

Abstract
Copy-number variations cause genomic disorders. Triplications, unlike deletions and duplications, are poorly understood because of challenges in molecular identification, the choice of a proper model system for study, and awareness of their phenotypic consequences. We investigated the genomic disorder Charcot-Marie-Tooth disease type 1A (CMT1A), a dominant peripheral neuropathy caused by a 1.4 Mb recurrent duplication occurring by nonallelic homologous recombination. We identified CMT1A triplications in families in which the duplication segregates. The triplications arose de novo from maternally transmitted duplications and caused a more severe distal symmetric polyneuropathy phenotype. The recombination that generated the triplication occurred between sister chromatids on the duplication-bearing chromosome and could accompany gene conversions with the homologous chromosome. Diagnostic testing for CMT1A (n = 20,661 individuals) identified 13% (n = 2,752 individuals) with duplication and 0.024% (n = 5 individuals) with segmental tetrasomy, suggesting that triplications emerge from duplications at a rate as high as ∼1:550, which is more frequent than the rate of de novo duplication. We propose that individuals with duplications are predisposed to acquiring triplications and that the population prevalence of triplication is underascertained.
Main Text
Genomic duplication was first visualized in the 1930s when Calvin Bridges used polytene chromosomes to show that a phenotype consisting of a reduction in the size of the eye in Drosophila melanogaster was due to a duplication of the Bar locus at cytological band X16A; the phenotype reverted to wild-type with loss of the duplication.1 Bar duplication and its reversion were hypothesized to occur by unequal crossing-over.2–4 Recent evidence has suggested BarH1 as the dosage-sensitive gene associated with the Bar phenotype.5 Bridges also noted that Ultra-Bar, a mutant isolated from the Bar stock and with a more severe phenotype (an even further slit eye), was associated with an apparent triplication at the locus.1 Questions were raised as to the frequency, molecular origin, and precise mechanism for triplication. However, efforts to reach conclusions experimentally were hampered by technological challenges and limitations in the knowledge of genomics and the mechanisms for genomic rearrangements.
After three quarters of a century, another chance to illuminate some of the questions raised by the observations of Bridges has been offered, although by work from a completely different model organism, Homo sapiens. Duplication at the human chromosomal region 17p12 causes Charcot-Marie-Tooth disease type 1A (CMT1A [MIM 118220]), the most common form of CMT1, but to date, triplications have not been observed. CMT1 is the most common inherited demyelinating peripheral neuropathy; characterized by distal muscle weakness and atrophy and reduced nerve conduction velocities (NCVs) of both motor and sensory nerves, it manifests clinically as a distal symmetric polyneuropathy (DSP).6 The disease is clinically heterogeneous with age-dependent penetrance. Abnormal NCV (conduction velocity of the motor median nerve < 38 m/s) is highly diagnostic of CMT1 and is a fully penetrant trait independent of age.7 The molecular mechanism for CMT1A has been extensively studied in affected individuals throughout the world, and the 1.4 Mb recurrent duplication has been shown to result from nonallelic homologous recombination (NAHR) between directly oriented ectopic substrate copies of flanking low-copy repeats (LCRs).8,9 This NAHR event is influenced by alleles of PRDM9 (MIM 609760), which encodes a protein important for homologous recombination.10 NAHR can occur either via interchromosomal crossover, i.e., NAHR between homologous chromosomes, or via intrachromosomal crossover, i.e., NAHR between sister chromatids. Theoretically, if NAHR occurs on chromosomes with preexisting duplications, recurrent tandem triplications can be produced.
To date, interstitial triplications have been observed in association with different human diseases, including genomic disorders (such as the 22q11.2 triplication) and common complex traits (such as Parkinson disease and hereditary pancreatitis).11–20 They often impart a more severe phenotype than do duplications because of further increments in gene dosage. Individuals who have segmental tetrasomy of the CMT1A-associated region as a result of homozygous duplication have been shown to exhibit severe neurological phenotypes.8,21 Evidence from a rat model for CMT1A has also suggested that an increase in copy number of the transgene PMP22 (MIM 601097) results in a more severe phenotype.22 However, it is not known whether CMT1A triplications could similarly convey a strong dosage effect, whether gene dosage effects in trans (i.e., homozygous duplication) versus cis (i.e., triplication) behave differently perhaps as a result of position effects, whether triplications arise de novo via a double crossover or arise from a pre-existing duplication, and what the prevalence of triplication is.
