

Abstract
The prevalence of asthma and obesity is increasing worldwide, and obesity is a well-documented risk factor for asthma. The mechanisms underlying this association and parallel time trends remain largely unknown but genetic factors may be involved. Here, we report on a common ∼0.45 Mb genomic inversion at 16p11.2 that can be accurately genotyped via SNP array data. We show that the inversion allele protects against the joint occurrence of asthma and obesity in five large independent studies (combined sample size of 317 cases and 543 controls drawn from a total of 5,809 samples; combined OR = 0.48, p = 5.5 × 10−6). Allele frequencies show remarkable worldwide population stratification, ranging from 10% in East Africa to 49% in Northern Europe, consistent with discordant and extreme genetic drifts or adaptive selections after human migration out of Africa. Inversion alleles strongly correlate with expression levels of neighboring genes, especially TUFM (p = 3.0 × 10−40) that encodes a mitochondrial protein regulator of energy balance and inhibitor of type 1 interferon, and other candidates for asthma (IL27) and obesity (APOB48R and SH2B1). Therefore, by affecting gene expression, the ∼0.45 Mb 16p11.2 inversion provides a genetic basis for the joint susceptibility to asthma and obesity, with a population attributable risk of 39.7%. Differential mitochondrial function and basal energy balance of inversion alleles might also underlie the potential selection signature that led to their uneven distribution in world populations.
Introduction
Epidemiological studies have shown that obese individuals are more likely to develop asthma than nonobese subjects and that asthma symptoms may improve after weight reduction.1 Obese individuals with asthma tend to have more severe symptoms with poorer response to glucocorticoid therapy. In addition, the epidemiological association between asthma and obesity seems to be modulated by gender and atopy,2,3 but the mechanisms underlying obesity in asthma remain largely unknown. Proposals for the joint occurrence of these two disorders include comorbidities such as gastresophageal reflux or sleep disordered breathing and common pathogenic mechanisms such as systemic inflammation, increased oxidative stress, or hormonal dysfunction.4,5 Genetic factors may also be involved in the coexistence of asthma and obesity. The best-fitting model of shared components of variance indicated that about 8% of the genetic component of obesity is shared with asthma.6
Recent candidate gene and genome-wide association studies (GWASs) have discovered polymorphisms in the 16p11.2 chromosomal region that are associated with obesity or body mass index (BMI)7,8 and also with asthma phenotypes.9 A functional SNP in the gene coding for the SH2B adaptor protein 1 (SH2B1 [MIM 608937]) is involved in the signaling of leptin,10 a hormone implicated in T cell immune response in addition to its major role in the regulation of body weight.11 SNPs located in the vicinity of the IL27 (interleukin 27 [MIM 608273]) locus have also been associated with susceptibility to asthma in adults and children.9,12 Recurrent copy-number variations in 16p11.2 such as the microdeletion and reciprocal microduplication (0.6 Mb, Mb 29.5–30.1, UCSC Genome Browser build hg18) are associated with extreme BMI and neuro-behavioral disorders.13,14 Deletions of the immediately distal region encompassing SH2B1 (0.22 Mb, Mb 28.75–28.95, hg18) have also been reported in early-onset obesity, as well as in developmental delay and schizophrenia.15–18 A more distal inversion variant (maximum size estimate ∼0.45 Mb: 28.25–28.7 Mb, hg18) was originally described during chromosome 16 assembly, where EIFVar1 and EIFVar2 are the reference noninverted (EIFVar1/NI) and the inverted (EIFVar2/I) alleles, respectively.19 Although the experimental detection of chromosomal inversions located in complex genomic regions is difficult, recently described methods can infer non- or low-recurrent inversions for which recombination between the inverted and noninverted segments is suppressed in heterozygous carriers via SNP genotype data.20–22 One such detectable inversions is the ∼0.45 Mb 16p11.2 inversion.21
Here we used SNP microarray data to detect and genotype the ∼0.45 Mb polymorphic inversion at 16p11.2 by using two algorithms20,21 and validated genotype predictions with allele-specific molecular assays. Association studies in a discovery case-control study for asthma (n = 2,205) and four validation data sets (n = 3,604) revealed that the ∼0.45 Mb 16p11.2 inversion provides a genetic basis for the joint susceptibility to asthma and obesity in European populations. We estimated the inversion prevalence in several world populations and compared the differences with demographic data showing an uneven distribution in world populations and suggesting either extraordinary genetic drift or adaptive selection. Functional impact of inversion alleles was analyzed in blood cell transcriptomes in two independent data sets pointing to candidate genes for this shared susceptibility.
Material and Methods
Subjects, Genotype, and Phenotype Data
We initially studied the Estonian Gene Expression Cohort, which is composed of 1,000 randomly selected samples (mean age 37 ± 16.6 years; 50% females) from a larger cohort of 53,000 samples of the Estonian Genome Center Biobank, University of Tartu (EGCUT). DNA was genotyped with the Human370CNV array (Illumina). Only samples with genotyping success rate above 95% were used. Cryptic relatedness was tested with the PLINK v.1.07 software. Only one of each detected relative pairs (up to second cousins) was randomly chosen for the subsequent analyses. Sample mix-ups were corrected by MixupMapper software.23
The 1,184 samples of the HapMap 3 panel had been genotyped with two platforms: the Illumina Human1M SNP array (by the Wellcome Trust Sanger Institute) and the Affymetrix SNP 6.0 (by the Broad Institute).24 Data from the two platforms corresponding to genome build 36 release 2 were merged and downloaded in PLINK format from the website. Data from 1000 Genomes were used to define genotypes in samples available for additional experimental validation. We then used DNAs from a total of 71 HapMap 3 individuals (23 European [CEU], 24 Yoruba, and 24 Asian [16 Chinese and 8 Japanese]), all obtained from Coriell.
For association studies with asthma and obesity, the discovery sample corresponded to the European Community Respiratory Health Survey I (ECRHS-I).25 This data set represents a nested asthma case-control sample from the cohort, including all subjects with asthma at baseline or follow-up with DNA and a random sample of controls (n = 2,205). Genotyping had been conducted with the Illumina Human 610 quad array at the French National Genotyping Centre (CNG) in the frame of the European GABRIEL consortium where quality control (QC) of genotyping was also conducted.
Four additional studies were used for association validation: (1) the population-based EGCUT data (n = 1,217), (2) the Epidemiological study on Genetics and Environment of Asthma (EGEA)26 (n = 1,252), (3) the Genetic Epidemiology of Chronic Obstructive Pulmonary Disease (COPD) cohort (n = 472) (COPDGene, dbGaP Study accession number phs000179.v3.p2), and (4) the Asthma Clinical Research Network (ACRN) (n = 663) from the National Heart, Lung, and Blood Institute SNP Health Association Asthma Resource Project (SHARP, dbGaP Study Accession number phs000166.v2.p1); studies 3 and 4 were from the database of Genotypes and Phenotypes (dbGAP)27.
