

Abstract
Four children in three unrelated families (one consanguineous) presented with lethargy, hyperlactatemia, and hyperammonemia of unexplained origin during the neonatal period and early childhood. We identified and validated three different CA5A alterations, including a homozygous missense mutation (c.697T>C) in two siblings, a homozygous splice site mutation (c.555G>A) leading to skipping of exon 4, and a homozygous 4 kb deletion of exon 6. The deleterious nature of the homozygous mutation c.697T>C (p.Ser233Pro) was demonstrated by reduced enzymatic activity and increased temperature sensitivity. Carbonic anhydrase VA (CA-VA) was absent in liver in the child with the homozygous exon 6 deletion. The metabolite profiles in the affected individuals fit CA-VA deficiency, showing evidence of impaired provision of bicarbonate to the four enzymes that participate in key pathways in intermediary metabolism: carbamoylphosphate synthetase 1 (urea cycle), pyruvate carboxylase (anaplerosis, gluconeogenesis), propionyl-CoA carboxylase, and 3-methylcrotonyl-CoA carboxylase (branched chain amino acids catabolism). In the three children who were administered carglumic acid, hyperammonemia resolved. CA-VA deficiency should therefore be added to urea cycle defects, organic acidurias, and pyruvate carboxylase deficiency as a treatable condition in the differential diagnosis of hyperammonemia in the neonate and young child.
Main Text
Hyperammonemia is a medical emergency that requires immediate and targeted treatment. Correct diagnosis is therefore essential, but it is challenging given heterogeneous etiologies, including genetic (inborn errors of metabolism), developmental (transient neonatal hyperammonemia), and environmental (infectious hepatitis, medication) causes.1 We present four children from three unrelated families with infantile hyperammonemic encephalopathy and hyperlactatemia. The underlying cause in each of these children was deficiency of carbonic anhydrase VA (CA-VA) (CA5A [MIM 114671]), an inborn error of metabolism broadening the differential diagnosis for hyperammonemia.
This study was initiated as part of the Treatable Intellectual Disability Endeavor in British Columbia and approved by the institutional review boards of BC Children’s Hospital and the University of British Columbia. Parents provided written informed consent.
In family 1, the female index (II-1 in Figure 1), her younger affected brother (II-2), and her unaffected sister (II-3) were born to healthy nonconsanguineous parents of Belgian-Scottish descent after uneventful pregnancies and deliveries. The index and her male sibling developed lethargy, tachypnea, hypoglycemia (2.2 and 2.9 μmol/l), hyperlactatemia (9.8 and 8.8 μmol/l), hypernatremia (Na 152 and 150 μmol/l), and hyperammonemia (780 and 238 μmol/l) with respiratory alkalosis (pH 7.48, pCO2 10.8 mm Hg, HCO3− 10.4 mEq/l; and pH 7.46, pCO2 21 mm Hg, HCO3− 23) within the first days of life. Liver transaminases, albumin, and clotting factors remained normal. Urine organic acids demonstrated high lactic, β-hydroxybutyric, and acetoacetic acid excretion, as well as increases in carboxylase substrates and related metabolites. Plasma amino acid analysis showed elevations of glutamine, alanine, and proline and reduction of citrulline and arginine. Further biochemical test results are shown in Table 1. Known urea cycle defects and primary causes of hyperlactatemia were excluded by sequencing and deletion/duplication analysis of N-acetylglutamate synthase (NAGS [MIM 608300]), carbamoylphosphate synthetase (CPS1 [MIM 608307), and pyruvate carboxylase (PC [MIM 608786]), as well as measurement of PC and biotinidase activities in fibroblasts and serum. Hyperammonia-hyperornithinemia-homocitrullinuria syndrome (MIM 238790) and lysinuric protein intolerance (MIM 222710) were excluded on the basis of a normal urine amino acid profile, and pyruvate dehydrogenase complex deficiency was excluded on the basis of an elevated lactate:pyruvate ratio (5.7 μmol/l:0.036 μmol/l = 138 [reference interval 10–20.7]) in the male sibling. Blue native gel analysis for respiratory chain complexes I–V was normal. Molecular analysis of nuclear-encoded ATPase deficiency (TMEM70 [MIM 612418]) did not reveal disease-causing mutations. Chromosomal microarray analysis (AffymetrixCytoscan HD) was unremarkable, and homozygosity analysis did not reveal evidence of consanguinity or uniparental disomy.
Figure 1.
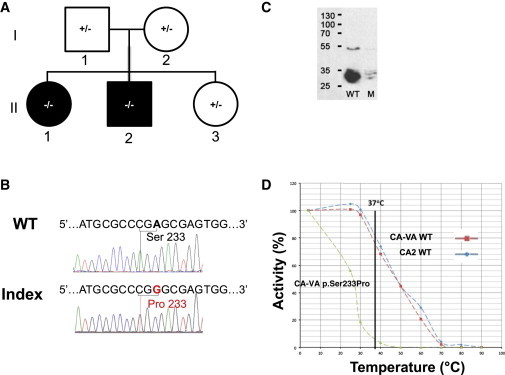
Family 1 with p.Ser233Pro Missense Variant
(A) Pedigree (black fill indicates clinically affected individuals, II-1 and II-2).
(B) Sanger sequence of CA5A from index (II-1) and control (wild-type sequence; WT) subjects; the variant nucleotide position and the corresponding codon alteration (p.Ser233Pro) are indicated.
