truncating variant in an X-linked family, and subsequent screening of an additional cohort of male individuals with ID (where X-linkage was not always obvious) identified two additional nonrecurrent missense variants.3 In two of these three families, the variants segregated as expected for X-linkage; in one case the inheritance could not be established (Figure S1 available online). The study was approved by local ethics committees and institutional review boards of each collaborating insititution, and informed consent for research was obtained from all individuals involved. In family 1, from France, a missense variant (c.6278T>A [RefSeq accession number NM_001039590.2], p.Leu2093His [RefSeq NP_001034679.2]) was found in a singleton affected male, with no other affected individuals known in the family (Figure S1). At 21 months of age, he displayed developmental delay, aggressive behavior, and hypotonia. Other features included relative macrocephaly, facial dysmorphism, broad thumbs and great toes, short stature, constipation, and hyperextensibility of joints and skin (Table 1). In family 2, from the USA, a second missense variant (c.6469C>A [p.Leu2157Ile]) was found in an affected male, his unaffected mother, and his unaffected grandmother (Figures S1 and S2). In addition, the affected individual was subsequently found to have a ∼790 kb deletion at 6q25.3, which includes ARID1B (MIM 614556). Haploinsufficiency of ARID1B is a common de novo cause of ID,6 and because his parents are unaffected, it is most likely that this deletion also occurred de novo (the parents could not be tested). Whether the ARID1B deletion itself is the only cause of ID in this patient or whether the USP9X variant provides an additional, second hit could not be easily determined. Prenatally, the affected male in family 2 displayed a raised maternal serum alpha-fetoprotein, intrauterine growth restriction, and an ectopic left kidney. Postnatally, he had feeding difficulties, hypotonia, tracheomalacia, gastro-esophageal reflux disease, and developmental delay, with speech development most affected. He also had broad thumbs, curving toe nails, and short stature. At 9 years of age, he had ID (he was nonverbal, speech having regressed at age 3), obsessive and autistic behaviors, elevated testosterone and decreased cholesterol levels, and short stature (Table 1). In the third family, from the UK, a single-nucleotide deletion was identified in two half brothers, their unaffected mother and affected uncle, and their unaffected grandmother (Figures S1 and S2). A single-nucleotide deletion in USP9X (c.7574delA [p.Gln2525Argfs∗18]) was the only plausible variant identified in the 700 genes on the X chromosome screened, and a subsequent systematic screening of 550 genes in which mutations are known (or suspected) to cause ID, via a customized exome pull-down platform, did not reveal any other plausible mutation.3 The deletion introduces a frameshift followed by 17 missense amino acids leading to a premature termination codon (PTC), and a 45 amino acid truncation. Because the PTC falls in the last exon of USP9X, transcripts are not likely to be degraded by the nonsense-mediated mRNA decay pathway. The affected individuals all had ID and hypotonia. In addition, one also displayed obsessive behaviors and another displayed autistic behaviors (Table 1). To summarize, the key clinical features shared by the five affected males from the three families with unique USP9X variants included ID, hypotonia, and short stature (where measured), with additional variable behavioral, gastroenterological, and dysmorphic features (Table 1).
Table 1.
Summary of Key Clinical Features of Affected Individuals
| Family 1 | Family 2 | Family 3 | |
|---|---|---|---|
| USP9X mutation | c.6278T>A (p.Leu2093His) | c.6469C>A (p.Leu2157Ile) | c.7574delA (p.Gln2525fs∗18) |
| Number of affected males | 1 | 1 | 3 |
| Neurological Features | |||
| ID (severity) | 1/1 (mild) | 1/1 (mild) | 3/3 (mild-moderate) |
| Autism | 0/1 | 1/1 | 1/3 |
| Aggression | 1/1 | 0/1 | 0/3 |
| Obsessiveness | 0/1 | 1/1 | 1/3 |
| Hypotonia | 1/1 | 1/1 | 3/3 |
| Dysmorphic Features | |||
| Craniofacial | relative macrocephaly and prominent forehead | 0/1 | 0/3 |
| Digital | broad thumbs and great toes | broad thumbs and curling toenails | 0/3 |
| Growth | |||
| age 21 months: Ht: 78.5 cm (3%) Wt: 11.4 kg (25%) HC: 50.5 cm (95%) |
age 12 months: Ht: 70.4 cm (3%) Wt: 7.87 Kg (<3%) HC: 44.7 cm (3%–25%) |
ND | |
| age 9 years: Ht: 124 cm (5%) Wt: 24.1 kg (10%) |
|||
| Other | hyperextensible joints and skin, constipation | IUGR, ectopic left kidney, tracheomalacia, gastresophageal reflux, upper airway congestion, hypospadia, retractable left testis | ND |
Abbreviations are as follows: Ht, height; Wt, weight, HC, head circumference; IUGR, intrauterine growth restriction; %, percentile; ND, no data.