All individuals selected in the discovery and validation studies were of European descent and all the analyses were adjusted for gender and principal components in order to address any possible confusion resulting from population stratification. From EGCUT, we considered a nested case-control study including adult individuals diagnosed with asthma belonging to the larger population-based cohort of 53,000 samples from Estonia. Controls were randomly selected from the general population without asthma diagnosis. DNA was genotyped as indicated above. EGEA combines a case-control study and a family study of asthmatic cases, but only adults were used for the present analysis (age ≥ 18 years old). Genotyping was conducted in a similar manner as in the ECRHS-I study, previously described. COPDGene is an adult case-control to study COPD that includes information about asthma and obesity (BMI). In order to eliminate possible confusion, the analysis only considered asthmatic and control individuals without COPD. Genotyping was performed with Illumina HumanOmni1 Quad v1-0 B platform. ACRN is part of SHARP, a study that provides a genome-wide analysis in adults and children who have participated in National Heart, Lung, and Blood Institute’s clinical research trials on asthma. Because all individuals included in the ACRN study were adult cases diagnosed with asthma, controls were obtained from the COPDGene study, assuming both studies had been carried out in populations with similar European ancestry. Notably, inversion genotype frequencies in the controls of the COPDGene study were almost identical to those in the discovery sample (ECRHS-I). Genotyping for the SHARP studies was performed with the Affymetrix 6.0 platform.
Asthma diagnosis was based on a positive response to the question whether it had been ever diagnosed, and obesity corresponded to a BMI ≥30 kg/m2. We used this sensitive definition for asthma (perhaps not the most specific) in order to have the lowest misclassification and less biased coefficients among studies. This definition was common for all studies and previously used in the GWASs performed with these data.28 Most individuals in the studies designed for asthma (ECRHS-I, EGEA, and ACRN) had a more specific diagnosis of current asthma at recruitment: attack of asthma during the last 12 months and/or current use of asthma medication. The total number of individuals included was 5,809, all adults of both genders: 1,872 cases with a diagnosis of asthma and 3,937 controls without asthma, including 317 cases with asthma and obesity and 543 controls with obesity and no asthma (Table S5 available online). All studies included in this manuscript had been performed in accordance with the ethical standards of the responsible committee on human experimentation and with proper informed consent from cases and controls.
Inversion Classification and Detection
We applied two algorithms that perform inversion calling from SNP data: inveRsion21 and Phase Free Inversion Detection Operator (PFIDO).20 We selected a region of interest defined by the interval between flanking blocks of segmental duplications (28.24–28.73 Mb, hg18) and let inveRsion look for a signal in the region of interest widen by 2 Mb, with a sliding window size of 0.39 Mb (∼80% the inverted interval). For the CEU data we used inveRsion on phased haplotype data21 but for all other cohorts the inversion signal could be detected with unphased genotype data. We selected the model for which the classification could explain most of the variance in the multidimensional scaling step of PFIDO. Here, we implemented a simple version of the method, where the clustering step is performed with a K-means algorithm by using multiple initial conditions. The final classification of inveRsion and PFIDO were contrasted with a Cohen’s kappa and with the variability explained by inveRsion’s classification by the multidimensional scaling analysis of PFIDO. We also computed linkage disequilibrium (LD) between the inversion and the SNPs of the region. For the ECRHS-I and EGEA samples, a total of 80 SNPs were analyzed, and for the EGCUT samples 132 SNPs were analyzed. Additional analyses searching for independent association of single SNPs in the region with either the asthma or BMI variables were also performed.
Comparative Mapping, Sequence, and Coalescence Analyses
In order to define the genomic architecture of the human genome region with respect to other species, we used the UCSC Genome Browser and compared the genomic structure, the relative gene order and orientation of the homologous regions, and the breaks of synteny between human, mouse, and other mammals. Complete alignment of the reference noninverted (EIFVar1/NI) and the inversion (EIFVar2/I) haplotypes was performed with the EMBOSS Stretcher algorithm. All single-nucleotide and insertion/deletion differences between both haplotypes were annotated covering both the single-copy region between segmental duplications (119 kb) and the flanking inverted repeats (134 kb). Sequence coalescence estimates were performed on the assumption of complete lack of recombination between both haplotypes over the 119 kb single-copy interval located between the flanking blocks of segmental duplications. The time to most recent common ancestor was calculated with MEGA5 by the Kimura 2-parameter method and evolutionary rate equality assessed with Tajima’s relative rate test.29 Divergence times between NI and I haplotypes were estimated with the formula T = K/2R (where T is time, K the Kimura 2-parameter, and R the average taxa substitution rate) with chimpanzee as an out-group. To minimize misalignment mistakes, we used 14,770 bp of chimpanzee sequence within the interval that pair with no gaps with the human sequence. These estimates were computed assuming a mutation rate of 2.5 × 10−8.
We identified the genotypes at the representative SNPs on strong LD with inversion haplotypes in the NI and I alleles. We then used the UCSC Genome Browser to define the genotypes at those representative loci in nonhuman primates, as well as in Neanderthal, Denisova, and several representative individuals from modern human populations.
Indel Polymorphism Genotyping
Two indel differences identified between the EIFVar1/NI and EIFVar2/I sequences were genotyped by PCR under standard conditions. For the 27 bp indel within APOB48R (apolipoprotein B48 receptor [MIM 605220]) (293 bp insertion allele, 266 bp deletion allele), we used the primers 5′-CTGTCTGACTCCCAGCAGGT-3′ and 5′-AGGCCAGGGCAATTTTAGAT-3′. For the intergenic 19 bp indel (224 bp insertion allele, 205 bp deletion allele), the primers were 5′-TACCGCACTAGGCCAATCTT-3′ and 5′-GTGTTTCTCCCTTGCCTGAG-3′. PCR products were visualized by ethidium bromide staining after electrophoresis on 2% agarose gels.
Gene Expression and Gene Ontology Enrichment Analyses
Transcriptomic data of RNA from lymphoblastoid cell lines of CEU individuals generated through the project E-MTAB-198 were obtained from the European Bioinformatics Institute at EMBL. Only data from 105 unrelated individuals were analyzed, removing samples from family relatives. We also assessed the association between inversion and gene expression levels in RNA from peripheral blood samples of the EGCUT sample. Whole-blood RNA was collected with Tempus Blood RNA Tubes (Life Technologies), RNA was extracted with Tempus Spin RNA Isolation Kit (Life Technologies), and quality was measured by NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific) and Agilent 2100 Bioanalyzer (Agilent Technologies). Whole-genome gene-expression levels were obtained by Illumina HT12v3 arrays (Illumina) according manufacturer’s protocols. Low-quality samples were excluded. All probes with primer polymorphisms were left out, leaving 34,282 probes. Raw gene expression data was Log-Quantile normalized with MixupMapper software.23 The final EGCUT sample size with both genotype and gene expression data was 882.
Gene ontology (GO) enrichment analysis was performed including all genes that were differentially expressed with respect to the inversion at a false discovery rate (fdr) <10−3. The analysis was performed with GOstats bioconductor package. GO terms were claimed as significantly associated with the inversion with an FDR <0.05. A concept-network with GeneAnswers bioconductor package was then estimated.
Statistical Analyses
Association analysis between phenotype and inversion genotype was performed with the SNPassoc R package, which provides p values from likelihood ratio test (LRT) as well as odds ratios (OR) and 95% confidence intervals (95% CI) for different genetic models (dominant, recessive, and additive).30 Multiple comparisons were addressed by correcting the p value for the exact number of tests, that was considered as 2.2.31 Models were adjusted for gender and the first two principal components to address population stratification. Models for EGEA also considered family structure.