(C) Immunoblot analyses by SDS-PAGE (ImageJ software) of WT and p.Ser233Pro (mutant; M) CA-VA protein levels in COS-7 cell lysates; the molecular weights (kDa) of protein standards are indicated on the left. Normal and mutant (c.697T>C) CA5A cDNAs, including the full mitochondrial targeting sequences, were synthesized via the NCBI reference sequence (NM_001739.1) by Genscript. The cDNAs were cloned into pUC57 at XhoI and BglII sites and verified by Sanger sequencing. Subsequently, the cDNA inserts were subcloned via the same restriction sites into the pCXN mammalian expression vector. COS-7 cells (ATCC CRL-1651) were transfected with wild-type or mutant CA5A plasmids with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol, as previously described.8 A β-glucuronidase expression plasmid was cotransfected as a marker of transfection efficiency.24 COS-7 cells were harvested 48–72 hr after transfection in lysis buffer and then lysed by sonication on ice.
(D) Thermal stability profiles for WT (red) and p.Ser233Pro mutant (green) CA-VA enzymes. Carbonic anhydrase II (CA2; blue) was used as a control.
Table 1.
Overview of Biochemical Abnormalities Resulting from CA-VA Deficiency: In Theory and for the Index Cases for Families 1, 2, and 3
|
Theoretical Possibilities |
Actual Resultsa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Possible Enzyme Deficiency | Predicted Secondary Biochemical Abnormalities | Metabolite |
Family 1 |
Family 2 |
Family 3 |
||||
| Female | Male | Normalb | Male | Normal | Male | Normal | |||
| carbamoyl phosphate synthetase | ↑ ammonia | plasma ammonia (μM) | 780∗ | 238∗ | <40 | 422∗ | <50 | 258∗ | <40 |
| ↓ citrulline | plasma citrulline (μM) | 5∗ | 18 | 8–47 | 17 | 3–36 | 10–45 | ||
| ↓ arginine | plasma arginine (μM) | 17∗ | NA | 32–142 | 35 | 17–119 | 22 | 12–133 | |
| ↑ glutamine | plasma glutamine (μM) | 1,051∗ | 1,237∗ | 457–746 | 2,606∗ | 243–822 | 571 | 254–823 | |
| N ornithine | plasma ornithine (μM) | 15∗ | 82 | 27–207 | 146 | 38–272 | 17 | 15–200 | |
| N orotate | urine orotate | non-det | 1.9 | <4.3 | 2.2 | <4.9 | 0.4 | <3 | |
| pyruvate carboxylase | ↓ gluconeogenesis (hypoglycemia) | serum glucose (mm) | 2.2∗ | 2.9∗ | 3.3–7.0 | 2.9∗ | 3.0–8.0 | 3.2∗ | 3.5–6.0 |
| serum lactate (mm) | 9.1∗ | 8.8∗ | 0.5–2.2 | 8.1∗ | 1.0–1.8 | 5.6∗ | 0.6–2.6 | ||
| plasma alanine (μm) | 1,531∗ | 609∗ | 148–475 | 1,078∗ | 132–455 | 603∗ | 143–439 | ||
| redox imbalance (↑ lactate & dicarboxylic acids) | plasma proline (μm) | 418∗ | 371∗ | 40–332 | 625∗ | 78–523 | 283∗ | 52–298 | |
| ↓ tricyclic acid cycle intermediates (cataplerosis) | urine lactate | 3,737∗ | 4,109∗ | <200 | 28,000∗ | <456 | grossly increased∗ | ||
| urine 3-OH-butryric acid | 7,902∗ | 2,657∗ | <21 | 7,060∗ | <22 | grossly increased∗ | |||
| urine aceto-acetic acid | 584∗ | 927∗ | non-det | ++c | grossly increased∗ | ||||
| urine fumaric acid | 13.8∗ | 38.5∗ | <10 | 8 | <13 | moderately increased | |||
| urine 2-α-ketoglutaric acid | 143∗ | 254.6∗ | <112 | 300∗ | <267 | slightly increased∗ | |||
| urine adipic acid | 55∗ | 219.8∗ | <29 | 340 | <25 | normal | |||
| urine suberic acid | 18.6∗ | 48.6∗ | <13 | 29 | <15 | normal | |||
| urine sebacic acid | NA | 18.7∗ | <10 | NA | normal | ||||
| ↑ lysine | plasma lysine (μm) | 87 | 161 | 71–272 | 306 | 71–272 | 94 | 71–272 | |
| proprionyl-CoA carboxylase | ↑ 3-OH-propionic acid | urine 3-OH-propionic acid | 79.38∗ | 54.57∗ | <16 | 59∗ | <21 | normal | |
| ↑ propionylglycine | urine proprionylglycine | 1.29∗ | 3.28∗ | non-det | 5.6∗ | <2 | trace∗ | ||
| ↑ methylcitrate | urine methylcitrate | 6.4∗ | non-det | non-det | normalc | trace∗ | |||
| 3-methylcrotonyl-CoA carboxylase | ↑ 3methylcrotonylglycine | urine 3-methylcrotonylglycine | 22.9∗ | non-det | <10 | 17∗ | <5 | trace∗ | |
| ↑ 3OH-isovaleric acid | urine 3-OH-isovaleric acid | 40.13∗ | 55.50∗ | <38 | 327∗ | <55 | increased∗ | ||
All values in urine are expressed as μmol/mmol of creatinine. Abbreviations are as follows: N, normal (within reference range); NA, not available; non-det, nondetectable.
In each individual, the value with maximal deviation from normal during crisis is provided. Asterisks (∗) indicate abnormal values.
Normal values differ for each family studied because values were measured in different laboratories.
Qualitative assessment.