USP9X is highly conserved7 with a residual variance intolerance score8 of −1.62, 2.93%, suggesting considerable intolerance to variation. The three USP9X variants are unique, not present in an additional 914 affected males in XLID families, 1,129 control X chromosomes,3 or dbSNP137, EVS, or the 1000 Genomes Project data sets. Furthermore, the variants were found to change highly conserved amino acid residues residing in the C-terminal region of the protein (Figure S1). In silico prediction programs (PolyPhen2, SIFT, and iPTREE) suggested that the c.6278T>A (p.Leu2093His) variant was deleterious, whereas for the c.6469C>A (p.Leu2157Ile) variant, only iPTREE reported an adverse effect (Figure S1). Additional prediction programs (PANTHER, Mutation Taster) corroborated this last result, suggesting that the p.Leu2157Ile variant was probably deleterious and disease causing.
USP9X encodes a very large substrate-specific deubiquitylating enzyme of 2,570 amino acids, with a largely unknown structure outside of its catalytic and ubiquitin-like domains.9 Although the variants did not locate within either of those domains, we tested whether the catalytic activity of the enzyme was affected. Overexpression of both USP9X and variant forms were able to stabilize the level of the known USP9X substrate MCL-110 (MIM 159552), suggesting that the variants did not affect the ability of USP9X to rescue MCL-1 from proteasomal degradation (Figure S3). We also observed no appreciative difference between the ubiquitylation status of proteins that interacted (i.e., coimmunoprecipitated) with USP9X or variant forms (Figure S3). Together, these data suggest that the variants do not affect the catalytic activity of USP9X. To provide further evidence that the variants are pathogenic, we employed our recently described Usp9x knockout mice11 to assay the variants’ effect on neurodevelopmental processes. Protocols relating to the use of animals were approved by the Women’s and Children’s Health Network Animal Ethics Committee. During embryogenesis, brain-specific deletion of Usp9x results in early postnatal death, whereas forebrain-specific deletion is compatible with survival to adulthood. In the absence of Usp9x the cortical architecture is disorganized, and neurons display reduced neurite growth in vivo and in vitro.11 We therefore asked whether the USP9X variants might also effect neurite growth, in particular axonal growth. We introduced the variants into a full-length human USP9X cDNA and placed it under control of an exogenous promoter providing uniform expression levels (Figure S4). We isolated primary hippocampal neurons from both wild-type (Usp9x+/Y) and knockout (Usp9x−/Y) embryonic mice and grew them in vitro. Prior to plating the neurons, they were transfected with expression plasmids encoding EGFP (to track transfected cells), together with either an empty expression plasmid or ones containing wild-type USP9X or USP9X with either of the three DNA variants. After 5 days of growth in vitro, we assayed the length of primary axons and the degree of arborization as reported by the number of axonal termini (Figure 1). The results show that overexpression of USP9X in wild-type neurons has no effect on either length or arborization but that the loss of Usp9x in knockout neurons resulted in a 43% reduction in both axon length and arborization, similar to previous reports.11 The re-expression of wild-type USP9X successfully rescued these defects, whereas the re-expression of the variant USP9X forms failed to do so (Figure 1). Together these data indicate that the three USP9X variants identified disrupt the ability of USP9X to function in axonal growth.
Figure 1.
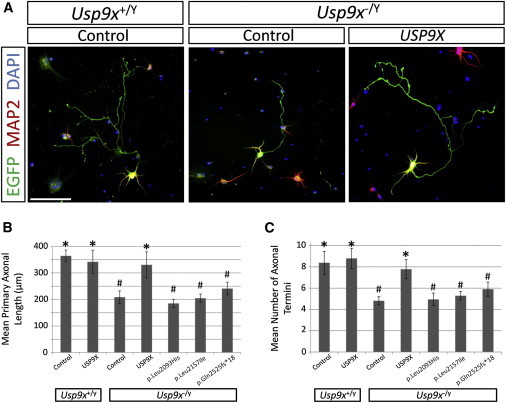
Expression of USP9X but Not USP9X Variants Rescues Axonal Defects in Hippocampal Neurons Isolated from Usp9x Knockout Mice
Usp9xloxP/LoxP female mice were crossed with Nestin-Cre males to delete Usp9x from the entire brain from E12.5 onward as previously described.11 Brains of male embryos from these matings are either wild-type (Usp9x+/Y) or knockout (Usp9x−/Y). For all experiments, hippocampal neurons were isolated from male embryos at E18.5 by established methods34 and nucleofected with 1 μg of pMAX-EGFP in addition to either 5 μg of the empty expression vector (pCMV; Control) or expression vectors containing USP9X (pCMV-USP9X (USP9X)) or USP9X variants (pCMV-USP9X_Leu2093His, pCMV-USP9X_Leu2157Ile, or pCMV-USP9X_Gln2525fs∗18) as previously described.34 After nucleofection, neurons were seeded in vitro and grown for 5 days, fixed and immunofluorescently stained, and subjected to morphometric analysis as previously described.35
(A) Representative immunofluorescent images of wild-type neurons nucleofected with control plasmids, compared with knockout neurons nucleofected with either control plasmid or USP9X expression plasmid. Nucleofected neurons are identified by EGFP expression (green). Scale bar represents 100 μm.