We identified and systematically studied families in which dominant CMT segregates and in which affected members from subsequent generations present with a more severe clinical neuropathy phenotype than do siblings or parents (an observation somewhat reminiscent of the genetic phenomenon of anticipation). All studies were approved by the institutional review boards of their respective institutions: Baylor College of Medicine, Tel Aviv Medical Center, and Vanderbilt University. Detailed clinical descriptions and nerve-conduction-study values can be found in the Supplemental Data, available online. CMT can have a widely varying severity of the clinical phenotype, and this variation can occur within the same family affected by the CMT1A duplication21 and even in identical twins.23 We identified two families in which one member appears to have disease at the extreme end of severity and out of proportion with the usual clinical variation seen within a family in which the CMT1A duplication segregates. These outlier individuals at the phenotypic extreme showed more severe DSP upon both clinical examination and objective electrophysiological measurements (Figures 1 and 2 and Table S1). Remarkably, the fact that these outlier individuals have phenotypes more severe than those of older family members suggests the possibility of a different underlying pathological mechanism.
Figure 1.
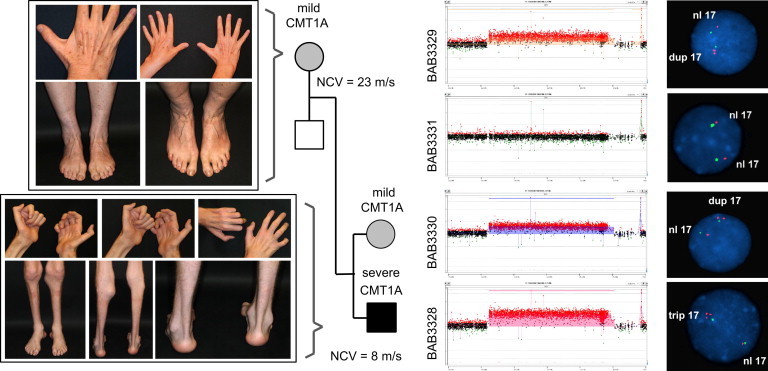
Clinical Phenotypes and Copy-Number Analyses in Family 1
On the left, pictures illustrate mild muscle atrophy of the mother (top left) and severe muscle atrophy of the lower leg and hand muscles and severe pes cavus deformity (extremely high-arched foot) in the index individual (bottom left), clinically indicating more severe DSP. NCVs of the median motor nerve for the mother and index individual are listed in the pedigree. On the right, array CGH and FISH results show duplication of the CMT1A-associated region in the mother (first panel) and sibling (third panel), neutral copy number in the father (second panel), and triplication in the index individual (fourth panel). In the FISH experiments, the target and control probes used were P150M12 and RP1-178F10, respectively.
Figure 2.
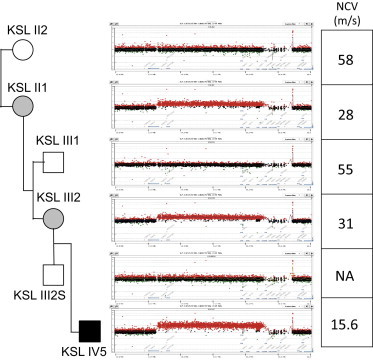
NCVs and Copy-Number Analyses in Family 2
Array-CGH analysis revealed that the index individual carries four copies, the mother and grandmother carry three copies, and the other family members carry two copies of the CMT1A-associated region. NCVs were lower in individuals with abnormal copy number than in healthy individuals. A very low NCV was observed in the index individual. The following abbreviation is used: NA, not assayed.
We hypothesized that the degree of severity of the clinical phenotype is associated with the copy number of the chromosomal region. To test this hypothesis, we performed locus-specific high-density array comparative genomic hybridization (CGH) and interphase fluorescence in situ hybridization (FISH). We used an Agilent 8X60k aCGH design with median probe spacing at ∼300 bp to interrogate the human 17p12 region. Array CGH detected copy number of the 1.4 Mb CMT1A-associated genomic interval of two, three, or four (Figures 1 and 2). The molecular copy-number gain correlated with the severity of clinical phenotype (Figures 1 and 2). FISH data suggested that the new rearrangement occurred on the chromosome with existing duplication, given the 3:1 ratio of experimental (red) fluorescence signals to control (green) signals on the rearranged chromosome (Figure 1, “trip 17”) versus the 1:1 ratio on the normal chromosome (Figure 1, “nl 17”) and the 2:1 ratio on both homologous chromosomes with homozygous duplication (Figure 7d in Lupski et al.8).