Clinical and metabolic findings normalized in both siblings with the administration of intravenous dextrose and bicarbonate, as well as enteral carglumic acid (Carbaglu). In our institution, carglumic acid is used to resolve hyperammonemia of unknown origin.2 Brain MRI and MRS on day 5 of life revealed a small periventricular petechial focus near the trigone of the right lateral ventricle, with a small lactate peak on spectroscopy in the male sibling; in the female sibling, both MRI and EEG were unremarkable.
At 2.5 and 3.5 years of age, during intercurrent illness, the index presented with lethargy, hyperammonemia, and hyperlactatemia. Hyperammonemia resolved with a single dose of carglumic acid. She has remained clinically stable with L-carnitine, vitamin C, coenzyme Q10, and “sick-day management” with high-caloric and lipid-rich formula during illnesses. At the age of 4.5 years, she exhibited mild axial hypotonia and septal thickening on cardiac ultrasound. Developmental and behavioral assessment at age 4.5 confirmed average functioning in all domains except for below-average motor coordination (Beery Buktenica Developmental Test of Visual-Motor Integration). The male sibling at age 2.3 years had difficulty engaging in an assessment as a result of heightened activity and self-directed behavior; scores on Bayley Scales of Infant and Toddler Development (Third Edition) were below average. Biochemical profiles and somatic and psychomotor development are unremarkable in the unaffected younger sibling, now 1.5 years of age.
WES was performed for the two affected siblings and their unaffected parents via the Agilent SureSelect kit and Illumina HiSeq 2000 (Perkin-Elmer). Rare variants were assessed for their potential to disrupt protein function and screened under a series of genetic models—primarily the Mendelian recessive mode of inheritance given the rarity of the phenotype and the pattern of inheritance of most IEMs.
Approximately 99% of the observed variations were classified as common (Table S1 available online). Eight rare candidate variants fit the autosomal-recessive model of homozygous (CA5A, ARSB [MIM 611542], CDKN2B [MIM 600431], CDKN2A [MIM 600160], OGG1 [MIM 601982]) or compound heterozygous (GRK4 [MIM 137026], SPG11 [MIM 610844], EPHX2 [MIM 132811]) variants in the affected siblings. The final set of rare variants (mean average frequency < 1%) was assessed for the potential to disrupt protein function via the Sift and PolyPhen2 software systems. Of these, only one variant (c.697T>C; RefSeq accession number NM_001739.1) in CA5A on chromosome 16 was considered a functional candidate; this variant was not reported in dbSNP (version 137), NHLBI ESP, or our in-house genome database (comprising 100 exomes and 10 whole genomes; anno December 2013). Integrative Genomics Viewer 2.0.34 was used to visualize the read alignment and assess variant quality prior to Sanger validation. Given the existence of a related pseudogene,3 the CA5A variant was confirmed by targeted Sanger sequencing via carefully designed primers to avoid amplification of the pseudogene sequences; this was achieved by review of paired-end sequence data and selection via a BLAST search of appropriate regions with least similarity, especially close to the 3′ end. Affected siblings are confirmed homozygous, whereas unaffected parents and the unaffected youngest female are heterozygous carriers (Figures 1A and 1B). This variant corresponds to a Ser to Pro substitution at position 233 that is predicted to disrupt structure around the conserved Thr235 residue that forms part of the substrate-binding region of the enzyme (Figure 2). Indeed, the p.Ser233 residue is highly conserved evolutionarily across species and in all 12 active human carbonic anhydrase isoforms. Studies of human CA-II have demonstrated that mutations in this hydrophobic patch in the active site destabilize the structure around the substrate-binding region and dramatically reduce the activity of the mutant CA-II.4
Figure 2.
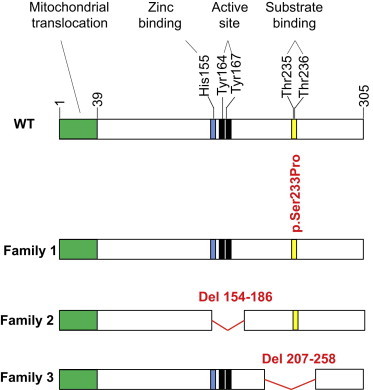
Effect of Genetic Variants Identified in CA-VA
Shown is a schematic diagram of the 305 amino acid wild-type (WT) CA-VA. Residues 1–39 encode a mitochondrial translocation signal (green). Homology predictions indicate that histidine 155 binds a zinc ion (blue), tyrosines 164 and 167 are active site residues (black), and threonines 235 and 236 comprise a substrate-binding region (yellow). Shown in red are the deduced CA-VA variants identified in this study. The index and affected brother of family 1 has a nonsynonymous Ser to Pro mutation at residue 233, adjacent to the substrate-binding region. The index of family 2 has a deletion of residues 154–186 (exon 4), thereby missing the metal-binding and active-site residues. The index of family 3 has a deletion of residues 207–258 (exon 6; the substrate-binding region), which results in absent protein.