(B) Loss of Usp9x reduces axonal length, which is rescued by overexpression of USP9X but not USP9X variants. Morphometric analysis of mean axon length conducted as previously described.35 Note that overexpression of USP9X in wild-type neurons has no significant effect.
(C) Loss of Usp9x reduces the number of axonal termini, which is rescued by overexpression of USP9X but not USP9X variants. Morphometric analysis of mean axonal termini was conducted as previously described.35 Note that overexpression of USP9X in wild-type neurons has no significant effect.
All experiments were conducted in triplicate, with at least 25 neurons scored per replicate (i.e., >75 neurons scored per experimental condition). Graphed values are the mean of the three average values derived from each replicate. Error bars are ±SD. Statistical analysis conducted by Student’s two-tailed, unpaired t test, assuming equal variance, and significance set as p < 0.05: ∗significantly different to “Usp9x−/Y + Control” condition; #significantly different to “Usp9x−/Y+ USP9X” condition.
Next we explored whether USP9X variants might alter other neurodevelopmental processes. The C terminus of USP9X (where the variants cluster) binds the regulator of neuronal cell migration Doublecortin (DCX [MIM 300121])12,13 and the related Doublecortin-like Kinase (DCLK1 [MIM 604742]).13 Furthermore, mutations in DCX cause XLID.14 Therefore, we asked whether the loss of Usp9x might also affect neuronal migration. We isolated neural progenitor cells (NPCs) from both wild-type and knockout embryonic brains and grew them in vitro as nonadherent neurosphere cultures. Next we employed an in vitro neuronal migration assay, wherein neurospheres are adhered to a poly-L-lysine substrate and the migration of neurons outward from the sphere boundary is recorded.15–17 We found a highly significant decrease in the migration of neurons from the neurospheres in the absence of Usp9x (Figure 2). Next, we tested whether re-expression of the wild-type USP9X and variant USP9X forms could rescue this defect. We transfected wild-type and knockout neurospheres by the same regime as described above for the axonal growth assay. We found that overexpression of wild-type USP9X had no effect, whereas loss of Usp9x resulted in a 42% reduction in neuronal migration, similar to our previous finding (Figure 2). This migration defect could be partially rescued when wild-type USP9X or the c.6278T>A (p.Leu2093His) variant were re-expressed in the knockout cells, but re-expression of c.6469C>A (p.Leu2157Ile) or c.7574delA (p.Gln2525Argfs∗18) USP9X variants failed to do so (Figure 2). These data reveal that USP9X is required for normal neuronal cell migration and that two of the three variant USP9X forms probably disrupt this process during brain development of the affected individuals.
Figure 2.
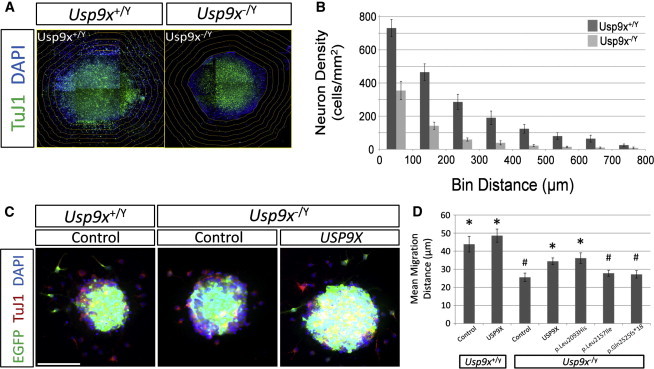
Expression of USP9X but Not USP9X Variants Rescues Neuronal Migration Defects in Cells Isolated from Usp9x Knockout Mice
Wild-type (Usp9x+/Y) or knockout (Usp9x−/Y) embryonic brains were generated and isolated as described earlier (see Figure 1). Neural progenitor cells (NPCs) were isolated from the dorsal cortex of E18.5 brains and grown as nonadherent neurospheres in culture as previously described.36
(A and B) Neuronal migration away from seeded neurospheres is inhibited in the absence of Usp9x. Passage 3 neurospheres were cultured for 5 days, adhered to a poly-L-lysine surface substrate (seeding) as previously described,17 cultured a further 5 days, fixed, and immunofluorescently stained as previously described.36
(A) Scoring system used to measure neuronal cell migration from seeded neurospheres. Composite images of immunofluorescently stained neurons present in neurosphere outgrowths (TuJ1: anti-βIII tubulin, green; DAPI: blue) were collated and the neurosphere periphery (as indicated by the edge of contiguous nuclei) outlined. The outline was then used to generate concentric bins outside of the neurosphere (i.e., in the migration zones) of increasing size (increases of 100 μm diameter) via ImageJ (NIH) software tools. This allowed for the calculation of neuronal cell density in each bin, which thus reports on migration independent of initial neurosphere size, and of bin volumes.