To investigate the mechanism of the crossovers, we designed microsatellite genotyping assays to phase haplotypes of the duplicated regions and flanking regions. We chose eight markers from the literature and seven newly developed markers with a preference for a high degree of heterozygosity and long repeat units (penta-, tetra-, or trinucleotides) (Figure 3A and Table S2).24,25 Taking into account the prior knowledge of array-CGH-ascertained copy number for each marker, analysis of the microsatellite genotyping data yielded size and copy-number information of the alleles at each locus. Haplotypes were constructed on the basis of the most parsimonious explanation of segregation and minimum number of recombination events required. In both families, the de novo triplication event arose from a pre-existing duplication and occurred during maternal meiosis (Figures 3B and 3C). This further confirmed that the segmental tetrasomy was a result of triplication instead of homozygous duplication. The duplications in both mothers were inferred to be products of interchromosomal NAHR in a previous generation because the duplicated alleles in cis were of differing sizes for most loci examined. In family 2, the triplication arose via intrachromosomal NAHR (Figure 3C). In the maternal meiosis that led to the index individual in family 1, crossover activity appeared to be more complicated. In addition to one NAHR crossover that created the triplication, a second gene-conversion event most likely exchanged alleles between the maternal homologous chromosomes (Figures 3B and 3D). Accompanying the transmission of duplication across generations in meiosis, crossing-over with the homologous chromosome was observed in regions distal to the duplication. This was evident in a comparison of the phased chromosomes between the mother (3329) and uncle in family 1, as well as those of the mother (KSLIII2), uncle (KSLIII1), and grandmother (KSLII1) in family 2.
Figure 3.
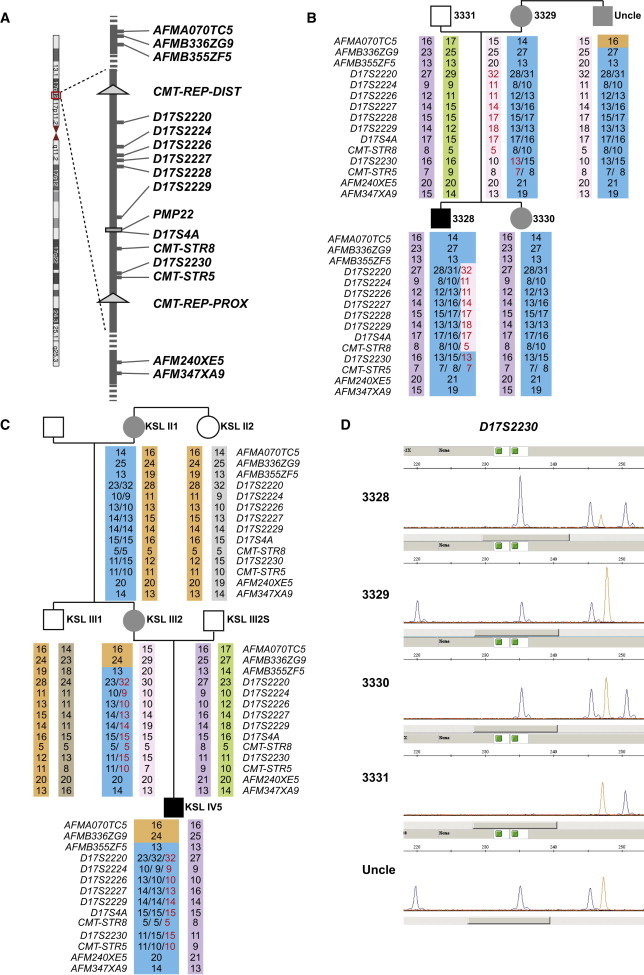
Microsatellite Analysis Results Suggest that De Novo Triplications Arose from Duplications by Different NAHR Mechanisms
Schematic representation of the CMT1A-associated region in 17p12. Positions of microsatellite markers used in this study are indicated in (A). Haplotypes were phased on the basis of microsatellite genotyping results in family 1 (B) and family 2 (C). Black indicates a severe CMT phenotype, and gray indicates the usual degree of neuropathy associated with a CMT1A-duplication phenotype. The numbers indicated for the microsatellite alleles reflect their numbers of repeats. In the index individual, new alleles reflecting the de novo triplication event are highlighted in red. Their inferred origin in the maternal chromosome is also colored in red. Alleles are in different color backgrounds to illustrate the inferred rearrangement paths. Of note, in the maternal meiosis that led to individual 3328, a gene-conversion event exchanged a segment within the triplication for the allele from the other homologous chromosome. Comparison of the phased haplotypes between individual 3329 and the uncle suggested that an allelic crossover occurred in the chromosomal region distal to the CMT1A duplication in the meiosis that led to either of these two individuals. One possibility, illustrated in the figure, is that the crossover occurred in the uncle. It is equally possible that the crossover was in individual 3329. A similar condition was observed in family 2 in individuals KSLIII1 and KSLIII2. Representative raw data of the microsatellite analysis resulting in interpretations in (B) and (C) are shown in (D). Relative peak heights were used for inferring the copy number of each allele.