Tissues with CA-VA (liver, kidney, skeletal muscle) were not available for the affected siblings. Therefore, the effect of the p.Ser233Pro substitution on CA-VA was characterized in cultured mammalian cells (COS-7). A marked reduction was observed in the steady-state levels of CA-VA p.Ser233Pro compared with wild-type protein despite similar transfection efficiencies (Figure 1C). As summarized in Table 2, CA-VA p.Ser233Pro-specific activity in total cell lysates is reduced to 20% of wild-type protein activity, whereas activities of the cotransfected marker enzyme β-glucuronidase were comparable. There was a negligible change in the activity of mutant enzyme in the presence of added zinc (22% of WT), suggesting that both WT and mutant enzymes in COS-7 cells are saturated with zinc (data not shown). Thermal stability of mutant recombinant human CA-VA was compared with the WT CA-VA and recombinant human Carbonic Anhydrase II as an additional control (Figure 1D). After a 30 min preincubation, the mutant enzyme had lost 80% of its activity at 30°C and almost all its activity at 40°C. By contrast, both WT CA-VA and human CA-II were much more stable at 30°C and 40°C, retaining approximately 100% and 70% residual activity, respectively. In a separate experiment (Table 3), preincubation of the mutant enzyme at 37°C (normal human body temperature) for 30 min retained only 5% of mutant enzyme activity, whereas wild-type protein activity was 80% relative to activity after a 4°C preincubation. The quantity of immune-reactive CA-VA p.Ser233Pro was demonstrably lower than that of wild-type CA-VA (37% of wild-type) at 72 hr after transient transfection of COS-7 cells (data not shown). Similar temperature-sensitive mutants are associated with disease in cases of medium chain acyl-coA dehydrogenase deficiency (ACADM [MIM 607008]) and recessively inherited parkinsonism (PINK1, a putative mitochondrial kinase [MIM 608309]).5,6
Table 2.
Steady-State Carbonic Anhydrase Activity in COS-7 Cells Cotransfected with Wild-Type and p.Ser233Pro Mutant CA-VA and β-Glucuronidase
| Enzymes | Specific Activity (EU/mg Cell Protein) | Specific Activity Minus Vector (EU/mg Cell Protein) | Percent of WT CA-VA | BGUS Activity (EU/mg) | Transfection Efficiency |
|---|---|---|---|---|---|
| PCXN vector only | 0.28 | 0.00 | 0 | 37.5 | 1 |
| WT | 1.88 | 1.60 | 100 | 7070 | 188.5 |
| MT | 0.60 | 0.32 | 20 | 8550 | 228 |
Values represent the average of two transfections; the enzyme activities were determined in duplicate and calculated as enzyme unit (EU) per milligram (mg) of enzyme protein. The specificity of the assay was verified with the CA-specific inhibitor acetazolamide (500 nM). Abbreviations are as follows: WT, wild-type; MT, mutant.
Table 3.
CA-VA Wild-Type and p.Ser233Pro Mutant Enzyme Activities in COS-7 Cell Lysates, before and after Incubation at Human Body Temperature
| Enzyme | EU/mg Cell Protein before Heating | EU/mg Cell Protein after Heating (30 min at 37°C) |
|---|---|---|
| WT | 1.02 (100%) | 1.25 (100%) |
| MT | 0.15 (14.7%) | 0.07 (5.4%) |
The enzyme activities were determined in duplicate and calculated as enzyme unit (EU) per milligram (mg) of enzyme protein. Incubation was at 37°C for 30 min. Abbreviations are as follows: WT, wild-type; MT, mutant.
In family 2, a male child (II-1 in Figure 3A) was born spontaneously at gestational age 36+2 weeks to nonconsanguineous Russian parents. On day 4 of life, he presented with lethargy, weight loss (15% below birth weight), jaundice, and tachypnea. Initial investigations showed hyperammonemia (316 and 422 μmol/l), hyperlactatemia (8.1 mmol/l), mild hypoglycaemia (2.9 mmol/l), metabolic acidosis (pH 7.16, pCO2 13 mm Hg, HCO3− 5 mEq/l), and ketonuria. Despite fluid resuscitation, sodium bicarbonate infusion, and antibiotics, the neonate’s clinical and biochemical status deteriorated; liver transaminases and synthetic function remained normal. Metabolic investigations are shown in Table 1; molecular analysis of CPS1 and NAGS did not reveal disease-causing mutations. Carglumic acid and biotin were initiated, along with protein-free formula and intravenous lipids; 12 hr later, the metabolic acidosis and hyperammonemia resolved. He resumed breastfeeding with normal weight gain, ammonia levels, and urine metabolites. Carglumic acid was stopped at 4 months of age, and the infant exhibited normal psychomotor development at age 6 months with the use of sick-day formula during illness.
Figure 3.
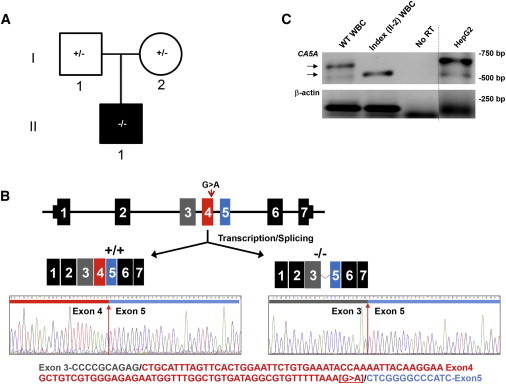
Family 2 with Exon 4 Splice Deletion
(A) Pedigree (black fill indicates clinically affected individual).
(B) Sanger sequencing of RT-PCR products generated in (B) with exons denoted by colors. Top: The CA5A structure (not to scale) and a schematic of the observed CA5A transcripts produced in a control subject and the index. Bottom: Sanger sequence of transcripts at exon 4 boundary in a control subject (+/+) and the index (−/−), along with WT CA5A cDNA sequence, color-coded as in the top panel.
(C) RT-PCR of CA5A mRNA from white blood cells (WBCs) or cultured liver cells (HepG2). Arrows indicate the products of differing size amplified from control subject (WT sequence) and index (II-1) WBCs. As controls, reverse transcriptase was omitted from the reaction (no RT) and a control gene (β-actin) was amplified in separate lane on a different cell type (denoted by the line).