(B) Quantitation of neuronal migration away from seeded neurospheres by the described scoring system. Neurospheres were isolated from three wild-type and three Usp9x knockout embryos. Scoring was restricted to spheres of similar size. At least five neurosphere outgrowths were scored per embryo (total outgrowths scored: wild-type, 32; knockout, 16). Graph values are means and error bars are ±SD, of pooled data.
(C and D) Expression of USP9X but not USP9X variants rescue neuronal migration defects. Neurospheres were nucleofected via the same regime described in Figure 1, with previously described nucleofection techniques.34 Neurospheres were allowed to recover for 48 hr prior to seeding and were cultured for an additional 48 hr to promote neuronal migration, prior to fixing for immunofluorescent staining as described above.
(C) Representative images showing migration from wild-type neurospheres nucleofected with control plasmids, compared with Usp9x knockout neurospheres nucleofected with either control plasmid or USP9X expression plasmid. Scale bar represents 100 μm.
(D) To measure the migration of nucleofected neurons after 2 days of adherent culture, an alternative scoring system was adopted. Individually nucleofected neurons (i.e., expressing EGFP [green] and labeled by TuJ1 antibody [red]) were scored for the length of their radial migration path extending from the periphery of their sphere of origin. The average migration distance of neurons was calculated per neurosphere, and the mean migration distance of all neurospheres per condition was calculated and graphed. At least 20 neurospheres were scored per condition. Error bars represent ±SD. Statistical analysis conducted by Student’s two-tailed, unpaired t test, assuming equal variance, and significance set as p < 0.05: ∗significantly different to “Usp9x−/Y + Control” condition; #significantly different to “Usp9x−/Y+ USP9X” condition.
Reduced axonal growth and neuronal migration are also features of neurons lacking DCX.18–22 Given this overlap, the shared aspects of respective knockout mice phenotypes,18,23,24 and the overlap of the variants with the known DCX-interacting domain of USP9X, we asked whether the variant USP9X proteins disrupted interactions with DCX. We first overexpressed USP9X and variant forms together with DCX in HEK293T cells and conducted coimmunoprecipitation experiments with USP9X as bait. DCX coimmunoprecipitated with USP9X and all three variant forms in this assay, suggesting that the overexpressed proteins could interact in these cells (Figure S5). DCX is, however, endogenously expressed only in the highly polarized newly born neurons, and DCX and USP9X are known to colocalize in the growth cones of extending axons in such cells.13 Therefore, we asked whether in these cells we could see evidence of altered interactions. Given the fact that DCX is not a USP9X substrate (rather, it is an interacting protein), we reasoned that this might present as an alteration in subcellular localization and/or changes in colocalization. In cultured Usp9x knockout neurons, the localization of Dcx was unaffected (Figure S6). Therefore, we asked whether the localization of USP9X was altered by the variants. We re-expressed either USP9X or the variant forms in knockout neurons as described above. We observed that Usp9x and Dcx colocalized in the cell soma and in the axonal growth cones of wild-type neurons as previously described13 (Figure 3). Likewise, when we re-expressed USP9X in knockout neurons, its localization overlapped substantially with Dcx in axonal growth cones (Figure 3). When we re-expressed the variant forms, however, we observed a clear reduction in the localization of USP9X in the axonal growth cones, whereas expression in the cell soma was unaffected (Figure 3). This in turn led to a drastic reduction in the level of colocalization of USP9X and Dcx in the axonal growth cones of neurons. These data suggest that variant USP9X proteins are unable to be targeted or maintained in the growth cones of axons, which probably hinders its ability to interact with DCX in these structures.
Figure 3.
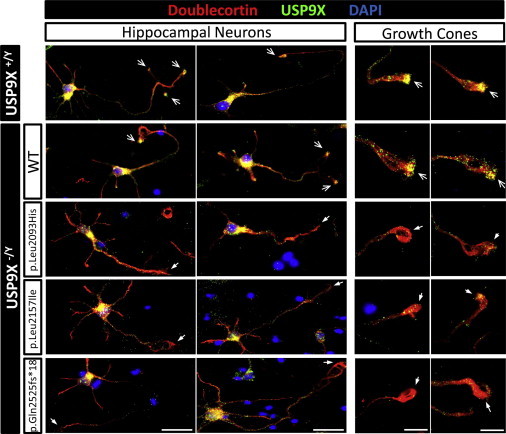
Reduced Localization of Variant USP9X in Axonal Growth Cones
Wild-type (Usp9x+/Y) or knockout (Usp9x−/Y) hippocampal neuronal cultures were generated and nucleofected with USP9X or variant forms as described previously (Figure 1). Immunoflourescent staining of Doublecortin (Dcx; red) and USP9X (green). Cell nuclei were stained with DAPI (blue). Open arrows indicate USP9X enrichment and closed arrows indicate depletion of USP9X in axonal growth cones. Scale bars represent 50 μm (left panels) and 20 μm (right panels).