FST values were calculated with the snpStats package v.1.8.1 from Bioconductor, and negative values were set to zero. To construct the empirical null distribution, FST values were calculated in all HapMap 3 populations for both the inversion alleles and a set of 1,024 chromosome 16 SNPs outside the inversion and not in LD (r2 < 0.2). A null distribution of each pairwise comparison was generated with the respective p values.
Population stratification of inversion genotypes of HapMap 3 samples was assessed via multiple correspondence analysis. Differential gene expression analysis between inversion genotypes and genes was performed via generalized linear models and LRT by means of the SNPassoc R package.30 LD (measured via R squared statistic) between the inversion genotype and the SNPs in the region of interest was computed with snpStats.
Results
Inversion Detection, Validation, and Comparative Mapping
The experimental detection of inversions located in complex genomic regions with large segmental duplications is technically difficult. It usually relies on individually applied cytogenetic, molecular cytogenetic, and more recently paired-end sequencing methods. Nonetheless, recently described and distinct methods such as Phase Free Inversion Detection Operator (PFIDO), inveRsion, and Principal Components Analysis (PCA) can infer inversion polymorphism from SNP genotype data.20–22 These methods mainly detect nonrecurrent inversions for which recombination between the inverted and noninverted segments is suppressed in inversion heterozygotes. One such detectable inversions is the ∼0.45 Mb distal 16p11.2 inversion.21 The inveRsion algorithm successfully detected signals of inversion in the interval between two blocks of segmental duplications at chromosome band 16p11.2 (28.39–28.50 Mb, hg18) (Figure 1). We used both inveRsion and PFIDO approaches to detect and classify individual chromosomes into the standard (EIFVar1/NI) or inverted (EIFVar2/I) subpopulations (Table S1), initially using the EGCUT (n = 882) and the HapMap 3 (n = 1,184) populations. A high calling concordance was observed between PFIDO and inveRsion (0.96–0.97) (Table S1).
Figure 1.
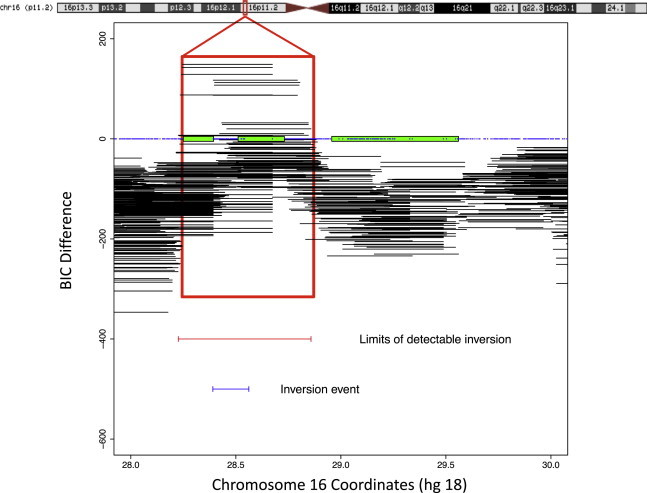
Evidence of an Inversion Polymorphism Signal at 16p11.2
The figure shows the result of a sliding window scan with inveRsion in the 16p11.2 chromosomal region (28–30 Mb, hg18), with a window size of 0.39 Mb in the CEU population. Segments with positive Bayesian Information Criterion (BIC) difference are considered informative for the presence of LD differences in the population resulting from an inversion event. A schematic representation of the genomic region with the blocks of segmental duplications as green filled rectangles is shown at the 0 value of the BIC difference. The region includes the predicted inversion polymorphism (minimal single-copy interval between two blocks of segmental duplications) and the limits of the detectable signal.
Comparative mapping of the human reference region with the orthologous region in mouse and other mammals revealed a redistribution of the human genome with breaks of synteny at the positions where the human segmental duplications have been generated. At least two evolutionary chromosomal inversion events must have occurred in the human reference chromosome with respect to the ancestral allele (Figure 2).
Figure 2.
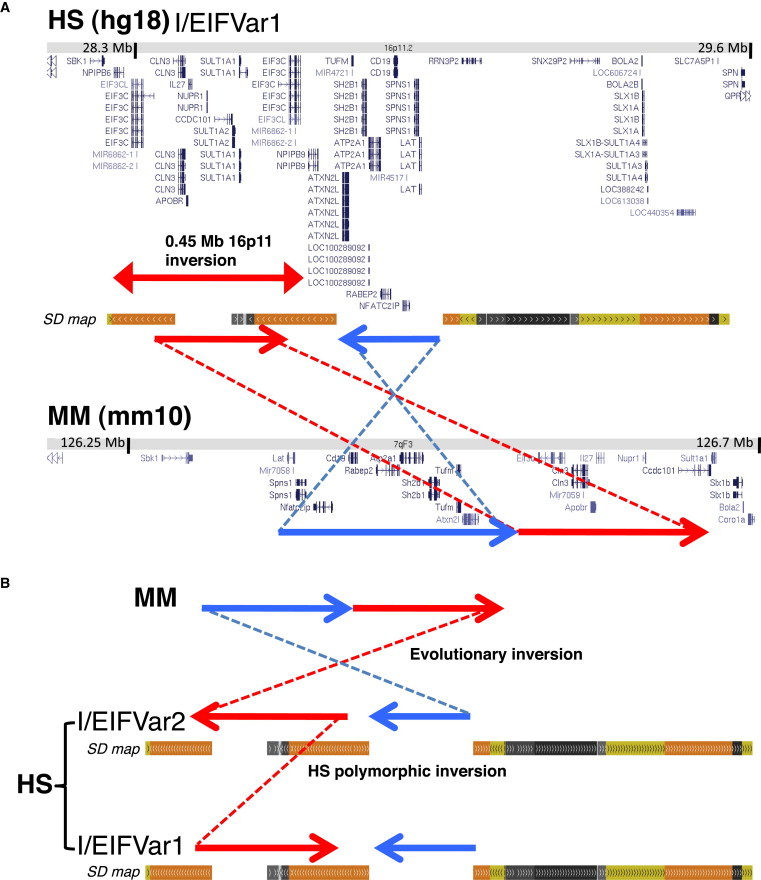
Comparative Mapping, Gene Content, and Predicted Evolution of the Inversion Polymorphism at 16p11.2
(A) Comparative mapping of the human (HS) reference region with the orthologous region in mouse (MM) showing the gene content and orientation according to the UCSC Genome Browser. The blue and red arrows with the connected lines illustrate the length and relative orientation of the blocks of conserved synteny between both species. Genomic structure in other sequenced mammals, such as cat, dog, and rat, is identical to that in mouse. Large and complex blocks of segmental duplications, absent in mouse, are observed in the human map at the breaks of synteny.
(B) Schematic representation of the predicted evolutionary inversion between the mouse genome (MM) and the likely most ancestral human allele (EIFVar2/I). The subsequent inversion between blocks of segmental duplications to generate the other allele (EIFVar1/NI) in humans (HS) is shown at the bottom.