Sanger sequencing of all seven exons of CA5A in the index identified a synonymous c.555G>A transition (RefSeq NM_001739.1) at the final base of exon 4 (Figure 3B). Given that guanine is the most common nucleotide found at this end of an exon in vertebrate genes, RT-PCR was undertaken to demonstrate an effect on mRNA splicing.7 RNA was purified from white blood cells of the index and a control subject. RT-PCR via primers designed to amplify exons 2–7 of CA5A generated distinctly different product sizes (approximately 550 bp and 650 bp, respectively; Figure 3B). Sanger sequencing of the ∼550 bp band revealed an in-frame deletion of exon 4 from the index RNA (Figure 3C). We cannot exclude the presence of other transcripts in vivo, which were missed because of the limited sensitivity of the assay combined with low CA5A expression in white blood cells. Alternatively, this single transcript may be explained by preferential formation during the transcription/splicing processes. Homology with carbonic anhydrase isoforms identifies three critical residues in the deleted CA-VA transcript (residues 154–185): His155, which binds to a catalytically essential zinc molecule, and Tyr164 and Tyr167, which form part of the active site of the CA-VA enzyme.8 Thus, this deletion is predicted to significantly impair CA-VA enzyme activity, if not lead to protein misfolding and degradation.
In family 3, a male child (II-5 in Figure 4A) was born at term by Caesarian section (because of placenta previa) as the youngest of five children to first-cousin consanguineous Pakistani parents. At 13 months of age, after unremarkable development, he presented with a 1-day history of visual unresponsiveness. At admission, he was encephalopathic with hyperammonemia (258 μmol/l) and hyperlactatemia (4.9 mmol/l), with a compensated metabolic acidosis (pH 7.43, pCO2 24.8 mm Hg, HCO3− 14 mEq/l). His encephalopathy improved after 48 hr of intravenous fluids and antibiotics administered for presumed meningo-encephalitis (cultures were negative). At the age of 16 months, he had a similar crisis; there were no signs of liver injury. Further metabolic investigations are shown in Table 1. Sodium benzoate and L-arginine were initiated with improvement after 48 hr, and he was discharged on a protein-restricted diet. Urea cycle defects (OTC [MIM 311250], CPS1 [MIM 237300], NAGS [MIM 237310] deficiencies) and PC (MIM 266150), citrin (MIM 605814), and biotinidase (MIM 253260) deficiencies were excluded by molecular or enzymatic analyses. After these two crises, he has demonstrated good developmental progress with only minor learning difficulties (no formal testing was available). He continues to have infrequent episodes of vomiting and ketoacidosis without hyperammonemia or lactic acidosis; the frequency of these episodes has not increased since the withdrawal of sodium benzoate and arginine therapy at the age of 7 years.
Figure 4.
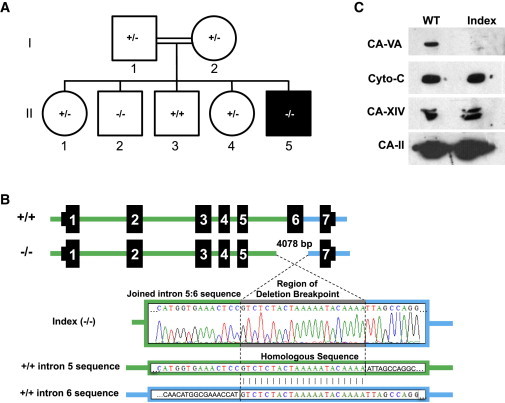
Family 3 with Exon 6 Deletion
(A) Pedigree (black fill indicates clinically affected individual, II-5).
(B) Top: Schematic representation of the 4,078 bp deletion that encompasses exon 6 of CA5A. Bottom: Sanger sequencing of PCR products generated from genomic DNA of the index (II-5) with a 21 bp repeated sequence (gray) at the breakpoint found in both intron 5 (green) and intron 6 (blue).
(C) Immunoblot analyses of control subject (WT) and index (II-5) liver homogenates. Samples were probed for CA-VA,25 cytochrome c (Cyto-c), carbonic anhydrase XIV (CA 14),26 and carbonic anhydrase II (CA 2)27 via specific antibodies.
Sanger sequencing of the CA5A exons for the index revealed a deletion of 4 kb encompassing exon 6 (Figure 4B). Relative copy number was assessed in the family via qPCR of exon 6 sequence, confirming the homozygous deletion in the two siblings and heterozygosity in the parents (Figure S1). Absence of CA-VA protein was confirmed by immunoblot in existing liver biopsy tissue (Figure 4C). Limited information is available on the neonatal and early childhood course in his four older siblings; parents reported no major health problems and normal learning ability. The oldest brother (II-2 in Figure 4) is homozygous for the CA5A deletion but declined further evaluation at age 17. Neonatal or childhood insults and a possible mild presentation cannot be excluded because of limited history. A benign clinical course after early childhood is consistent with the index cases of each of the three families after initial metabolic decompensations. The condition may exhibit intrafamilial variability of the phenotype, as reported for other inborn errors of metabolism.9,10
CA-VA deficiency is a human inborn error of metabolism presenting with hyperammonemic encephalopathy in early life. Initial evidence of causality for the identified CA5A alterations is provided by their similar biochemical phenotypes during metabolic crises. Findings are consistent with dysfunction of all four enzymes to which CA-VA provides bicarbonate as substrate in mitochondria (CPS1 and three biotin-dependent carboxylases: propionyl-CoA [PCC], 3-methylcrotonyl-CoA [3MCC], and pyruvate carboxylase [PC]).
