

Abstract
Desbuquois dysplasia (DBQD) is a severe condition characterized by short stature, joint laxity, and advanced carpal ossification. Based on the presence of additional hand anomalies, we have previously distinguished DBQD type 1 and identified CANT1 (calcium activated nucleotidase 1) mutations as responsible for DBQD type 1. We report here the identification of five distinct homozygous xylosyltransferase 1 (XYLT1) mutations in seven DBQD type 2 subjects from six consanguineous families. Among the five mutations, four were expected to result in loss of function and a drastic reduction of XYLT1 cDNA level was demonstrated in two cultured individual fibroblasts. Because xylosyltransferase 1 (XT-I) catalyzes the very first step in proteoglycan (PG) biosynthesis, we further demonstrated in the two individual fibroblasts a significant reduction of cellular PG content. Our findings of XYLT1 mutations in DBQD type 2 further support a common physiological basis involving PG synthesis in the multiple dislocation group of disorders. This observation sheds light on the key role of the XT-I during the ossification process.
Introduction
Desbuquois dysplasia (DBQD [MIM 251450]) belongs to the multiple dislocation group of disorders (group 20, international classification of skeletal disorders).1 It is characterized by dislocations of large joints, severe pre- and postnatal growth retardation (−5 SD), joint laxity, and flat face with prominent eyes. Radiological features include short long bones with a monkey wrench appearance of the proximal femora (exaggerated trochanter) and advanced carpal and tarsal ossification. Based on the presence or absence of additional hand anomalies (ranging from extra ossification center distal to the second metacarpal, delta phalanx, or bifid distal phalanx of the thumb), we have distinguished DBQD type 1 from DBQD type 2.2,3
In 2009, we identified CANT1 (calcium activated nucleotidase 1 [MIM 613165]) mutations as responsible for DBQD type 1.4 CANT1 mutations have then been reported in the “Kim variant” of DBQD, characterized by short metacarpals and elongated middle and proximal phalanges but very short distal phalanges.5,6 All subjects with the Kim variant were of Japanese or Korean origin and had a p.Val226Met substitution on at least one allele, with a shared common haplotype, supporting a founder effect in this population.
After our initial study, we screened CANT1 in a series of 38 DBQD-affected individuals and found CANT1 mutations not only in all subjects with DBQD type 1 but also in a Kim variant subject and in one atypical DBQD-affected individual with thumb anomaly and major joint dislocations.7 However, none of our 30 DBQD type 2 individuals was found to carry a CANT1 mutation.7
DBQD types 1 and 2 have overlapping features with the autosomal-dominant form of Larsen syndrome (MIM 150250) resulting from FLNB mutations (MIM 603381),8 the spondylo-epiphyseal dyplasia form with dislocations (MIM 143095) resulting from chondroitin 6-O-sulfotransferase 3 (C6ST-1) (CHST3 [MIM 603799]) mutations,9 the diastrophic dysplasia (DTD [MIM 222600]) resulting from solute carrier family 26 (sulfate transporter) member 2 (SLC26A2 [MIM 606718]) mutations,10 and a chondrodysplasia with joint dislocations resulting from IMPAD1 (inositol monophosphatase domain-containing protein 1 [MIM 614010]) mutations.11 Apart from Larsen syndrome, all these disorders are characterized by a reduced amount of proteoglycans (PGs), which are known to be essential macromolecules with a large panel of functions, including extracellular matrix organizers and cell signaling mediators.12 PGs are composed of a core protein with at least one glycosaminoglycan (GAG) chain attached via a Ser-Gly consensus motif and can be either located at the cell surface or secreted in the extracellular matrix.12
Because of the clinical overlap of this group of disorders, we screened CHST3, SLC26A2, and IMPAD1 in the remaining DBQD type 2 individuals and found a homozygous CHST3 mutation in 1/3013 and IMPAD1 mutations in 2/30 subjects.