BLAST alignment between the reference EIFVar1 and inverted EIFVar2 sequences revealed 99.89% sequence identity throughout the single-copy interval (119 Kb) flanked by two large blocks of inverted paralogous segmental duplications (134 Kb, 99.7% sequence identity) (Figure 1). Interallelic sequence changes included multiple SNPs, microsatellite length changes, and two small insertion/deletion (indel) changes (Figure S1). We designed a PCR-based method to genotype these two indel polymorphisms and correlated the results with the algorithm-based inversion classification (Figure 3A). Out of 71 selected HapMap individuals (142 chromosomes), a single nonconcordant chromosome was identified at the 19 bp indel locus, reinforcing the accurate prediction of the calling methods (99% concordance). High concordance (90%) although with some more discrepancies (14 out of 136 chromosomes) was also found at the 27 bp indel. These discrepancies are most probably due to the higher mutation rate at this variable number of tandem repeat locus, including a few chromosomes with a different allele (Table S2). Additional haplotypes with structural and/or sequence changes that cannot be currently resolved appear to exist within the NI and I lineages, as noted by the presence of six clearly distinguishable clusters by a multidimensional scaling analysis of the SNPs in the inverted region for the 1000 Genomes populations (Figure 3B).
Figure 3.
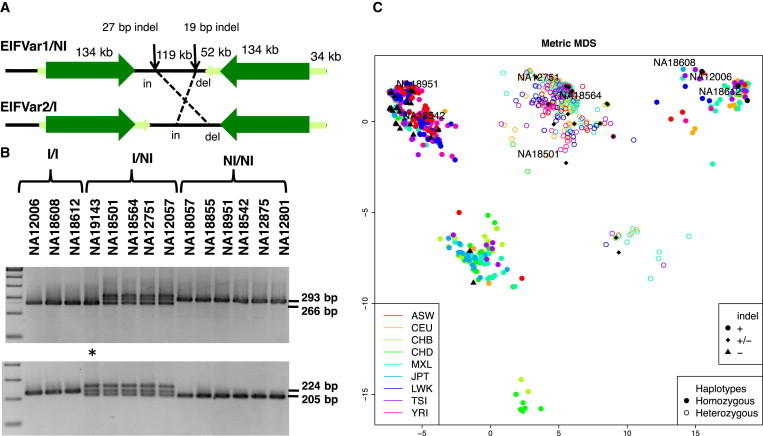
Definition of Haplotype Markers, Experimental Validation, and Genotype Clustering
(A) Schematic representation of the EIFVar1/NI and EIFVar2/I alleles at 16p11.2 with the flanking segmental duplications (inverted dark green arrows) and the single-copy interval in between. Other smaller blocks of segmental duplications are shown in light green. The location of the two insertion-deletion polymorphisms and the specific alleles per haplotype are indicated.
(B) Results of genotyping by PCR these two indels in 14 HapMap individuals. Predicted inversion haplotypes of the same individuals by inveRsion and PFIDO are shown on top of each line and represented in the plot below. An asterisk denotes the only individual with discordant results.
(C) Plot showing the clustering of the different haplotypes predicted by PFIDO via multidimensional scaling (MDS) in some of the 1000 Genomes populations. Each circle (open for heterozygotes and filled for homozygotes) represents an individual from a population, differentiated by colors as indicated. Abbreviations are as follows: ASW, African ancestry in Southwest USA; CEU, Utah residents with Northern and Western European ancestry; CHB, Han Chinese in Beijing, China; CHD, Chinese in metropolitan Denver, Colorado; MXL, Mexican ancestry in Los Angeles, California; JPT, Japanese in Tokyo, Japan; LWK, Luhya in Webuye, Kenya; TSI, Toscans in Italy; YRI, Yoruba in Ibadan, Nigeria. The samples that have also been studied by PCR (Table S2) are represented by black circles, diamonds, or triangles according their 19 bp indel genotype. A few samples also included in (B) are identified.
Inversion Origin and Population Distribution
We identified the genotypes at 24 representative SNPs on strong LD (r2 > 0.8) with inversion haplotypes in the NI and I alleles. At most of these loci, all nonhuman primates as well as the Neandertal and Denisova genomes carry the I-allele nucleotide (Table S3). Therefore, although the EIFVar1/NI is the most common allele in the human populations studied, comparative mapping data suggest that the I allele is indeed the most ancient, and the allele represented in the human reference genome must have been generated more recently in human evolution. Assuming a mutation rate of 2.5 × 10−8, we estimated an ancient coalescence of approximately 1.35 million years ago.
The I-allele frequency ranged from 0.10 in Eastern Africans (Massai from Kenya) to 0.49 in Northern Europeans (Estonia), showing significant stratification in HapMap 3 populations (Figure 4, Table S4). Allele frequencies were found at Hardy-Weinberg equilibrium in each population, except for the Gujarati Indians in Houston, using a relatively conservative threshold for equilibrium rejection (p < 0.05) (Table S4). The distribution of frequencies showed a systematic gradation over space. However, the inversion distribution does not appear to correlate with demographic factors because pairwise FST values did not fully correlate with the geographic distance between sampled populations (Figure S2). Therefore, these data highly indicate either a remarkable drift or signatures of selection for the inversion alleles in different populations.
Figure 4.
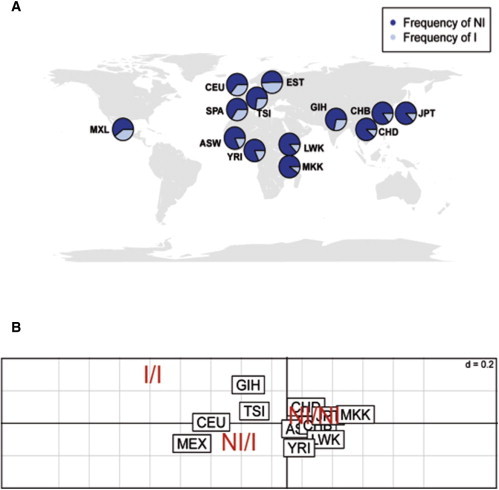
Worldwide Population Stratification of 16p11.2 Inversion
(A) Frequency of ∼0.45 Mb 16p11.2 inversion alleles in different populations, with the circles located on the map at their approximate geographic origin. The figure shows very low frequencies of the I allele in Africa and Asia populations and gradual increase in European populations with higher frequencies in the north.
(B) The two principal components obtained after performing a correspondence analysis via inversion polymorphism genotypes of HapMap 3 samples are represented. Prevalence of the inversion clusters changes according to the geographical population origin, indicating strong population stratification.
The populations included in the analysis are the following: ASW (n = 83); CEU (n = 165); CHB (n = 84); CHD (n = 85); JPT (n = 86); LWK (n = 90); MXL (n = 77); TSI (n = 88); YRI (n = 167); MKK, Massai in Kinyawa, Kenya (n = 171); GIH, Gujarati Indians in Houston, Texas (n = 88); SPA, Spaniards in Spain (n = 765); EST, Estonian in Estonia (n = 882).
Association with the Joint Occurrence of Asthma and Obesity
A decreased risk of asthma associated with the I allele was found in the ECRHS-I data set (333 asthma cases and 1,527 controls), borderline significant under a dominant model (OR = 0.75, 95% CI = 0.59–0.96, p = 0.0231, corrected p = 0.0508) (Table S5). This risk was further and significantly decreased for the joint occurrence of asthma and obesity also under a dominant model (OR = 0.35, 95% CI = 0.14–0.84, p = 0.0178, corrected p = 0.039). The additive model was also borderline significant (OR = 0.40, p = 0.0379, corrected p = 0.0834) whereas recessive was not (p = 0.1410).