Although CMT1A duplications usually arise from interchromosomal NAHR (Figure 4A), estimated to be at least 50-fold greater than intrachromosomal NAHR,26 we propose that triplications can arise from duplications through interchromosomal (Figure 4B) or intrachromosomal (Figures 4D–4F) NAHR. The rearrangements in family 2 can be explained by intrachromosomal NAHR between flanking LCRs or the duplicated segments (Figures 4D–4F). The triplication in family 1 was also produced by intrachromosomal NAHR. However, a gene conversion potentially exchanged part of the triplicated CMT1A-associated segment for the chromosome in trans. The combination of intrachromosomal NAHR and interchromosomal allelic recombination brings about a variety of possible mechanisms, four of which are shown in Figures 4G–4J. These two events can occur concurrently or in two separate cell divisions.
Figure 4.
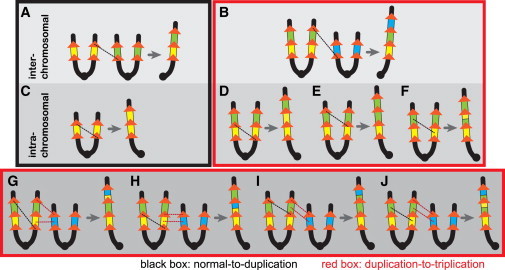
More NAHR Pathway Choices Are Available for the Duplication-to-Triplication Process than for the Normal-to-Duplication Process
For simplicity, only chromosome short arms are shown. The NAHR products consist of one chromosome with copy-number gain and the other chromosome with the reciprocal loss; only the chromosome with gain is shown. Yellow, green, or blue boxes represent the 1.4 Mb CMT1A-associated segment. Orange arrows indicate LCRs (the 24 kb segmental duplications that are known to mediate the de novo CMT1A duplication). Dashed lines indicate sites of recombination. Interchromosomal NAHR can produce duplication (A) and triplication (B) similarly. When NAHR occurs between sister chromatids (intrachromosomal), only one pathway (C) exists for de novo duplication, whereas three pathways (D–F) are available for the production of triplication. Additional (but not exhaustive) possibilities of triplication formation in which one NAHR (black dashed lines) and one gene conversion (red dashed lines) are involved are shown in (G)–(J) and potentially explain the rearrangement outcome in family 1. A distinctive possibility where a quadruplication is first made by an intrachromosomal NAHR but subsequently reverted to triplication by a gene conversion is shown in (G).
The unexpected allelic crossovers and/or gene conversions accompanying transmissions of duplications or the de novo duplication-to-triplication process observed in the two families can be potentially explained by the asymmetry between homologous chromosomes posed by the duplication and the resulting increased probability of ectopic synapsis27 facilitating subsequent recombination. This hypothesis is supported by the “oblique synapsis” in Bar chromosomes, as noted by Bridges, and is further reinforced by his observation that such a trend is more frequent in Ultra-Bar than in Bar.1 From another perspective, during the de novo duplication-to-triplication process, a chromosome with reversion to normal copy status or deletion can be produced concurrently with the triplication chromosome—a normal dosage and absence of a phenotype would not be readily ascertained in a human population. So far, one case with somatic CMT1A duplication reversion has been reported.28 Given the above evidence, duplications could introduce instability into meiosis. Duplication could give rise to triplications, allelic recombinations, reversions, deletions, or a combination thereof, potentially contributing to clinical variability within the pedigree affected by duplication. Furthermore, increased genomic instability might result in susceptibility to postzygotic mitotic events producing mosaic states29 that could influence disease severity and potentially account for clinical variability within some families in which CMT neuropathy segregates.