The carbonic anhydrase family of zinc-containing metallo-enzymes all catalyze the reversible conversion of carbon dioxide to bicarbonate (CO2 + H2O ↔ HCO3− + H+). Mitochondria are impermeable to HCO3−, and thus the two intramitochondrial carbonic anhydrases (CA-VA and CA-VB) are pivotal in providing HCO3− for multiple mitochondrial enzymes that catalyze the formation of essential metabolites of intermediary metabolism in the urea and Krebs cycles (Figure 5).11,12
Figure 5.
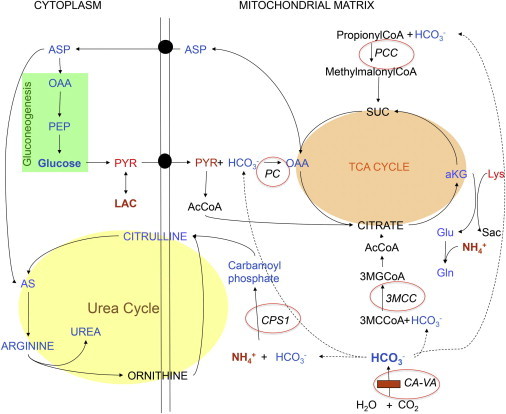
Biochemical Pathways Affected by CA-VA Deficiency
CA-VA deficiency is indicated by the horizontal red bar (bottom right). Dotted lines link CA-VA-mediated bicarbonate production and intramitochondrial donation to the bicarbonate-dependent carboxylases; the affected enzyme deficiencies are denoted with red circles. Enzymes: CPS1, carbamoylphosphate synthetase I; PC, pyruvate carboxylase; PCC, propionyl CoA carboxylase; 3MCC, 3-methylcrotonyl CoA carboxylase. Metabolites: SUC, succinyl CoA; αKG, α-ketoglutarate; OAA, oxaloacetate; Glu, glutamate; Gln, glutamine; Sac, saccharopine; Lys, lysine; ASP, aspartate; PEP, phosphoenolpyruvate; PYR, pyruvate; LAC, lactate; AcCoA, acetyl CoA; AS, argininosuccinate; 3MGCoA, 3-methylglutaconyl CoA; 3MCCoA, 3-methylcrotonyl CoA.
These include CPS1, which catalyzes the synthesis of carbamoylphosphate from ammonia and bicarbonate, a rate-limiting step in the conversion of waste nitrogen into urea, and pyruvate carboxylase (PC), which stands at the interface between glycolysis and the Krebs cycle, facilitating the production of oxaloacetate from pyruvate, the former being a major anaplerotic substrate essential for the maintenance of the Krebs cycle.13 Primary PC deficiency is biochemically characterized by lactic acidosis, hyperammonemia, and hypoglycemia, as was observed in our CA-VA-deficient individuals with presumed secondary PC deficiency leading to hypoglycemia through impaired gluconeogenesis.14
CA-VA deficiency provides the unique opportunity to study the opposite consequences of CPS1 deficiency (hyperammonemia, elevated glutamine, and decreased citrulline and other amino acids distal to the block) and PC deficiency (hyperammonemia, normal to decreased glutamine, increased citrulline and lysine). Given that glutamine was elevated in three of the four affected individuals and that the citrulline was low to normal in all, it appears that an impaired urea cycle, rather than deficient glutamate, was the major cause of hyperammonemia. Combined with the normal lysine levels and relatively mild elevations of 2-α-ketoglutaric acid and other Krebs cycle intermediates, the biochemical profiles in our CA-VA deficiency individuals support a predominant effect of (secondary) CPS1 versus PC deficiency.
The observed moderate increase of urinary 3-OH propionic acid, propionyl glycine, and methylcitrate along with traces of 3-methylcrotonylglycine are consistent with the expected reduced activities of PCC and 3MCC in CA-VA deficiency. Although biotinidase and holocarboxylase synthetase (HCS [MIM 253270]) deficiencies share the metabolite profiles of secondary PC, PCC, and 3MCC deficiencies, there are three major differences relative to primary CA-VA deficiency: (1) the significantly higher level of PCC and 3MCC metabolites in (even well-controlled) individuals with the two former disorders compared to those with CA-VA deficiency during metabolic decompensation; (2) the presence of CPS1 deficiency as (likely) major cause of hyperammonemia in CA-VA deficiency; and (3) the presence of acetyl-CoA carboxylase deficiency (MIM 613933) in HCS and biotinidase deficiency.15,16 Our CA-VA-deficient individuals exhibited normal levels of free fatty acids and of total and free carnitine, as well as normal acylcarnitine profiles (data not shown), mostly probably as a result of the activity of the cytosolic acetyl-CoA carboxylase 2 isoform that is not affected by impaired provision of mitochondrial HCO3−.17
Although not approved for this indication, carglumic acid may be of benefit as synthetic treatment of partial CPS1 deficiency through its known ability to enhance the activity of this enzyme.2 This could increase production of carbamoyl phosphate even at HCO3− levels below the Km for the enzyme. Although the hyperammonemia resulted from a reduction of substrate to CPS1, rather than a primary CPS1 deficiency, three individuals (families 1 and 2) showed a good response to carglumic acid (normalization of ammonia and for the female of family 1, a mild increase of citrulline), consistent with the suggestion that enhanced CPS1 activity can partially compensate for reduced HCO3− resulting from CA-VA deficiency. This hypothesis should be tested in the Car5A-null mouse model (Car5Adl1Sws),18 especially given the high cost of this medication and uncontrolled circumstances in which these children were managed with multiple therapeutic modalities.