Given that dominant was the best model of inheritance, it was selected for replication in four additional independent studies. A protection for the asthma and obesity co-occurrence with the I haplotype was also found in each study (OR = 0.33–0.62). A meta-analysis of the results of the five studies improved the significant evidence for a decreased risk of asthma in obese people with the I allele (OR = 0.48, 95% CI = 0.34–0.66, p = 5.5 × 10−6) (Figure 5, Table S5). The estimated population attributable risk of the inversion polymorphism for the asthma and obesity phenotype was 39.7%. The association of the I allele with asthma alone in the meta-analysis was also significant but weaker (OR = 0.82, 95% CI = 0.73–0.93, p = 0.0026) (Figure S4), whereas there was no significant association between the inversion polymorphism and obesity in nonasthmatic subjects (Table S5).
Figure 5.
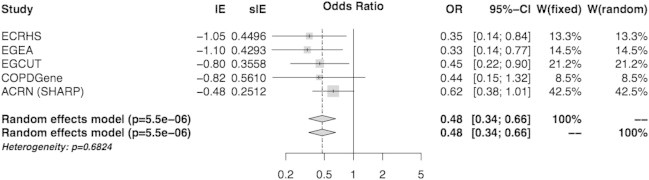
Meta-analysis Forest Plot for the Association Studies of Inversion with Phenotypes
The results of the association analysis between the ∼0.45 Mb 16p11.2 distal inversion with asthma, stratified by obesity phenotype (BMI ≥ 30 kg/m2) in the five independent studies are shown. The inversion-allele effect (IE), the standard error of the effect (sIE), the odds ratio (OR) with confidence intervals, and the relative weight (W) of each study correspond to the inversion allele effects under a dominant model on asthma in obese individuals.
We then investigated whether a set of SNPs can tag the inversion polymorphism by computing LD between the inversion and the SNPs of the region. We observed clusters of high LD composed of 7 SNPs in a small 30 Kb interval within the inversion (28.42–28.45 Mb) (r2 > 0.8) and 24 SNPs in a 70 Kb region (28.73–28.8 Mb) immediately proximal to the inversion with r2 > 0.94 (Figure 6A). Interestingly, SNP rs26528 is within the IL27 locus and rs7498665 in SH2B1. The search for independent association of SNPs in the region with either the asthma or BMI variables did not yield significant results in any of the studied sample data sets, indicating that the observed effect on asthma and obesity is more probably due to the entire inversion haplotype or a combination of SNPs than to individual SNPs.
Figure 6.
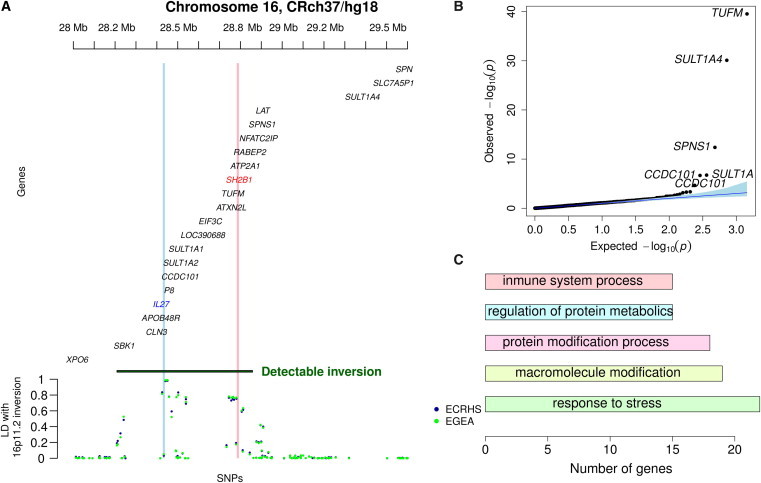
Functional Correlations of the 16p11.2 Inversion
(A) LD between the ∼0.45 Mb 16p11.2 inversion polymorphism and SNPs in the 28.2–29.5 Mb region of chromosome 16. Gene annotation was taken from the UCSC Genome Browser, hg18 assembly. IL27 and SH2B1 are highlighted in blue and red, respectively, and by vertical shading, indicating they have been already reported as asthma and obesity susceptibility genes, respectively, by association studies.
(B) Quantile-quantile plot of genes corresponding to gene expression analysis in the Estonian samples.
(C) Barplot showing the top five gene ontology (GO) category distribution of the genes differentially expressed (FDR < 10−3) with respect to the inversion in Estonian samples.
Functional Characterization
In order to characterize the functional consequences of the inversion polymorphism and define candidate genes for the associated phenotypes, we analyzed gene expression levels in peripheral blood of EGCUT and lymphoblastoid cell lines of HapMap CEU samples (Figure 6B). Several genes located in the chr16 inversion or immediately flanking intervals were identified in both data sets among the top ten most differentially expressed genes per inversion genotype (Table 1). In particular, expression of TUFM (mitochondrial Tu translation elongation factor [MIM 602389]), SULT1A4 (sulfotransferase family, cytosolic, 1A, phenol-preferring, member 4), SPNS1 (spinster homolog 1 [MIM 612583]), SULT1A1 (sulfotransferase family, cytosolic, 1A, phenol-preferring, member 1 [MIM 171150]), and CCDC101 (coiled-coil domain containing 101 [MIM 613374]) highly correlated with the inversion genotypes (p ranging from 10−40 to 10−7) in EGCUT (Table S5). In the HapMap CEU samples, SNPS1, EIF3C (eukaryotic translation initiation factor 3, subunit C [MIM 603916]), and TUFM were also among the most differentially expressed genes (p < 1 × 10−2), and IL27 expression also showed some correlation with the inversion (p = 1.0 × 10−2) (Table S6). Expression effects were observed on single-copy genes located within the inversion (IL27 and CCDC101) and immediately proximal interval (TUFM and SPNS1), as well as on multiple-copy genes located in the flanking segmental duplications (EIF3C, SULT1A1, and SULT1A4). In the case of TUFM, SPNS1, EIF3C, SULT1A1, and APOB48R, expression levels were higher in I alleles; for SULT1A4 and CCDC101, expression levels were higher in NI alleles; and in the case of IL27, the higher expression was in NI/I heterozygotes (Figures S4–S8 and Tables S6, S7 and S8).
Table 1.
Top Five Deregulated Genes of the Chromosome 16 in Association with 16p11.2 Inversion Alleles
| Probe Set | p Value | Start (bp) | End (bp) | Gene | NI/NI | NI/I | I/I |
|---|---|---|---|---|---|---|---|
| ILMN_1738369 | 3.0 × 10−40 | 28,761,849 | 28,761,898 | TUFM | − | ref | + |
| ILMN_2336133 | 7.8 × 10−31 | 29,467,127 | 29,476,300 | SULT1A4 | + | ref | − |
| ILMN_1681016 | 4.2 × 10−13 | 28,903,301 | 28,903,350 | SPNS1 | − | ref | + |
| ILMN_1657302 | 1.8 × 10−7 | 28,538,926 | 28,538,955 | SULT1A1 | − | ref | + |
| ILMN_1684789 | 2.2 × 10−7 | 28,510,516 | 28,510,565 | CCDC101 | + | ref | − |
Annotation taken from UCSC Genome Browser, hg18 assembly. Columns NI/NI, NI/I, I/I provide information about the mean value of gene expression of each genotype with respect to the heterozygous NI/I, provided as reference. Plus sign (+) indicates more; minus sign (−) indicates less.