Theoretically, there are more ways for a chromosome to acquire a gain in copy number through intrachromosomal NAHR after a duplication has been introduced into a chromosome (Figure 4). This is partly because the newly introduced duplication creates substrate choices that were not previously available for intrachromosomal NAHR and thus leads to triplication. For example, at the CMT1A-associated locus, only one set of a 24 kb flanking LCR pair is available for de novo duplication formation (Figure 4C), whereas three sets (including two homologous recombination substrate pairs of the 24 kb LCRs [Figures 4D and 4E] and one pair of a much larger segment, the 1.4 Mb CMT1A-associated region [Figure 4F]) are available for the duplication-to-triplication process. In support of the contention that a chromosome with a duplication has increased probability of intrachromosomal NAHR, both de novo triplications examined in this study occurred by intrachromosomal NAHR, despite the fact that de novo CMT1A duplications were previously shown to occur preferentially by interchromosomal NAHR (at least 50-fold greater than intrachromosomal NAHR).26 Other examples of duplication creating new substrates for NAHR can be found in the human trypsinogen locus20 and Parkinsonism locus,30 the mouse Wallerian degeneration locus,31 and the fly Bar locus.1 In support of our hypothesis that intrachromosomal NAHR is favored for the duplication-to-triplication process, the Parkinson triplication identified in a Lister kindred was shown to be a result of intrachromosomal rearrangement occurring on the foundation of an existing duplication.30
On the basis of these mechanistic predictions, we propose that triplications are produced at a rate higher than that of de novo duplications. Re-examination and estimation of frequencies of de novo duplications and triplications at the fly Bar locus provided empirical data supporting this hypothesis.1 Two Ultra-Bar mutants were identified after facet count in 14,000 Bar flies, whereas no Bar mutation was discovered after eye-size scrutiny in 46,290 full-eye flies (triplication at 2/14,000 versus duplication at 0/46,290).32 Thus, triplications might be more prevalent in the clinical population than expected, especially for a condition such as CMT1A, wherein transmission of duplication is frequently observed and ascertainment bias between duplication and triplication is trivial.
To this end, we examined the database of a clinical diagnostic laboratory performing CMT1A-duplication testing. The assay applied multiplex-ligation-dependent probe amplification (MLPA) to examine copy-number or gene dosage in the 17p12 interval, where the CMT1A-associated locus resides. MLPA can distinguish the common CMT1A duplications generated by NAHR from rare smaller and larger duplications and can also reveal dosage differences of one, two, three, or four copies.33 We followed the MLPA data for six years (2007–2012) and used the frequency of identifying four copies versus three copies (the latter of which was observed with CMT1A duplication) as a surrogate measure to estimate the frequency of triplication. Among a total of 20,661 individuals referred for MLPA testing, 2,758 were identified to have the common recurrent CMT1A gain. Re-examination and individual evaluation of the raw data from these 2,758 positive tests confirmed six cases with four copies of the CMT1A-associated genomic segment on 17p12 (Table S3). Examination of demographics and molecular investigation of microsatellite markers revealed that five out of the six samples correspond to unique individuals and most likely independent families (data not shown). These five samples predicted by the MLPA assay to carry four copies of the CMT1A-associated region were unanimously confirmed by array CGH to have equivalent dosage and breakpoint boundaries consistent with recurrent triplications (Figure S1). The recurrent nature (flanked by LCRs) of these rearrangements suggests that they were produced by NAHR. Theoretically, a recurrent tandem triplication is thought to be generated from a recurrent duplication through one NAHR event. Although it has been shown in other loci that triplication can emerge directly from a copy-number-neutral region,15,34 such a phenomenon was always associated with a genomic architecture of inverted LCRs and the insertion of one of the triplicated segments in an inverted orientation (rather than in tandem), and these do not apply to the CMT1A-associated region. Therefore, we assumed that each of the five samples reflects a potential duplication-to-triplication event. The five potential triplication carriers were not reported as consanguineous; genotyping analysis did not reveal an absence of heterozygosity within the triplication, suggesting that the probability of a homozygous duplication is slim. However, because of their anonymous identity, we could not obtain parental samples to completely rule out the possibility that homozygous duplication contributed to these five cases. Overall, our data suggest that the de novo rate of CMT1A triplication arising from duplication is at least 1.8 × 10−3 if calculated on the basis that all five MLPA-detected cases with segmental tetrasomy represent triplication. The false-negative rate of the MLPA assay for detecting tetrasomy is unknown. Because of the limitations mentioned above, we might have overestimated (if one or more of the five candidates carry homozygous duplications rather than a triplication) or underestimated (false-negative detection of triplication via MLPA) the triplication formation rate. Further investigation is warranted for a more comprehensive estimation.