Among the remaining 27 DBQD type 2 subjects with no known molecular basis, only 20 with detailed clinical and radiological data and regular follow-up were included in our study. To identify the DBQD type 2 gene, we selected two siblings from consanguineous parents for whole-exome sequencing analysis and eventually identified homozygous mutations in seven individuals from six families in the xylosyltransferase 1 gene (XYLT1 [MIM 608124]). XYLT1 encodes xylosyltransferase 1 (XT-I, EC 2.4.2.26), which is involved in PG synthesis.12 Indeed, the very first step of this process consists of the initiation of the glycosaminoglycan (GAG) chains and can be catalyzed by the two paralogs XT-I and XT-II.14
Material and Methods
Individuals
The 20 DBQD-affected individuals selected for this the study fulfilled the diagnostic criteria for DBQD type 2, namely severe pre- and postnatal short stature, short extremities, dislocations with monkey wrench appearance of the femora, short long bones with metaphyseal widening, epiphyseal dysplasia, and advanced carpal and tarsal ossification (Figure 1). Among them, the two siblings selected for whole-exome sequencing analysis were born to healthy consanguineous Tunisian parents (f = 1/32). At birth, they both had a severe short stature, hypotonia, flat face with prominent eyes, hyperlaxity, hip dislocation, and narrow thorax leading to a respiratory distress that spontaneously resolved in the first year of life. Skeleton X-rays showed a monkey wrench appearance of the femoral neck, epiphyseal dysplasia, knee dislocation, and advanced carpal bone age (Figure 2, Table 1).
Figure 1.
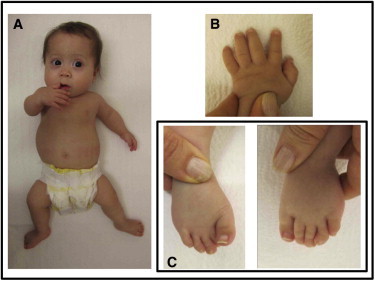
Clinical Features of One DBQD Type 2 Subject with XYLT1 Mutation
Individual 7 (family 6) at 12 months. Note the flat face, narrow thorax, short limbs (A) with hip dislocation, hyperlaxity of fingers (B), and deviation of toes (C).
Figure 2.
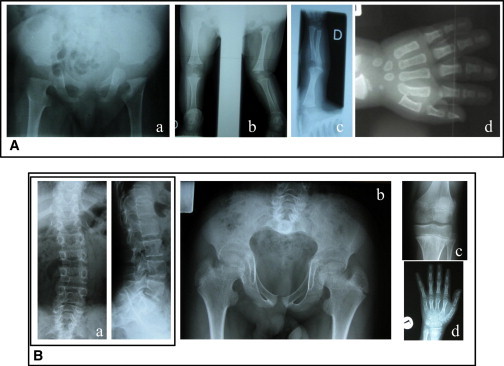
Radiological Features of Two DBQD Type 2 Subjects with XYLT1 Mutations
(A) Subject 1 (family 1) at 8 months. Note the monkey wrench appearance of the proximal femora, absence of upper femoral epiphyseal centers (a), knee dislocation (b), short long bones (c), and advanced carpal bone age (d).
(B) Subject 4 (family 3) at 12 years of age. Note the scoliosis with irregular vertebral endplates (a), monkey wrench appearance of the proximal femora with short femoral necks, short and broad iliac wings (b), flat knee epiphyses (c), short metacarpals, and advanced carpal bone age and prominent wrist epiphyses (d).
Table 1.