Newborn screening profiles (Tandem MS/MS), specifically C3 and C5OH levels, were unremarkable in all four cases, probably because of the relatively mild biochemical profile for the carboxylase-related metabolites along with the low sensitivity of acylcarnitine analyses compared to urine organic acids. Furthermore, outside of acute events, biochemical parameters remained normal in all affected children except for mildly elevated blood lactate and/or ketonuria. We propose several explanations for the relatively benign clinical course in these individuals and lack of apparent phenotype in the oldest male sibling in family 3. First, overlapping function of CA-VB may help prevent deleterious sequelae of reduced CA-VA activity.19 In the mouse, Car5A is mainly localized in liver and its deficiency results in profound hyperammonemia. Car5B, though almost undetectable in liver, is predominant in mitochondria of many other tissues. Nonetheless, Car5B deficiency alone has no obvious phenotype. However, when superimposed on Car5A deficiency in the doubly deficient mouse (Car5Adl1Sws/Car5Bdl1Sws), Car5B deficiency aggravated the hyperammonemia and hypoglycemia and shortened survival.18 Thus, Car5B does contribute to handling the metabolic load, though its action is evident only in the absence of Car5A. Second, although carbonic anhydrases accelerate the conversion of CO2 to HCO3− by 1,000-fold or greater, some bicarbonate is produced via the nonenzymatic reaction, even in the absence of carbonic anhydrases.20 Sufficiency for the product of this limited nonenzymatic source of bicarbonate may differ for the four different bicarbonate-requiring enzymes depending on their individual Kms.
Thus, CA-VA deficiency should be considered among urea cycle defects, organic acidurias, and PC deficiency in the differential diagnosis for hyperammonemia and hyperlactatemia in the neonate and young child. Whereas many of the biochemical markers are nonspecific, some metabolites such as propionylglycine and 3-methylcrotonylglycine are more specific for deficiencies of PCC and 3MCC deficiencies that occur secondary to CA-VA deficiency. However, the increase in these metabolites is subtle compared with individuals who have a primary defect of these enzymes. Furthermore, these abnormalities may be present only during symptomatic episodes. Therefore, diagnostic molecular analysis of CA5A should be considered in persons with suggestive biochemical findings.
CA-VA deficiency expands the list of treatable inborn errors of metabolism potentially causing intellectual disability.21 Effective therapy in the affected individuals comprised (1) preventive sick-day management during intercurrent illnesses, including a high-caloric, lipid-rich formula restricted in protein but normal in carbohydrates; and possibly (2) carglumic acid to enhance the activity of the first step in the urea cycle as a treatment for the hyperammonemia.
Other experimental therapies deserving exploration include oral zinc supplementation (a CA-VA cofactor that may increase residual activity in persons with mutations outside the zinc-binding site) and interventions to enhance or replace anaplerosis to alleviate metabolic symptoms of PC dysfunction, such as the administration of aspartate and citrate (but not triheptanoin, given the secondary PCC deficiency22), as demonstrated by improved growth and survival in CA-VA-null mice receiving citrate water.18,23
Acknowledgments
We gratefully acknowledge the families for their participation in this study, Xiahua Han for expert performance of CA5A molecular analyses, M. Higginson and M. Zhou for DNA extraction, sample handling, and technical data, M. Thomas and M. Lafek for consenting and data management, K. MacKenzie for psychological testing of the siblings, and J. Rilstone for text editing. This study is part of the Treatable Intellectual Disability Endeavour in British Columbia. This work was supported by funding from the B.C. Children’s Hospital Foundation as “1st Collaborative Area of Innovation,” the British Columbia Clinical Genomics Network (grant number BCCGN00031), Genome British Columbia (grant number SOF5-021), the Rare Diseases Foundation (grant number 2012-8) to C.D.v.K., National Institutes of Health (grant number R01GM084875) to W.W.W., and National Institutes of Health (grant number DK40163) to W.S.S.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
TIDE-BC, http://www.tidebc.org
Treatable Intellectual Disability App, http://www.treatable-id.org
UniProt Sequence Annotation (Features), http://www.uniprot.org/uniprot/P35218#section_features
Accession Numbers
The Leiden Open Variation Databank (http://databases.lovd.nl/shared/genes/CA5A) accession number for the c.697T>C mutation reported in this paper is CA5A_000001; for the c.555G>A mutation, CA5A_000002; and for the 4,078 bp exon 6 deletion is CA5A_000003.