We then estimated the percentage of observed variability in gene expression of the top five genes explained by the inversion genotype, gender, and BMI in EGCUT samples. We observed that in all five genes, the inversion is the factor explaining the highest percentage of variability, ranging from 3.1% (CCDC101) to 17% (TUFM) (Table S9). Therefore, the expression variation of these genes is most likely related to the inversion itself, driven either by direct mechanisms such as chromatin structure or positional change of promoter or enhancer elements or by indirect effects secondary to functional SNPs fixed on the different haplotypes with no recombination.
In order to define in a more global way the transcriptomic effects of the 16p11.2 inversion, gene ontology (GO) enrichment analysis was performed including all genes differentially expressed in both data sets with respect to the inversion (p < 10−3). A concept-network was obtained with remarkable enrichment of terms related to regulation and activation of immune response, positive regulation of leukocyte, lymphocyte, and T cell activation, NK-T cell proliferation, and immune response to virus, as well as regulation of response to stress and biotic stimulus (Figure S9).
Discussion
We report here that the ∼0.45 Mb 16p11.2 inversion is a common polymorphism that can be efficiently inferred from SNP data in different populations as a result of the inversion-mediated suppression of recombination. This also indicates that its origin is ancient with very low recurrence, similar to other reported genomic inversions that can be tagged by SNP haplotypes.20,32 However, the classification is not perfect as noted by the presence of distinguishable clusters in the different populations, which are probably due to additional structural or sequence changes within and/or in the flanking regions not yet resolved (Figure 3C). The inversion contains a single-copy inverted interval (119 Kb) flanked by two large blocks of highly similar segmental duplications that most likely mediated the evolutionary inversion by nonhomologous unequal recombination. This complex structure precludes the genotyping of the inverted alleles by standard methods such as interphase FISH, long PCR, or paired-end mapping by sequencing, even via large insert clones. Therefore, the experimental validation of the predicted inversion genotypes or haplotypes was achieved by using an indel polymorphism that almost perfectly tagged both the I and NI alleles. This marker can also be used for high-scale genotyping in other populations when SNP data from the region are not available.
Contrary to expected, comparative mapping indicated that the I allele is more ancient and the reference and most common allele in all populations studied must have been generated more recently in human evolution. However, I allele frequencies were very low in Africans and eastern Asians, being higher in Europeans with a gradation increasing from south to north. The poor correlation of allelic frequencies with the demographic history of the human populations suggests either an extraordinary genetic drift or, most likely, selective sweeps for the 16p11.2 inversion alleles. When the NI allele appeared in humans around 1.35 million years ago, it might have provided a strong fitness bonus in order to become the most common over time in Africa. However, after the migrations out of Africa 50,000 to 100,000 years ago, the I allele may have provided its own advantage mainly to the populations migrating farther north in Europe. A similar pattern of population stratification with African origin and remarkable genetic drift or selective sweeps has been reported for the 17q21.31 inversion polymorphism.32
We found a strong correlation of inversion genotypes with expression effects on several regional genes. These expression effects in cis could be driven either by direct mechanisms, such as chromatin structure or positional change of promoter or enhancer elements, or by indirect effects secondary to functional SNPs fixed on the different inversion haplotypes with no recombination. In agreement with our data, a cluster of five SNPs of the region has been previously associated with the expression of CLN3 (ceroid-lipofuscinosis, neuronal 3 [MIM 607042]), and three SNPs at SH2B1 were associated with EIF3C and TUFM levels in lymphocytes.33 In addition, a single SNP (rs7498665) in strong LD with inversion alleles was highly correlated with the expression of several genes (TUFM, SULT1A1, SULT1A2, SH2B1, APOB48R, and EIF3C) in blood and/or adipose tissue of the Icelandic population.8 Given that all these SNPs are in strong LD with inversion alleles, some relevant changes on gene expression are probably due to indirect effects of the maintenance of allelic combination influencing levels of several transcripts, although direct effects of the inversion can not be discarded.
The I allele of the 16p11.2 inversion has a significant protective effect on the risk of asthma, stronger for the co-occurrence of asthma and obesity, as documented in five independent studies. It could also read in the opposite direction, implying that the most common NI allele contributes to the susceptibility for the joint occurrence of asthma and obesity. Remarkably, this inversion polymorphism accounts for 3% of the shared components of variance between asthma and obesity with a population attributable risk of 39.7%, thus representing a large proportion of the estimated 8% of variance explained by genetic factors.6 The influence of the inversion on this shared susceptibility is probably mediated by its proven strong effect on the expression of several neighboring genes with regulatory functions of both energy balance, such as APOB48R, SH2B1, and TUFM, and immunity, such as IL27 and also TUFM.
APOB48R encodes the apolipoprotein B48 macrophage receptor that binds to the apolipoprotein B48 of dietary triglyceride-rich lipoproteins and provides essential lipids, lipid-soluble vitamins, and other nutrients to reticuloendothelial cells. Dietary fat induces lipid accumulation along with APOB48R transcription in circulating monocytes.34 Variations close to or in the SH2B1 have been found to associate with obesity in two very large GWASs,7,8 but the risk of obesity is conferred by the SH2B1 variant in LD with the I allele, which is protective for the association of asthma and obesity in our data. TUFM, whose recessive mutations cause combined oxidative phosphorylation deficiency (MIM 610678) resulting in lactic acidosis and fatal encephalopathy, participates in protein translation in mitochondria as well as in the regulation of type 1 interferon and autophagy attenuating the inflammatory response to infections.35 In further agreement with a potential role of these last two genes in the regulation of energy balance, TUFM has been found upregulated in adipose tissue, muscle, and liver, and SH2B1 was upregulated in muscle and downregulated in hypothalamus of Long-Evans rats fed on high-fat diet.36
IL27 is one of the critical cytokines that mediates between the innate and adaptive immune system. IL27 downregulates airway hyperreactivity and lung inflammation during the development of allergic asthma through its suppressive effect on cytokine production. IL27 also cooperates with IFN-γ to inhibit glucocorticoid-induced translocation of the glucocorticoid receptor to the nucleus of macrophages inducing glucocorticoid-resistant airway hyperresponsiveness in a mouse model.37 Expression of IL27 and IFN-γ is increased in the induced sputum of steroid-refractory asthmatics. Inflammation and airway hyperresponsiveness are hallmark features of asthma and deficient type I interferon induction has been reported in atopic asthma exacerbations.38 Therefore, deregulation of IL27 and impaired interferon induction mediated by TUFM with secondary dysfunction of the innate host defense pathways might underlie the higher susceptibility to severe, and possibly steroid-refractory, asthma associated with obesity in NI-allele carriers.