Clinical Features of the Seven DBQD Type 2-Affected Individuals from Six Families
| Ethnic Origin | Csg | Parameters at Birth | Clinical Features at First Exama | Radiological Features | Follow-up: Growth and Skeleton | Other | |
|---|---|---|---|---|---|---|---|
| Family 1, sibling 1 (female) | Tunisian | 1/32 | length 37 cm (term) | hyperlaxity respiratory distress hypotonia flat face |
monkey wrench of the femoral neck epiphyseal dysplasia knee dislocation advanced carpal bone age |
at 24 years of age: height 111.5 cm (< −6 SD) | mild intellectual disability |
| Family 1, sibling 2 (male) | Tunisian | 1/32 | length 41 cm (term) | hypotonia narrow thorax hip dislocation flat face |
monkey wrench of the femoral neck epiphyseal dysplasia knee dislocation advanced carpal bone age |
at 20 years of age: height 121 cm (< −6 SD) | intellectual disability |
| Family 2 (female) | Mauritian | 1st cousins | weight 2,000 g | lower limb deformity multiple dislocations (hip, knee) |
monkey wrench of the femoral neck brachymetacarpy epiphyseal dysplasia |
at 13 years of age: flessum of hips and knees valgus deformation of the lower limbs patella instability multiple surgeries weight 35 kg (−1 SD) height 98 cm (< -6 SD) toe deformations |
mild intellectual disability |
| Family 3 (male) | Belgian | 1st cousins | weight 2,570 g, length 39 cm, HC 33 cm (term); transient respiratory problems in the neonatal period | flat face low nasal bridge blue sclerae cleft palate short neck narrow thorax short limbs |
coronal clefts in the neonatal period, thereafter mild platyspondyly shortening of tubular bones absent ossification of distal femoral epiphyses at birth |
at 12 years, 9 months of age: weight 23.7 kg (−3.5 SD) height 109.5 cm (−6 SD) span 111 cm HC 50.8 cm (−2 SD) flat face, prominent eyes, low nasal bridge pectus carinatum, narrow thorax hyperlaxity of fingers and knees (genua valga) broad feet, toe clinodactyly |
intellectual disability |
| Family 4 | Turkish | 1st cousins | length 44 cm (term) | at 3.5 months: height 48.5 cm head control: 2 months hip dislocation (right) knee dislocation simian creases hypermobile fingers flat face blue sclerae |
neonatal period: advanced carpal ossification right hip and bilateral knee dislocation at 8 months: advanced bone age elbow dislocation |
at 11 months of age: height 56 cm at 5.5 years of age: height 84 cm (−5.5 SD) coarse and round face full cheek, long philtrum, mild micrognathia hypermobile fingers moderate truncal obesity pectus excavatum |
sitting at 9 months walking at 3 years |
| Family 5 | Turkish | 1st cousins | length 43 cm (term) | at 52 days: height 46 cm round and flat face epicanthal folds short extremities and hands bilateral simian crease |
neonatal period: monkey wrench advanced carpal ossification, short metacarpals and phalanges widened anterior ribs at 9 years: patella and elbow subluxation short iliac wings |
at 13 months of age: height 59 cm at 9 years of age: height 99 cm at 13 years of age: height 109 cm (−9 SD) HC 53 cm coarse and round face, blue sclera, proptotic eyes short extremities increased lumbar lordosis hypermobile joints pectus excavatum pes planus truncal obesity |
mild intellectual disability sitting at 8 months walking at 24 months |
| Family 6 | Turkish | from the same village | length 33 cm, weight 1,200 g (born at 36 WG) | cleft palate subluxation of right knee |
monkey wrench advanced carpal ossification and tarsal extra ossification double proximal femoral epiphyses short phalanges with short 1st metacarpal |
at 12 months of age: height 50 cm (<−6 SD) hypermobile joints respiratory problems 2 first months of life |
normal motor development at age 2 |
Abbreviations are as follows: Csg, consanguinity; WG, weeks of gestation; HC, head circumference.
First exam was performed at birth unless noted otherwise.
Samples
Informed consent for participation and sample collection were obtained via protocols approved by the Necker Hospital ethics board committee. Venous blood was obtained for DNA extraction from DBQD-affected individuals (QIAamp DNA blood Maxi kit, QIAGEN). Fibroblast cultures were established from skin biopsies obtained from scalp incision.
Exome Sequencing
Exome capture was performed at the genomic platform of Foundation IMAGINE (Paris, France) with the SureSelect Human All Exon kit (Agilent Technologies).15 Single-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina) generating 72-base reads. Sequence data were analyzed by the Bioinformatic platform and visualized via the interface created by the Bioinformatic platform (Université Paris Descartes, Paris). For sequence alignment, variant calling, and annotation, the sequences were aligned to the human genome reference sequence (UCSC Genome Browser, hg18 build) by BWA aligner.16 Downstream processing was carried out with the Genome Analysis Toolkit (GATK),17 SAMtools18 and Picard Tools. Substitution calls were made with GATK Unified Genotyper, whereas indel calls were made with a GATK IndelGenotyperV2. All calls with a read coverage ≤2× and a Phred-scaled SNP quality of ≤20 were filtered out. All the variants were annotated with an in-house-developed annotation software system.