References
- 1.Häberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch. Biochem. Biophys. 2013;536:101–108. doi: 10.1016/j.abb.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Daniotti M., la Marca G., Fiorini P., Filippi L. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int. J. Gen. Med. 2011;4:21–28. doi: 10.2147/IJGM.S10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagao Y., Batanian J.R., Clemente M.F., Sly W.S. Genomic organization of the human gene (CA5) and pseudogene for mitochondrial carbonic anhydrase V and their localization to chromosomes 16q and 16p. Genomics. 1995;28:477–484. doi: 10.1006/geno.1995.1177. [DOI] [PubMed] [Google Scholar]
- 4.Krebs J.F., Fierke C.A. Determinants of catalytic activity and stability of carbonic anhydrase II as revealed by random mutagenesis. J. Biol. Chem. 1993;268:948–954. [PubMed] [Google Scholar]
- 5.Andresen B.S., Dobrowolski S.F., O’Reilly L., Muenzer J., McCandless S.E., Frazier D.M., Udvari S., Bross P., Knudsen I., Banas R. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001;68:1408–1418. doi: 10.1086/320602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narendra D.P., Wang C., Youle R.J., Walker J.E. PINK1 rendered temperature sensitive by disease-associated and engineered mutations. Hum. Mol. Genet. 2013;22:2572–2589. doi: 10.1093/hmg/ddt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krawczak M., Thomas N.S.T., Hundrieser B., Mort M., Wittig M., Hampe J., Cooper D.N. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum. Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 8.Nagao Y., Platero J.S., Waheed A., Sly W.S. Human mitochondrial carbonic anhydrase: cDNA cloning, expression, subcellular localization, and mapping to chromosome 16. Proc. Natl. Acad. Sci. USA. 1993;90:7623–7627. doi: 10.1073/pnas.90.16.7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debray F.G., Lambert M., Lemieux B., Soucy J.F., Drouin R., Fenyves D., Dubé J., Maranda B., Laframboise R., Mitchell G.A. Phenotypic variability among patients with hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome homozygous for the delF188 mutation in SLC25A15. J. Med. Genet. 2008;45:759–764. doi: 10.1136/jmg.2008.059097. [DOI] [PubMed] [Google Scholar]
- 10.Klaus V., Vermeulen T., Minassian B., Israelian N., Engel K., Lund A.M., Roebrock K., Christensen E., Häberle J. Highly variable clinical phenotype of carbamylphosphate synthetase 1 deficiency in one family: an effect of allelic variation in gene expression? Clin. Genet. 2009;76:263–269. doi: 10.1111/j.1399-0004.2009.01216.x. [DOI] [PubMed] [Google Scholar]
- 11.Dodgson S.J., Forster R.E., 2nd, Storey B.T. The role of carbonic anhydrase in hepatocyte metabolism. Ann. N Y Acad. Sci. 1984;429:516–524. doi: 10.1111/j.1749-6632.1984.tb12380.x. [DOI] [PubMed] [Google Scholar]
- 12.Tong L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 2013;70:863–891. doi: 10.1007/s00018-012-1096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martínez A.I., Pérez-Arellano I., Pekkala S., Barcelona B., Cervera J. Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency. Mol. Genet. Metab. 2010;101:311–323. doi: 10.1016/j.ymgme.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 14.García-Cazorla A., Rabier D., Touati G., Chadefaux-Vekemans B., Marsac C., de Lonlay P., Saudubray J.M. Pyruvate carboxylase deficiency: metabolic characteristics and new neurological aspects. Ann. Neurol. 2006;59:121–127. doi: 10.1002/ana.20709. [DOI] [PubMed] [Google Scholar]
- 15.Wolf B. Biotinidase deficiency. In: Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. GeneReviews, 1993-2000. University of Washington; Seattle: 2011. [Google Scholar]
- 16.Suzuki Y., Yang X., Aoki Y., Kure S., Matsubara Y. Mutations in the holocarboxylase synthetase gene HLCS. Hum. Mutat. 2005;26:285–290. doi: 10.1002/humu.20204. [DOI] [PubMed] [Google Scholar]
- 17.Brownsey R.W., Zhande R., Boone A.N. Isoforms of acetyl-CoA carboxylase: structures, regulatory properties and metabolic functions. Biochem. Soc. Trans. 1997;25:1232–1238. doi: 10.1042/bst0251232. [DOI] [PubMed] [Google Scholar]
- 18.Shah G.N., Rubbelke T.S., Hendin J., Nguyen H., Waheed A., Shoemaker J.D., Sly W.S. Targeted mutagenesis of mitochondrial carbonic anhydrases VA and VB implicates both enzymes in ammonia detoxification and glucose metabolism. Proc. Natl. Acad. Sci. USA. 2013;110:7423–7428. doi: 10.1073/pnas.1305805110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah G.N., Hewett-Emmett D., Grubb J.H., Migas M.C., Fleming R.E., Waheed A., Sly W.S. Mitochondrial carbonic anhydrase CA VB: differences in tissue distribution and pattern of evolution from those of CA VA suggest distinct physiological roles. Proc. Natl. Acad. Sci. USA. 2000;97:1677–1682. doi: 10.1073/pnas.97.4.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sly W.S., Hu P.Y. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu. Rev. Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- 21.van Karnebeek C.D., Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol. Genet. Metab. 2012;105:368–381. doi: 10.1016/j.ymgme.2011.11.191. [DOI] [PubMed] [Google Scholar]
- 22.Mochel F., DeLonlay P., Touati G., Brunengraber H., Kinman R.P., Rabier D., Roe C.R., Saudubray J.M. Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol. Genet. Metab. 2005;84:305–312. doi: 10.1016/j.ymgme.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Marin-Valencia I., Roe C.R., Pascual J.M. Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. Mol. Genet. Metab. 2010;101:9–17. doi: 10.1016/j.ymgme.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Shipley J.M., Grubb J.H., Sly W.S. The role of glycosylation and phosphorylation in the expression of active human beta-glucuronidase. J. Biol. Chem. 1993;268:12193–12198. [PubMed] [Google Scholar]
- 25.Nagao Y., Srinivasan M., Platero J.S., Svendrowski M., Waheed A., Sly W.S. Mitochondrial carbonic anhydrase (isozyme V) in mouse and rat: cDNA cloning, expression, subcellular localization, processing, and tissue distribution. Proc. Natl. Acad. Sci. USA. 1994;91:10330–10334. doi: 10.1073/pnas.91.22.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochrietor J.D., Clamp M.F., Moroz T.P., Grubb J.H., Shah G.N., Waheed A., Sly W.S., Linser P.J. Carbonic anhydrase XIV identified as the membrane CA in mouse retina: strong expression in Müller cells and the RPE. Exp. Eye Res. 2005;81:492–500. doi: 10.1016/j.exer.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Sundaram V., Rumbolo P., Grubb J.M., Strisciuglio P., Sly W.S. Carbonic anhydrase II deficiency: diagnosis and carrier detection using differential enzyme inhibition and inactivation. Am. J. Hum. Genet. 1986;38:125–136. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.