We sought to compare the frequency of observing triplication at the CMT1A-associated locus to the de novo duplication rate. Previous theoretical estimates from population prevalence data have suggested a locus-specific mutation rate between 1.7 × 10−5 and 2.6 × 10−5.35 Remarkably, direct experimental studies using pooled sperm PCR demonstrated a de novo duplication mutation rate of 1.73 × 10−5 (±4.88 × 10−6).26 The agreement of rates estimated from population frequencies and empirical studies using direct germ cell measurements is consistent with the idea of an absence of selection against the CMT1A duplication and no reduced fecundity. Therefore, the de novo rate of the duplication-to-triplication process is approximately 100-fold higher than that of the normal-to-duplication process, supporting our hypothesis that the presence of a duplication in one chromosome facilitates NAHR to cause a further copy-number gain.
Recurrent triplications are often misdiagnosed as duplications because of technical challenges in discovery and validation. In recent years, it has been demonstrated apodictically that high-density array CGH can robustly diagnose triplications and differentiate them from duplications.13,15,16,36 We suggest that such experience and knowledge about duplication-to-triplication frequencies can be applied clinically to NAHR-prone regions of the human genome, where triplications can result in a clinical phenotype, but not one too severe to be lethal. Several candidate loci were reported by Dittwald et al., who found triplications or homozygous duplications of regions including NPHP1 (MIM 607100) in 2q13 (n = 4), BP1 and BP2 in 15q11.2 (n = 1), CHRNA7 (MIM 118511) in 15q13.3 (n = 2), the DiGeorge region in 22q11.21 (n = 3), and the region associated with Prader-Willi and Angelman syndromes in 15q11.2q13 (n = 1).37 Such regions might include genomic-disorder-associated loci similar to the CMT1A-associated locus, such as tetrasomy of the DiGeorge region.38 Additionally, increasing numbers of dosage-sensitive regions have been identified by clinical microarray screening wherein deletion of the region is clearly associated with disease but the reciprocal duplication is still of borderline or unknown clinical significance, presumably because the duplication causes a milder dosage deviation from normal than does the deletion. According to our hypothesis, these duplications, which are considered benign variants contributing to a fraction of polymorphisms in the general population, could amplify at an unexpectedly high rate and eventually lead to multiplication, resulting in a deleterious phenotype. This prediction resembles the genetic phenomenon of anticipation. For example, duplication of the human Xp22.31 region is considered to not be sufficient to result in an abnormal phenotype, whereas triplication of the same region is suggested to cause neurodevelopmental problems.16 The proposed trend of amplification could have an impact on various human genomic disorders and copy-number evolution in the human genome.
We have demonstrated that recurrent CMT1A triplication arose from duplications in two families and that the frequency of such events was orders of magnitude greater than that of de novo duplication. The CMT1A triplication causes a severe clinical phenotype, and its prevalence has been substantially underestimated in the CMT population. Therefore, individuals with a family history of CMT1A duplication should be evaluated and counseled for potential triplications, especially in a CMT1A-duplication-affected family with an individual considered a “clinical outlier” or presenting with a phenotypic extreme. Molecularly, we have shown that triplications can be produced through a variety of NAHR mechanisms. We also provide evidence suggesting that duplications might be unstable and undergo allelic recombination when transmitted. Our study further illustrates the concept of clan genomics, wherein new mutations within a family or clan contribute to clinically relevant phenotypes against which evolutionary forces have not yet had the time to select.39
Acknowledgments