With the exception of the c.6278T>A (p.Leu2093His) in the migration assay, the variant forms of USP9X behaved similar to a complete loss of function in our neuronal growth and migration assays. To gain further insight into the molecular pathways behind the neuronal cell defects, we employed a global, unbiased approach. Because Usp9x is a substrate-specific deubiquitylating enzyme, and thus regulates the proteome, we sought to identify proteins that were differentially expressed in Usp9x knockout neurons. We isolated and grew primary cortical neuronal cultures from four wild-type and four knockout E18.5 embryos. Lysates were isolated at day 5 of culture and subjected to two-dimensional difference in gel electrophoresis (2D-DIGE; Figure S7). We identified 50 protein spots that were differentially represented in the gels (p < 0.05; DeCyder Software Module, GE Health). We manually inspected all protein spots and removed those with un-spot-like appearance, noise spikes, or poor resolution. The remaining spots were excised from the gel and proteins identified by liquid chromatography-electrospray ionization tandem mass spectroscopy (LC-ESI-MS/MS). From this analysis, we identified 28 proteins that were differentially expressed. We did not identify any known substrates of USP9X, but we found evidence for all identified proteins being regulated by ubiquitylation (Table S1). Furthermore, 27/28 proteins were downregulated in the absence of Usp9x, consistent with its role in antagonizing the ubiquitin-proteasome pathway (Figure 4). Therefore, the list may contain potential substrates of Usp9x. It is, however, likely that at least some of these deregulated proteins are indirectly affected by the absence of Usp9x, and instead report on the molecular pathways deregulated in Usp9x knockout neurons. The deregulated gene list was submitted to the DAVID annotation as well,25 and significant (p < 0.05) gene ontology and pathway terms were identified. The highest ranking gene ontology terms and PANTHER pathways reveal that loss of Usp9x affected proteins involved in regulation and structure of the cytoskeleton (Figure 4 and Table S2). Together, the data suggest that the neuronal migration and axonal growth defects observed in the absence of Usp9x, and also probably in neurons of the affected individuals, are based on a disruption of the neuronal cytoskeleton.
Figure 4.

Loss of Usp9x Disrupts Cytoskeleton Components
Cortical neurons were isolated from either wild-type (Usp9x+/Y; n = 4) or knockout (Usp9x−/Y; n = 4) E18.5 embryos and cultured in vitro as previously described.36 After 5 days, cell lysates were generated and differentially expressed proteins identified as described in the text.
(A) Loss of Usp9x downregulates proteins. A total of 28 unique proteins were identified as being deregulated, with 27 of them downregulated in the absence of Usp9x.
(B) Gene ontology and PANTHER pathway analysis summary. The list of deregulated genes was analyzed by DAVID.25
The recent advances in sequencing and genome annotation are revolutionizing the discovery of genetic causes of disease. These approaches have found many variants that appear likely, based on initial genetic evidence, to be pathogenic. However, their acceptance as disease-causing mutations should be treated with caution, and supported by additional evidence, including variant frequency, segregation, genotype-phenotype correlations, and in silico and wet laboratory functional investigation. Here we focused on validating three unique variants discovered in USP9X that associated with ID in three unrelated families.3 For family 3, the clear X-linked mode of inheritance of a truncation type USP9X variant, coupled with the lack of any other plausible genetic alterations, provided persuasive genetic support of pathogenicity. Further evidence of the involvement of this USP9X variant in ID was derived from the variants discovered in families 1 and 2, but X-linkage in these families is less clear, and additional genetic contributions from other X-linked and autosomal regions cannot be entirely excluded (e.g., as in the case for family 2; see below). Our subsequent analyses are, however, consistent with the pathogenicity of all three USP9X variants. Although the families were ascertained on the basis of ID, our follow-up investigations extended the clinical phenotypes of the affected individuals to also include hypotonia in all cases, short stature (in all where information was available), and a spectrum of additional problems that were present in multiple affected individuals. Our in silico analysis predicted the USP9X variants to be deleterious, and functional evidence revealed that the variants of USP9X were unable to rescue the neuronal migration and/or axon growth defects observed in Usp9x knockout neurons. Finally, we show that all three variants of USP9X were unable to efficiently localize to axonal growth cones. Together these data provide genetic, clinical, and functional evidence in support of these USP9X variants being pathogenic mutations.