Microsatellite Analysis of XYLT1 Locus
Microsatellite analysis was performed in consanguineous families at the XYLT1 locus on chromosome 16p12.3, by means of four repeat-containing microsatellite markers: D16S405, D16S499 located on both sides of XYLT1, and two intragenic microsatellites, D16S103 and D16S3017. FAM-labeled PCR products were run on an ABI 3130 sequencer and analyzed with GeneMapper (Applied Biosystems).
Sequencing Analysis of XYLT1
The exons and exon-intron boundaries of XYLT1 were amplified with specific primers (available upon request). Amplification products were purified by ExoSapIT (Amersham) and directly sequenced with the Big Dye Terminator Cycle Sequencing Ready Reaction kit v.1.1 on an automatic sequencer (ABI3130xl; PE Applied Biosystems). Sequence analyses were performed with Seqscape software v.2.5 (Applied Biosystems).
RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR
Skin primary fibroblasts were cultured in DMEM medium supplemented with 10% fetal calf serum and antibiotics at 37°C in a humidified atmosphere containing 5% CO2. For real-time PCR analyses, fibroblasts were seeded onto 96-well plates after standardization via the cell counter CASY Model TT (Roche). After reverse transcription of mRNA with M-MLV reverse transcriptase kit (Invitrogen), PCR analyses were performed on the 7300 Real Time PCR System (Roche). The cDNA level of XYLT1, XYLT2, and B4GALT7 was normalized to GAPDH.
Data were generated from three independent experiments performed in quadruplate and compared to control data with one-way ANOVA followed by Tukey's post hoc correction test with GraphPad Prism 6.0 software (GraphPad).
Analysis of PG Biosynthesis and Profile after Metabolic Radiolabeling in Cultured Fibroblasts
Fibroblasts were seeded onto 6-well plates and incubated the following day in Fischer’s medium containing 10 μCi/ml Na2[35S]SO4 in the presence of 5 μM of methylumbelliferyl-β-D-xylopyranoside (4-MUX) or vehicle (DMSO) as described previously.19 After radiolabeling, each cell media or cellular fractions were applied to G-50 columns (GE Healthcare, VWR) and quantified by scintillation counting on Packard 1600TR Tri Carb Liquid Scintillation Analyzer.
To establish the PG profile, the PG fractions were digested for 4 hr at 37°C by either the Chondroitinase ABC (cABC) from Proteus vulgaris (Sigma-Aldrich) or by a mixture containing both the Heparinases II and III from Flavobacterium heparinum (Sigma-Aldrich). All samples were resolved by SDS-PAGE with Criterion Precast gels (4%–15% Bis-Tris, Bio-Rad) and visualized by autoradiography. In order to compare PG distribution, the same amount of radioactivity was loaded in each well.
Results
Identification of a Homozygous XYLT1 Mutation by Whole-Exome Sequencing in Two Siblings with DBQD Type 2
For exome-sequencing analysis, we first focused our analyses on nonsynonymous variants, splice acceptor and donor site mutations, and coding indels, anticipating that synonymous variants were far less likely to cause disease (Table 2). We also defined variants as previously unidentified if they were absent from both control populations and data sets including dbSNP129, the 1000 Genomes Project, and in-house exome data.
Table 2.
Filtering Procedure for Bioinformatic Analysis
| Individuals |
Single-Nucleotide Variations |
||||
|---|---|---|---|---|---|
| Total | Without Duplicatesa | Not in Databasesb | In Essential Splicing and Coding Regionsc | Deleterious/Damaging or Unknownd | |
| Family 1, sibling 1 (Csg) | 11,534 | 5,560 | 216 | 60 | 11 |
| Family 1, sibling 2 (Csg) | 11,324 | 5,245 | 159 | 35 | 4 |
| Family 1, siblings 1+2 | 8,881 | 4,590 | 70 | 18 | 2 |
Abbreviation is as follows: Csg, consanguinity.
Without duplicates resulting from the homozygous status of subjects.
Databases searched were dbSNP, 1000 Genomes, Exome Variant Server, and in-house exomes.
Variants included were essential splicing and nonsynonymous (missense, nonsense, deletion and insertion); eliminated variants were intergenic or intronic noncoding RNA or UTR splicing.