We thank Ping Fang and Weihong Jin for technical support and all the participating subjects and families for their time and effort. This work was supported in part by National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke grant R01NS058529 to J.R.L. and grant R01NS066927 to J.L., Texas Children’s Hospital General Clinical Research Center grant M01RR00188, Intellectual and Developmental Disabilities Research Centers grant P30HD024064, and Vanderbilt Institute for Clinical and Translational Research fund VR1687. J.R.L. holds stock ownership of 23andMe and Ion Torrent Systems and is a coinventor on multiple United States and European patents for DNA diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine receives clinical service revenue for genomic analyses by array comparative genomic hybridization and exome sequencing. J.J.H. holds stock options from Quest Diagnostics and receives research support from the Hartwell Foundation, March of Dimes, and NIH grant NS075397. J.J.H. is a voluntary professor in the Division of Pediatric Neurology at Weill Cornell Medical College. A.C.M., R.J.M., C.D., and J.J.H. are employed by Quest Diagnostics.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Bridges C.B. The Bar “Gene” a Duplication. Science. 1936;83:210–211. doi: 10.1126/science.83.2148.210. [DOI] [PubMed] [Google Scholar]
- 2.Sturtevant A.H., Morgan T.H. Reverse Mutation of the Bar Gene Correlated with Crossing Over. Science. 1923;57:746–747. doi: 10.1126/science.57.1487.746. [DOI] [PubMed] [Google Scholar]
- 3.Sturtevant A.H. The Effects of Unequal Crossing over at the Bar Locus in Drosophila. Genetics. 1925;10:117–147. doi: 10.1093/genetics/10.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturtevant A.H. A Further Study of the so-Called Mutation at the Bar Locus of Drosophila. Genetics. 1928;13:401–409. doi: 10.1093/genetics/13.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kojima T., Ishimaru S., Higashijima S., Takayama E., Akimaru H., Sone M., Emori Y., Saigo K. Identification of a different-type homeobox gene, BarH1, possibly causing Bar (B) and Om(1D) mutations in Drosophila. Proc. Natl. Acad. Sci. USA. 1991;88:4343–4347. doi: 10.1073/pnas.88.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.England J.D., Gronseth G.S., Franklin G., Carter G.T., Kinsella L.J., Cohen J.A., Asbury A.K., Szigeti K., Lupski J.R., Latov N., American Academy of Neurology Practice Parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72:185–192. doi: 10.1212/01.wnl.0000336370.51010.a1. [DOI] [PubMed] [Google Scholar]
- 7.Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259–280. doi: 10.1093/brain/103.2.259. [DOI] [PubMed] [Google Scholar]
- 8.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 9.Pentao L., Wise C.A., Chinault A.C., Patel P.I., Lupski J.R. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- 10.Berg I.L., Neumann R., Lam K.W., Sarbajna S., Odenthal-Hesse L., May C.A., Jeffreys A.J. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 12.Wolf N.I., Sistermans E.A., Cundall M., Hobson G.M., Davis-Williams A.P., Palmer R., Stubbs P., Davies S., Endziniene M., Wu Y. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain. 2005;128:743–751. doi: 10.1093/brain/awh409. [DOI] [PubMed] [Google Scholar]
- 13.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beunders G., van de Kamp J.M., Veenhoven R.H., van Hagen J.M., Nieuwint A.W., Sistermans E.A. A triplication of the Williams-Beuren syndrome region in a patient with mental retardation, a severe expressive language delay, behavioural problems and dysmorphisms. J. Med. Genet. 2010;47:271–275. doi: 10.1136/jmg.2009.070490. [DOI] [PubMed] [Google Scholar]
- 15.Carvalho C.M., Ramocki M.B., Pehlivan D., Franco L.M., Gonzaga-Jauregui C., Fang P., McCall A., Pivnick E.K., Hines-Dowell S., Seaver L.H. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat. Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P., Erez A., Nagamani S.C., Bi W., Carvalho C.M., Simmons A.D., Wiszniewska J., Fang P., Eng P.A., Cooper M.L. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications. Hum. Mol. Genet. 2011;20:1975–1988. doi: 10.1093/hmg/ddr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi W., Sapir T., Shchelochkov O.A., Zhang F., Withers M.A., Hunter J.V., Levy T., Shinder V., Peiffer D.A., Gunderson K.L. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yobb T.M., Somerville M.J., Willatt L., Firth H.V., Harrison K., MacKenzie J., Gallo N., Morrow B.E., Shaffer L.G., Babcock M. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am. J. Hum. Genet. 2005;76:865–876. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schinzel A.A., Brecevic L., Bernasconi F., Binkert F., Berthet F., Wuilloud A., Robinson W.P. Intrachromosomal triplication of 15q11-q13. J. Med. Genet. 1994;31:798–803. doi: 10.1136/jmg.31.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chauvin A., Chen J.M., Quemener S., Masson E., Kehrer-Sawatzki H., Ohmle B., Cooper D.N., Le Maréchal C., Férec C. Elucidation of the complex structure and origin of the human trypsinogen locus triplication. Hum. Mol. Genet. 2009;18:3605–3614. doi: 10.1093/hmg/ddp308. [DOI] [PubMed] [Google Scholar]