Although no other plausible pathogenic genetic alterations were found in family 3, less exhaustive genetic studies had been applied to families 1 and 2.3 A subsequent CNV screen of genomic DNA from the affected individual in family 2 identified a microdeletion encompassing ARID1B, a known cause of ID.6 Although this finding initially complicated our interpretation of the pathogenicity of the c.6469C>A (p.Leu2157Ile) USP9X variant, combined genetic and experimental evidence suggests that this variant is most likely damaging to USP9X function. In addition, close assessment of the available clinical information showed that the affected individual in family 2 has (1) some features that are consistent with the most severe phenotypes reported for individuals with altered ARID1B function, e.g., complete loss of speech, short stature, and light weight (at 12 months, height and weight both ≤3%); (2) numerous features that are typically only variably present, e.g., hypotonia, autism, genital deformities, digital anomalies, feeding difficulties; and (3) some that are yet to be described, e.g., obsessiveness, ectopic kidney, gastro-esophageal reflux disease, together suggesting that the USP9X mutation has resulted in a severe or aggravated phenotype normally associated with ARID1B haploinsufficiency.6 The presence of multiple hits with additive effect or independently causing ID or other disorders has been documented previously.26
Our study indicates potential pathological mechanisms including perturbed neuronal cytoskeletal dynamics, resulting in defective migration and axonal growth, that may underlie ID associated with USP9X mutations. Resolving full cellular and molecular pathways altered by USP9X mutations is expected to be important for the understanding of how ID develops. Because oligonucleotide-driven knockdown of Usp9x mRNA during preimplantation development in mouse leads to a failure of blastocyst development,27 it is unlikely that the USP9X mutations result in a complete loss of function, because this might not be compatible with life. Consistently, we did not find evidence that the variants affected the overall catalytic activity of the enzyme. Instead, it is likely that the mutations effect specific aspects of USP9X function required for brain development. Intriguingly, all the mutations altered the C-terminal region of USP9X, which is known to interact with DCX.13 Mutations in DCX cause XLID that stems from the neuronal migration disorders lissencephaly in males and subcortical band heterotopia in females (MIM 300067).12,14 The interaction of USP9X and DCX appears to be important for the pathology, because a mutation in DCX has been shown to abolish the interaction specifically with USP9X but not other DCX-interacting proteins.13 Thus the mutations in USP9X may perturb DCX function. In line with this hypothesis, the brain-specific knockout of Usp9x shows many correlates with the Dcx/Dclk double knockout, including reduced hippocampal volume, agenesis of the corpus callosum, and delamination of CA3 neurons.18,24 It is noteworthy that Dclk, which appears to be functionally redundant to Dcx in mouse,18,22 is also reported to be a USP9X-interacting protein.13 Loss of either Dcx or Usp9x also share common neuronal cell affects in vitro, including reduced neuronal migration and axon growth.18–21 Although overexpressed mutants of USP9X were able to interact with overexpressed DCX in nonpolarized HEK293T cells, we show that in immature polarized neurons (where endogenous DCX is expressed), their colocalizations were drastically reduced owing to the fact that USP9X mutants were not efficiently targeted or maintained in axonal growth cone structures. DCX is a microtubule-associated protein involved in vesicle transport, microtubule dynamics, and actin structure, and interestingly, our proteomic analysis highlighted that the loss of Usp9x resulted in changes to the neuronal cytoskeleton. Among the proteins deregulated are Tubulin subunits (Tubulin βIII, βIIB, βIIC, and α1a), Tubulin-regulating proteins (Stathmin and the Dihydropyriminidase-related proteins 2 and 3), and Actin filament-regulating proteins (Cofilin and Actin related protein 2/3 complex subunit 2). All of these proteins play integral roles in cytoskeletal dynamics and have established roles in neuronal migration, neuronal polarity, and axonal growth.28,29 Furthermore, among the deregulated proteins are those whose orthologous human genes have mutations that cause other forms of ID, including the neuronal migration disorders lissencephaly 3 ([MIM 611603]; TUBA1A [MIM 602529]), complex cortical dysplasia ([MIM 614039]; TUBB3 [MIM 602661]), and polymicogyria ([MIM 610031]; TUBB2B [MIM 612850]), as well as other neurological disorders ([MIM 610992]; PSAT1 [MIM 610936]). Individuals with these neuronal migration disorders display signature changes in MRI scans. Only the affected male in family 2 has undergone such investigation, with no obvious abnormalities reported. Therefore, it will be of interest to investigate additional individuals as they become available. Still, the overall underlying pathological mechanisms stemming from USP9X mutations are predicted to be more complex than a simple disruption of DCX and cytoskeletal functions owing to the fact that USP9X has more than 30 known substrates, many with bona fide roles in neural development. Of note, the axon growth defect in Usp9x-null neurons has been linked to a failure of TGF-β signaling response.11 Usp9x has two substrates known to regulate TGF-β signaling, including the common TGF-β signal transduction molecule SMAD4 (MIM 600993) and the ubiquitin ligase SMURF1 (MIM 605568).30–32 Interestingly, SMURF1 is known to bind the C terminus of USP9X and has established roles in cell migration.32 The probable pathogenic role of USP9X during the development of the brain is also not expected to be restricted to neurons. For example, it is known that Usp9x can affect the polarity and self-renewal of neural progenitor cells and has been implicated in the regulation of synaptic transmission.23,33 Thus, resolving which cells and key cellular processes are altered and which key substrates and molecular pathways are disrupted by USP9X mutations will provide the high-ranking candidate mechanisms underlying ID and associated clinical features. Our collective data suggest that mutations in USP9X have a functional impact and therefore are implicated in intellectual disability. It is now important to identify additional individuals with USP9X variants that will validate or challenge our data.