Evidence for adaptive elevations in basal metabolic rate has been reported in northern (circumpolar) populations.39 Given the positive selection of I alleles with an apparent south to north gradient in Europeans and the remarkable contribution to TUFM expression (explaining 17% of gene expression variability), improved mitochondrial function and basal energy balance associated with higher TUFM expression of the I alleles might have conferred an adaptive advantage to the cold climate for the northern populations. This potential selection signature is in line with the pattern of selection of the human mitochondrial DNA and other genes of mitochondrial function.40 In addition, it has been shown that overexpression of TUFM can rescue mutated mitochondrial tRNAs from degradation, acting as suppressor of mitochondrial defects.41,42 This is another potential contributor mechanism to positive selection because it is becoming evident that somatic mutations in human mitochondrial tRNA genes are associated with degenerative disorders, aging, and cancer.43
The analysis of all genes differentially expressed with respect to the inversion also revealed a remarkable enrichment of terms related to regulation and activation of immune response, response to stress, and biotic stimulus. This pattern is consistent with a disturbance of immune response and energy metabolism and further support the observation that genes deregulated by the inversion polymorphism or associated haplotypes may be responsible for the joint protection against asthma and obesity, along with potentially conferring divergent adaptive advantages to different climates.
In summary, by affecting gene expression, the 16p11.2 inversion polymorphism provides a shared genetic basis for the joint susceptibility to asthma and obesity, with uneven distribution in different world populations resulting from either extraordinary genetic drift or adaptive selection. Our data reinforce the concept that the association of asthma and obesity share some common genetic basis as part of their multifactorial etiology, providing additional clues to understand the shared physiopathology and guide specific therapies. The ∼0.45 Mb 16p11.2 inversion joins other genomic inversions also associated with disease susceptibility that can be tagged by SNP haplotypes.20,44 Because genome-wide genomic inversions have been poorly explored to date, additional studies to better define their frequency, evolutionary history, and potential contribution to human genetic variation and disease are required.
Acknowledgments
We thank David Izquierdo and Raquel Flores for technical help and Joel Martin for kindly providing the entire sequence of the EIFVar2 haplotype. Work was supported by the Spanish Ministry of Science and Innovation (MTM2008-02457) and Statistical Genetics Network – GENOMET (MTM2010-09526-E) to J.R.G., the European Research Council (ERC) Starting Grant (243212-INVFEST) to M.C., and the Spanish Ministry of Science and Innovation-ISCIII-FEDER (FIS PI10/2512 and PI13/02481) and the Catalan Department of Economy and Knowledge (2009SGR1274 and ICREA Acadèmia) to L.A.P.-J. Supplemental Data contains detailed information about acknowledgements of EGCUT, ECRHS, EGEA, and dbGAP studies.
Contributor Information
Juan R. González, Email: jrgonzalez@creal.cat.
Luis A. Pérez-Jurado, Email: luis.perez@upf.edu.
Supplemental Data
Each column provides the −log p value of association between gene expression and inversion in the EGCUT sample. p values of association are also given stratified by gender (males and females) and BMI (lean, <18; normal, 18–25; overweight, 25–30; obese, ≥30).
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
ArrayExpress, http://www.ebi.ac.uk/arrayexpress/
Emboss Stretcher, http://www.ebi.ac.uk/Tools/psa/emboss_stretcher/
Estonian Genome Center, University of Tartu, http://www.geenivaramu.ee/en/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Beuther D.A., Sutherland E.R. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am. J. Respir. Crit. Care Med. 2007;175:661–666. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chinn S., Downs S.H., Anto J.M., Gerbase M.W., Leynaert B., de Marco R., Janson C., Jarvis D., Künzli N., Sunyer J., ECRHS. SAPALDIA Incidence of asthma and net change in symptoms in relation to changes in obesity. Eur. Respir. J. 2006;28:763–771. doi: 10.1183/09031936.06.00150505. [DOI] [PubMed] [Google Scholar]
- 3.Beuther D.A. Recent insight into obesity and asthma. Curr. Opin. Pulm. Med. 2010;16:64–70. doi: 10.1097/MCP.0b013e3283338fa7. [DOI] [PubMed] [Google Scholar]
- 4.Shore S.A. Obesity and asthma: possible mechanisms. J. Allergy Clin. Immunol. 2008;121:1087–1093. doi: 10.1016/j.jaci.2008.03.004. quiz 1094–1095. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg H., Cohen R.I. Nocturnal asthma. Curr. Opin. Pulm. Med. 2012;18:57–62. [Google Scholar]
- 6.Hallstrand T.S., Fischer M.E., Wurfel M.M., Afari N., Buchwald D., Goldberg J. Genetic pleiotropy between asthma and obesity in a community-based sample of twins. J. Allergy Clin. Immunol. 2005;116:1235–1241. doi: 10.1016/j.jaci.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A., Styrkarsdottir U., Gretarsdottir S., Thorlacius S., Jonsdottir I. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 8.Speliotes E.K., Willer C.J., Berndt S.I., Monda K.L., Thorleifsson G., Jackson A.U., Lango Allen H., Lindgren C.M., Luan J., Mägi R., MAGIC. Procardis Consortium Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chae S.C., Li C.S., Kim K.M., Yang J.Y., Zhang Q., Lee Y.C., Yang Y.S., Chung H.T. Identification of polymorphisms in human interleukin-27 and their association with asthma in a Korean population. J. Hum. Genet. 2007;52:355–361. doi: 10.1007/s10038-007-0123-8. [DOI] [PubMed] [Google Scholar]
- 10.Li Z., Zhou Y., Carter-Su C., Myers M.G., Jr., Rui L. SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol. Endocrinol. 2007;21:2270–2281. doi: 10.1210/me.2007-0111. [DOI] [PubMed] [Google Scholar]
- 11.Lord G.M., Matarese G., Howard J.K., Baker R.J., Bloom S.R., Lechler R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 12.Chen J., Yuan Y.S., Li Q., Liu M., Zhou S.H., Jin W. [Interleukin-27 affects IFN-gamma and IL-4 activities in children with asthma] Zhongguo Dang Dai Er Ke Za Zhi. 2010;12:521–523. [PubMed] [Google Scholar]
- 13.Walters R.G., Jacquemont S., Valsesia A., de Smith A.J., Martinet D., Andersson J., Falchi M., Chen F., Andrieux J., Lobbens S. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacquemont S., Reymond A., Zufferey F., Harewood L., Walters R.G., Kutalik Z., Martinet D., Shen Y., Valsesia A., Beckmann N.D. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachmann-Gagescu R., Mefford H.C., Cowan C., Glew G.M., Hing A.V., Wallace S., Bader P.I., Hamati A., Reitnauer P.J., Smith R. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet. Med. 2010;12:641–647. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 16.Bochukova E.G., Huang N., Keogh J., Henning E., Purmann C., Blaszczyk K., Saeed S., Hamilton-Shield J., Clayton-Smith J., O’Rahilly S. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barge-Schaapveld D.Q., Maas S.M., Polstra A., Knegt L.C., Hennekam R.C. The atypical 16p11.2 deletion: a not so atypical microdeletion syndrome? Am. J. Med. Genet. A. 2011;155A:1066–1072. doi: 10.1002/ajmg.a.33991. [DOI] [PubMed] [Google Scholar]