- 21.Kaku D.A., Parry G.J., Malamut R., Lupski J.R., Garcia C.A. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806–1808. doi: 10.1212/wnl.43.9.1806. [DOI] [PubMed] [Google Scholar]
- 22.Sereda M., Griffiths I., Pühlhofer A., Stewart H., Rossner M.J., Zimmerman F., Magyar J.P., Schneider A., Hund E., Meinck H.M. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 23.Garcia C.A., Malamut R.E., England J.D., Parry G.S., Liu P., Lupski J.R. Clinical variability in two pairs of identical twins with the Charcot-Marie-Tooth disease type 1A duplication. Neurology. 1995;45:2090–2093. doi: 10.1212/wnl.45.11.2090. [DOI] [PubMed] [Google Scholar]
- 24.Badano J.L., Inoue K., Katsanis N., Lupski J.R. New polymorphic short tandem repeats for PCR-based Charcot-Marie-Tooth disease type 1A duplication diagnosis. Clin. Chem. 2001;47:838–843. [PubMed] [Google Scholar]
- 25.Latour P., Boutrand L., Levy N., Bernard R., Boyer A., Claustrat F., Chazot G., Boucherat M., Vandenberghe A. Polymorphic short tandem repeats for diagnosis of the Charcot-Marie-Tooth 1A duplication. Clin. Chem. 2001;47:829–837. [PubMed] [Google Scholar]
- 26.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P., Lacaria M., Zhang F., Withers M., Hastings P.J., Lupski J.R. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am. J. Hum. Genet. 2011;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rautenstrauss B., Liehr T., Fuchs C., Bevot A., Bornemann A., Postler E., Meyermann R., Uhlhaas S., Friedl W., Michaelis R. Mosaicism for Charcot-Marie-Tooth disease type 1A: onset in childhood suggests somatic reversion in early developmental stages. Int. J. Mol. Med. 1998;1:333–337. doi: 10.3892/ijmm.1.2.333. [DOI] [PubMed] [Google Scholar]
- 29.Lupski J.R. Genetics. Genome mosaicism—one human, multiple genomes. Science. 2013;341:358–359. doi: 10.1126/science.1239503. [DOI] [PubMed] [Google Scholar]
- 30.Ross O.A., Braithwaite A.T., Skipper L.M., Kachergus J., Hulihan M.M., Middleton F.A., Nishioka K., Fuchs J., Gasser T., Maraganore D.M. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008;63:743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman M.P., Conforti L., Buckmaster E.A., Tarlton A., Ewing R.M., Brown M.C., Lyon M.F., Perry V.H. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc. Natl. Acad. Sci. USA. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeleny C. The direction and frequency of mutation in the bar-eye series of multiple allelomorphs of Drosophila. J. Exp. Zool. 1921;34:202–233. [Google Scholar]
- 33.Zhang F., Seeman P., Liu P., Weterman M.A., Gonzaga-Jauregui C., Towne C.F., Batish S.D., De Vriendt E., De Jonghe P., Rautenstrauss B. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giorda R., Ciccone R., Gimelli G., Pramparo T., Beri S., Bonaglia M.C., Giglio S., Genuardi M., Argente J., Rocchi M., Zuffardi O. Two classes of low-copy repeats comediate a new recurrent rearrangement consisting of duplication at 8p23.1 and triplication at 8p23.2. Hum. Mutat. 2007;28:459–468. doi: 10.1002/humu.20465. [DOI] [PubMed] [Google Scholar]
- 35.Lupski J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007;39(Suppl):S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 36.Liu P., Erez A., Nagamani S.C., Dhar S.U., Kołodziejska K.E., Dharmadhikari A.V., Cooper M.L., Wiszniewska J., Zhang F., Withers M.A. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dittwald P., Gambin T., Szafranski P., Li J., Amato S., Divon M.Y., Rodríguez Rojas L.X., Elton L.E., Scott D.A., Schaaf C.P. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013;23:1395–1409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bi W., Probst F.J., Wiszniewska J., Plunkett K., Roney E.K., Carter B.S., Williams M.D., Stankiewicz P., Patel A., Stevens C.A. Co-occurrence of recurrent duplications of the DiGeorge syndrome region on both chromosome 22 homologues due to inherited and de novo events. J. Med. Genet. 2012;49:681–688. doi: 10.1136/jmedgenet-2012-101002. [DOI] [PubMed] [Google Scholar]
- 39.Lupski J.R., Belmont J.W., Boerwinkle E., Gibbs R.A. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.