As determined by SIFT or PolyPhen.
Based on the recessive mode of inheritance of DBQD, two genes were found to harbor an identical homozygous substitution in the two siblings: the structural maintenance of chromosome 1B (SMC1B [MIM 608685], RefSeq accession number NM_148674.3) and the xylosyltransferase 1 gene (XYLT1, RefSeq NM_022166.3) (Table 2). These results were confirmed by direct sequencing.20 The SMC1B (c.11T>G [p.Leu4Arg]) and XYLT1 (c.1792C>T [p.Arg598Cys]) substitutions cosegregated with the disease, were present at the heterozygous state in the parents, and were considered pathogenic in the PolyPhen and Sift analytical programs. SMC1B belongs to the cohesin family required for chromatid cohesion and DNA recombination during meiosis and mitosis. Nevertheless, because XT-I catalyzes the very first step of PG biosynthesis (Figure 3) and owing to functional relevance of PGs in extracellular matrix, we considered XYLT1 as our best candidate gene and undertook its study in the 19 remaining families affected by DBQD type 2. XYLT1 is composed of 12 exons encoding a protein of 959 amino acids (aa) consisting of two domains, the glycosyltransferase family 14 domain (aa 328–581) and the xylosyltransferase domain (aa 613–794).
Figure 3.
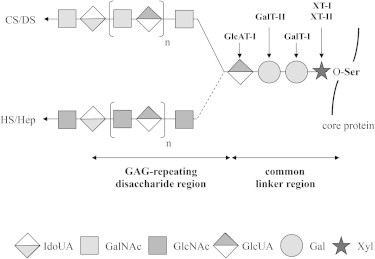
Schematic Representation of the PG Biosynthesis
XT-I and XT-II participate in the initiation of the PG biosynthesis by transferring a Xyl to specific Ser residues of the core protein. Two Gal and one GlcUA will be sequentially added via the action of GalT-I, GalT-II, and GlcAT-I to form the common linker region. The addition of a GalNAc will then initiate the assembly of chondroitin-sulfate/dermatan-sulfate (CS/DS) chains while addition of a GlcNAc will initiate the synthesis toward heparan-sulfate/heparin (HS/Hep) chains. Mutations in genes involved in the assembly of the common linker region have been associated with several disorders, namely DBQD type 2 (XYLT1), Ehlers-Danlos progeroid syndrome, type 1 (B4GALT7), Ehlers-Danlos progeroid syndrome, type 2/spondyloepimetaphyseal dysplasia with joint laxity type 1 (SEMD-JL1) (B3GALT6), and Larsen-like syndrome (B3GAT3).
Among the 19 remaining families affected by DBQD type 2, 5/13 inbred individuals were homozygous at the XYLT1 locus and direct sequencing was finally performed in 11 families (including 6 nonconsanguineous families). A total of five distinct XYLT1 mutations in seven individuals (six families) with DBQD type 2 were identified (Figures 1 and 2, Tables 1 and 3). Two resulted in a premature stop codon (c.276dupG [p.Pro93Alafs∗69], c.439C>T [p.Arg147∗]) located N-terminal to the glycosyltransferase family 14 and xylosyltransferase domains; two were splice site mutations (c.1290−2A>C, c.1588−3C>T) located in the donor sites of exons 6 and 8, respectively, involved in the glycosyltransferase family 14 domain; and one was a missense substitution (c.1792C>T [p.Arg598Cys]) located in the xylosyltransferase domain, considered as damaging in PolyPhen and Sift analytical programs (Figure 4, Table 3). All mutations segregated with the disease and were not identified in 200 control chromosomes. SMC1B was excluded by linkage analysis in the remaining five consanguineous families with XYLT1 mutations.
Table 3.