Acknowledgments
We are grateful for the cooperation of the families involved in this study. We thank Vishva M. Dixit, Gentech, Genentech, Inc. (San Francisco, CA, USA) for the provision of the pRK5-USP9X plasmid; Orly Reiner, Weizmann Institute of Science (Rehovot, Israel) for providing the Flag-Dcx plasmid; and Steven Reed, The Scripps Research Institute (San Diego, CA, USA) for the 6xHA-Ubiquitin plasmid. This work was supported by the Women’s and Children’s Hospital Foundation Grant to L.A.J.; the National Health and Medical Research Council of Australia grants APP628952, APP1041920, and APP1008077 to J.G. and APP1009248 to S.A.W.; National Institutes of Health 2R01HD026202 (NICHD) and 1R01NS73854 (NINDS) to C.S.; and an Australian Postgraduate Award Scholarship to C.C.H.
Contributor Information
Jozef Gecz, Email: jozef.gecz@adelaide.edu.au.
Lachlan A. Jolly, Email: lachlan.jolly@adelaide.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MutationTaster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PANTHER, http://www.pantherdb.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 2.Gécz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O’Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whibley A.C., Plagnol V., Tarpey P.S., Abidi F., Fullston T., Choma M.K., Boucher C.A., Shepherd L., Willatt L., Parkin G. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 2010;87:173–188. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoubridge C., Tarpey P.S., Abidi F., Ramsden S.L., Rujirabanjerd S., Murphy J.A., Boyle J., Shaw M., Gardner A., Proos A. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat. Genet. 2010;42:486–488. doi: 10.1038/ng.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoyer J., Ekici A.B., Endele S., Popp B., Zweier C., Wiesener A., Wohlleber E., Dufke A., Rossier E., Petsch C. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khut P.Y., Tucker B., Lardelli M., Wood S.A. Evolutionary and expression analysis of the zebrafish deubiquitylating enzyme, usp9. Zebrafish. 2007;4:95–101. doi: 10.1089/zeb.2006.0502. [DOI] [PubMed] [Google Scholar]
- 8.Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood S.A., Pascoe W.S., Ru K., Yamada T., Hirchenhain J., Kemler R., Mattick J.S. Cloning and expression analysis of a novel mouse gene with sequence similarity to the Drosophila fat facets gene. Mech. Dev. 1997;63:29–38. doi: 10.1016/s0925-4773(97)00672-2. [DOI] [PubMed] [Google Scholar]
- 10.Schwickart M., Huang X., Lill J.R., Liu J., Ferrando R., French D.M., Maecker H., O’Rourke K., Bazan F., Eastham-Anderson J. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 11.Stegeman S., Jolly L.A., Premarathne S., Gecz J., Richards L.J., Mackay-Sim A., Wood S.A. Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFβ-mediated axonogenesis. PLoS ONE. 2013;8:e68287. doi: 10.1371/journal.pone.0068287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gleeson J.G., Lin P.T., Flanagan L.A., Walsh C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 13.Friocourt G., Kappeler C., Saillour Y., Fauchereau F., Rodriguez M.S., Bahi N., Vinet M.C., Chafey P., Poirier K., Taya S. Doublecortin interacts with the ubiquitin protease DFFRX, which associates with microtubules in neuronal processes. Mol. Cell. Neurosci. 2005;28:153–164. doi: 10.1016/j.mcn.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 14.des Portes V., Francis F., Pinard J.M., Desguerre I., Moutard M.L., Snoeck I., Meiners L.C., Capron F., Cusmai R., Ricci S. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH) Hum. Mol. Genet. 1998;7:1063–1070. doi: 10.1093/hmg/7.7.1063. [DOI] [PubMed] [Google Scholar]
- 15.Angata K., Fukuda M. Roles of polysialic acid in migration and differentiation of neural stem cells. Methods Enzymol. 2010;479:25–36. doi: 10.1016/S0076-6879(10)79002-9. [DOI] [PubMed] [Google Scholar]
- 16.Ocbina P.J., Dizon M.L., Shin L., Szele F.G. Doublecortin is necessary for the migration of adult subventricular zone cells from neurospheres. Mol. Cell. Neurosci. 2006;33:126–135. doi: 10.1016/j.mcn.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Durbec P., Franceschini I., Lazarini F., Dubois-Dalcq M. In vitro migration assays of neural stem cells. Methods Mol. Biol. 2008;438:213–225. doi: 10.1007/978-1-59745-133-8_18. [DOI] [PubMed] [Google Scholar]