- 18.Guha S., Rees E., Darvasi A., Ivanov D., Ikeda M., Bergen S.E., Magnusson P.K., Cormican P., Morris D., Gill M., Molecular Genetics of Schizophrenia Consortium. Wellcome Trust Case Control Consortium 2 Implication of a rare deletion at distal 16p11.2 in schizophrenia. JAMA Psychiatry. 2013;70:253–260. doi: 10.1001/2013.jamapsychiatry.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin J., Han C., Gordon L.A., Terry A., Prabhakar S., She X., Xie G., Hellsten U., Chan Y.M., Altherr M. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004;432:988–994. doi: 10.1038/nature03187. [DOI] [PubMed] [Google Scholar]
- 20.Salm M.P., Horswell S.D., Hutchison C.E., Speedy H.E., Yang X., Liang L., Schadt E.E., Cookson W.O., Wierzbicki A.S., Naoumova R.P., Shoulders C.C. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 2012;22:1144–1153. doi: 10.1101/gr.126037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cáceres A., Sindi S.S., Raphael B.J., Cáceres M., González J.R. Identification of polymorphic inversions from genotypes. BMC Bioinformatics. 2012;13:28. doi: 10.1186/1471-2105-13-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J., Amos C.I. Investigation of inversion polymorphisms in the human genome using principal components analysis. PLoS ONE. 2012;7:e40224. doi: 10.1371/journal.pone.0040224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westra H.J., Jansen R.C., Fehrmann R.S., te Meerman G.J., van Heel D., Wijmenga C., Franke L. MixupMapper: correcting sample mix-ups in genome-wide datasets increases power to detect small genetic effects. Bioinformatics. 2011;27:2104–2111. doi: 10.1093/bioinformatics/btr323. [DOI] [PubMed] [Google Scholar]
- 24.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burney P.G., Luczynska C., Chinn S., Jarvis D. The European Community Respiratory Health Survey. Eur. Respir. J. 1994;7:954–960. doi: 10.1183/09031936.94.07050954. [DOI] [PubMed] [Google Scholar]
- 26.Kauffmann F., Dizier M.H., EGEA Co-operative Group EGEA (Epidemiological study on the Genetics and Environment of Asthma, bronchial hyperresponsiveness and atopy)—design issues. Clin. Exp. Allergy. 1995;25(Suppl 2):19–22. doi: 10.1111/j.1365-2222.1995.tb00413.x. [DOI] [PubMed] [Google Scholar]
- 27.Mailman M.D., Feolo M., Jin Y., Kimura M., Tryka K., Bagoutdinov R., Hao L., Kiang A., Paschall J., Phan L. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castro-Giner F., Bustamante M., Ramon González J., Kogevinas M., Jarvis D., Heinrich J., Antó J.M., Wjst M., Estivill X., de Cid R. A pooling-based genome-wide analysis identifies new potential candidate genes for atopy in the European Community Respiratory Health Survey (ECRHS) BMC Med. Genet. 2009;10:128. doi: 10.1186/1471-2350-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.González J.R., Armengol L., Solé X., Guinó E., Mercader J.M., Estivill X., Moreno V. SNPassoc: an R package to perform whole genome association studies. Bioinformatics. 2007;23:644–645. doi: 10.1093/bioinformatics/btm025. [DOI] [PubMed] [Google Scholar]
- 31.González J.R., Carrasco J.L., Dudbridge F., Armengol L., Estivill X., Moreno V. Maximizing association statistics over genetic models. Genet. Epidemiol. 2008;32:246–254. doi: 10.1002/gepi.20299. [DOI] [PubMed] [Google Scholar]
- 32.Steinberg K.M., Antonacci F., Sudmant P.H., Kidd J.M., Campbell C.D., Vives L., Malig M., Scheinfeldt L., Beggs W., Ibrahim M. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet. 2012;44:872–880. doi: 10.1038/ng.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge B., Pokholok D.K., Kwan T., Grundberg E., Morcos L., Verlaan D.J., Le J., Koka V., Lam K.C., Gagné V. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat. Genet. 2009;41:1216–1222. doi: 10.1038/ng.473. [DOI] [PubMed] [Google Scholar]
- 34.Varela L.M., Ortega A., Bermúdez B., López S., Pacheco Y.M., Villar J., Abia R., Muriana F.J. A high-fat meal promotes lipid-load and apolipoprotein B-48 receptor transcriptional activity in circulating monocytes. Am. J. Clin. Nutr. 2011;93:918–925. doi: 10.3945/ajcn.110.007765. [DOI] [PubMed] [Google Scholar]
- 35.Lei Y., Wen H., Yu Y., Taxman D.J., Zhang L., Widman D.G., Swanson K.V., Wen K.W., Damania B., Moore C.B. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36:933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gutierrez-Aguilar R., Kim D.H., Woods S.C., Seeley R.J. Expression of new loci associated with obesity in diet-induced obese rats: from genetics to physiology. Obesity (Silver Spring) 2012;20:306–312. doi: 10.1038/oby.2011.236. [DOI] [PubMed] [Google Scholar]
- 37.Li J.J., Wang W., Baines K.J., Bowden N.A., Hansbro P.M., Gibson P.G., Kumar R.K., Foster P.S., Yang M. IL-27/IFN-γ induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J. Immunol. 2010;185:4401–4409. doi: 10.4049/jimmunol.1001039. [DOI] [PubMed] [Google Scholar]
- 38.Bartlett N.W., Slater L., Glanville N., Haas J.J., Caramori G., Casolari P., Clarke D.L., Message S.D., Aniscenko J., Kebadze T. Defining critical roles for NF-κB p65 and type I interferon in innate immunity to rhinovirus. EMBO Mol. Med. 2012;4:1244–1260. doi: 10.1002/emmm.201201650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard W.R., Sorensen M.V., Galloway V.A., Spencer G.J., Mosher M.J., Osipova L., Spitsyn V.A. Climatic influences on basal metabolic rates among circumpolar populations. Am. J. Hum. Biol. 2002;14:609–620. doi: 10.1002/ajhb.10072. [DOI] [PubMed] [Google Scholar]
- 40.Wallace D.C. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 2):8947–8953. doi: 10.1073/pnas.0914635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belostotsky R., Frishberg Y., Entelis N. Human mitochondrial tRNA quality control in health and disease: a channelling mechanism? RNA Biol. 2012;9:33–39. doi: 10.4161/rna.9.1.18009. [DOI] [PubMed] [Google Scholar]
- 42.Montanari A., Zhou Y.F., D’Orsi M.F., Bolotin-Fukuhara M., Frontali L., Francisci S. Analyzing the suppression of respiratory defects in the yeast model of human mitochondrial tRNA diseases. Gene. 2013;527:1–9. doi: 10.1016/j.gene.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 43.Schon E.A., DiMauro S., Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stefansson H., Helgason A., Thorleifsson G., Steinthorsdottir V., Masson G., Barnard J., Baker A., Jonasdottir A., Ingason A., Gudnadottir V.G. A common inversion under selection in Europeans. Nat. Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Each column provides the −log p value of association between gene expression and inversion in the EGCUT sample. p values of association are also given stratified by gender (males and females) and BMI (lean, <18; normal, 18–25; overweight, 25–30; obese, ≥30).