XYLT1 Mutations in the Six DBQD Type 2-Affected Families
| Family | Ethnic Origin | Csg | No. of Affected Children | Nucleotide Change | Status | Amino Acid Change | Location |
|---|---|---|---|---|---|---|---|
| 1 | Tunisian | yes | 2 | c.1792C>T | Ho | p.Arg598Cys | Ex9 |
| 2 | Mauritian | yes | 1 | c.439C>T | Ho | p.Arg147∗ | Ex3 |
| 3 | Belgian | yes | 1 | c.276dupG | Ho | p.Pro93Alafs∗69 | Ex1 |
| 4 | Turkish | yes | 1 | c.1588−3C>T | Ho | ? | In7 |
| 5 | Turkish | yes | 1 | c.1290−2A>C | Ho | ? | In5 |
| 6 | Turkish | yes | 1 | c.1290−2A>C | Ho | ? | In5 |
Abbreviations are as follows: Csg, consanguinity; ho, homozygote; ?, mutation not localized in a protein domain.
Figure 4.
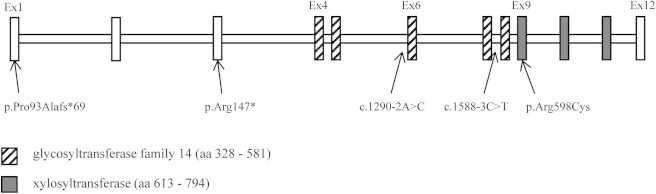
Location of the Five XYLT1 Mutations
Organization of XYLT1 gene and protein with the two glycosyltransferase family 14 and xylosyltransferase domains.
XYLT1 Mutations Are Correlated with a Decrease of cDNA Level in Two Cultured Individual Fibroblasts
By quantifying the cDNA level of XYLT1 in the two cultured individual fibroblasts (p.Arg147∗, female aged 14; and p.Pro93Alafs∗69, male aged 23), we found a dramatic reduction of XYLT1 cDNA level (Figure 5A). Because the two xylosyltransferases XT-I and XT-II14 can both initiate the synthesis of GAG chains to PG core proteins, we also measured the cDNA level of XYLT2 and found a significant decrease of XYLT2 expression in the two individual cell lines when compared to control 2 (male, aged 12). No significant differences were found when individual data were compared to control 1 (female, aged 2.5; Figure S1 available online).
Figure 5.
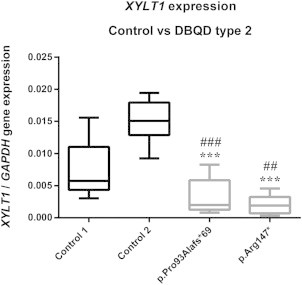
cDNA Level of XYLT1 in Controls and Two Cultured Individual Fibroblasts
cDNA levels were normalized to GAPDH. Individuals p.Arg147∗ and p.Pro93Alafs∗69 are female aged 14 and male aged 23 years, respectively. Control 1 (female) and 2 (male) are aged 2.5 and 12 years, respectively. Control (black boxes) and individual (gray boxes) data are presented as box plots and are the mean of three independent experiments performed in quadruplate. ##p < 0.01, ###p < 0.001 compared to control 1, ∗∗∗p < 0.001 compared to control 2 by one-way ANOVA with Tukey’s post hoc test.
Note that mutations in XYLT1 affect cDNA level of XYLT1.
By contrast, the cDNA level of B4GALT7, which encodes the glycosyltransferase (GT) that adds the second sugar galactose (Gal) to the xylose (Xyl), was not affected by XYLT1 mutations (Figures 3 and S1).
The PG Biosynthesis Is Strongly Affected by XYLT1 Mutations in the Two Cultured Individual Fibroblasts
To evaluate the impact of the loss of function of XYLT1, the cells were treated with the 4-MUX, an exogenous acceptor substrate of the GalT-I that can increase GAG synthesis (Figure 3). No major differences in the sulfate incorporation level were found for the extracellular fractions in the absence of 4-MUX. In the presence of 4-MUX, a significant increase of sulfate incorporation was observed, indicating that GAG biosynthesis was initiated and enhanced by the 4-MUX via the GalT-I activity (Figure S2). In contrast, the incorporated sulfate level associated with the cellular fractions was significantly lower in the two cultured individual fibroblasts and no increase was observed after the addition of 4-MUX (Figure 6).
Figure 6.