- 18.Deuel T.A., Liu J.S., Corbo J.C., Yoo S.Y., Rorke-Adams L.B., Walsh C.A. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Fu X., Brown K.J., Yap C.C., Winckler B., Jaiswal J.K., Liu J.S. Doublecortin (Dcx) family proteins regulate filamentous actin structure in developing neurons. J. Neurosci. 2013;33:709–721. doi: 10.1523/JNEUROSCI.4603-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jean D.C., Baas P.W., Black M.M. A novel role for doublecortin and doublecortin-like kinase in regulating growth cone microtubules. Hum. Mol. Genet. 2012;21:5511–5527. doi: 10.1093/hmg/dds395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koizumi H., Higginbotham H., Poon T., Tanaka T., Brinkman B.C., Gleeson J.G. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat. Neurosci. 2006;9:779–786. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- 22.Koizumi H., Tanaka T., Gleeson J.G. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 23.Jolly L.A., Taylor V., Wood S.A. USP9X enhances the polarity and self-renewal of embryonic stem cell-derived neural progenitors. Mol. Biol. Cell. 2009;20:2015–2029. doi: 10.1091/mbc.E08-06-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka T., Koizumi H., Gleeson J.G. The doublecortin and doublecortin-like kinase 1 genes cooperate in murine hippocampal development. Cereb. Cortex. 2006;16(Suppl 1):i69–i73. doi: 10.1093/cercor/bhk005. [DOI] [PubMed] [Google Scholar]
- 25.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 26.Girirajan S., Eichler E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010;19(R2):R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pantaleon M., Kanai-Azuma M., Mattick J.S., Kaibuchi K., Kaye P.L., Wood S.A. FAM deubiquitylating enzyme is essential for preimplantation mouse embryo development. Mech. Dev. 2001;109:151–160. doi: 10.1016/s0925-4773(01)00551-2. [DOI] [PubMed] [Google Scholar]
- 28.Feng Y., Walsh C.A. Protein-protein interactions, cytoskeletal regulation and neuronal migration. Nat. Rev. Neurosci. 2001;2:408–416. doi: 10.1038/35077559. [DOI] [PubMed] [Google Scholar]
- 29.Liu J.S. Molecular genetics of neuronal migration disorders. Curr. Neurol. Neurosci. Rep. 2011;11:171–178. doi: 10.1007/s11910-010-0176-5. [DOI] [PubMed] [Google Scholar]
- 30.Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M.J., Soligo S., Morsut L. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 31.Stinchfield M.J., Takaesu N.T., Quijano J.C., Castillo A.M., Tiusanen N., Shimmi O., Enzo E., Dupont S., Piccolo S., Newfeld S.J. Fat facets deubiquitylation of Medea/Smad4 modulates interpretation of a Dpp morphogen gradient. Development. 2012;139:2721–2729. doi: 10.1242/dev.077206. [DOI] [PubMed] [Google Scholar]
- 32.Xie Y., Avello M., Schirle M., McWhinnie E., Feng Y., Bric-Furlong E., Wilson C., Nathans R., Zhang J., Kirschner M.W. Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem. 2013;288:2976–2985. doi: 10.1074/jbc.M112.430066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer J.A., Overstreet E. Fat facets does a Highwire act at the synapse. Bioessays. 2002;24:13–16. doi: 10.1002/bies.10030. [DOI] [PubMed] [Google Scholar]
- 34.Huang L., Jolly L.A., Willis-Owen S., Gardner A., Kumar R., Douglas E., Shoubridge C., Wieczorek D., Tzschach A., Cohen M. A noncoding, regulatory mutation implicates HCFC1 in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2012;91:694–702. doi: 10.1016/j.ajhg.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbett M.A., Bahlo M., Jolly L., Afawi Z., Gardner A.E., Oliver K.L., Tan S., Coffey A., Mulley J.C., Dibbens L.M. A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am. J. Hum. Genet. 2010;87:371–375. doi: 10.1016/j.ajhg.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jolly L.A., Homan C.C., Jacob R., Barry S., Gecz J. The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum. Mol. Genet. 2013;22:4673–4687. doi: 10.1093/hmg/ddt315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.