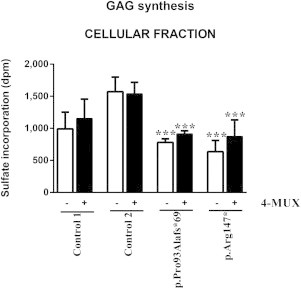
Sulfate Incorporation of Cellular Fractions in Controls and the Two Cultured Individual Fibroblasts in the Presence of 4-MUX or Vehicle
The black (+4-MUX, 5 μM) and white (DMSO vehicle) bars represent the mean ± SEM of two independent experiments performed in triplicate. ∗∗∗p < 0.001 when compared to the control 2 by one-way ANOVA with Tukey’s post hoc test.
Note that XYLT1 mutations have an impact on the biosynthesis of cellular PGs in the two cultured individual fibroblasts.
Modifications in PG Pattern Are a Consequence of XYLT1 Mutations
An important reduction of secreted and large chondroitin-sulfate proteoglycan (CSPG) production was observed in the two cultured individual fibroblasts (lanes 4, 6, 7, and 9, Figure 7). In the p.Arg147∗ cell line, the presence of a stronger second band at 90 kDa (lanes 7 and 9, Figure 7) was observed, presumably corresponding to the accumulation in the extracellular media of other small CSPGs such as decorin. For individual and control fibroblast cell lines, the addition of 4-MUX led to the production of GAG chains with an increased ratio of lower-molecular-weight GAGs. The sensitivity to Chondroitinase ABC treatment supported that these GAG chains were mainly chondroitin-sulfate (CS) (lanes 11, 14, and 17, Figure 7). In the p.Arg147∗ cultured fibroblasts, the accumulation of CSPGs at 90 kDa was still observed after the xyloside addition (lanes 16 and 18, Figure 7).
Figure 7.
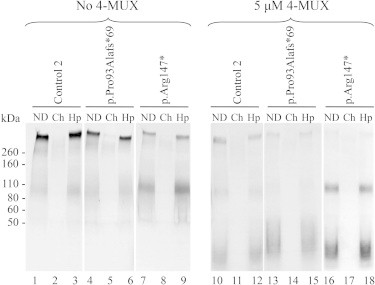
Gel Electrophoresis of Radiolabeled GAG Chains Extracted from Control 2 and the Two Cultured Individual Fibroblasts Media in the Presence 4-MUX or Vehicle
For each sample, three lanes corresponding to nondigested (ND), digested by the Chondroitinase ABC (Ch), and digested by the Heparinases II and III (Hp) were loaded onto the gel and the volumes were adjusted to dpm values to visualize GAG profile and distribution. Control 2 (male) is aged 12.
Note that mutations in XYLT1 change the distribution and profile of secreted PGs in the two cultured individual fibroblasts.
For the cellular fractions, mainly heparan-sulfate proteoglycans (HSPGs) with high molecular weight (>200 kDa) were detected and no major differences in PG profiles and distribution were observed between control 2 and individual fibroblasts in the absence or presence of 4-MUX (data not shown).
Discussion
We report here the identification of XYLT1 mutations in seven individuals from six families with DBQD type 2. All individuals presented severe pre- and postnatal short stature below –6 SD, flat face with prominent eyes, short extremities, dislocations with monkey wrench appearance of the femora, short long bones with metaphyseal widening, epiphyseal dysplasia, and advanced carpal and tarsal ossification. No significant clinical or radiological differences could be found with the remaining 14 DBQD type 2 subjects with unknown molecular bases. However, long-term follow-up of XYLT1-mutated individuals emphasizes the severity of the short stature (<−6 SD) contrasting with obesity, lower limb and foot deformities requiring often repeated surgeries, and intellectual disability (5/7). Interestingly, respiratory distress was present at birth in 4/7 subjects and spontaneously resolved in the first years of life but thorax narrowness persisted in the eldest children. No major scoliosis was observed.
Among the five mutations identified, four were expected to result in loss of function, confirmed in two by the demonstration of a drastic reduction of gene expression in cultured fibroblasts. The substitution p.Arg598Cys identified is located in a highly conserved region of the protein, between the glycosyltransferase family 14 and the catalytic domains of the enzyme,21 pointing out possible changes in the protein folding and/or stability as the positively charged amino acid Arg is replaced by a polar and neutral Cys residue. Very recently, another substitution of XT-I (p.Arg481Trp) has been reported in Turkish siblings presenting short stature and minor skeleton features associated with intellectual disability.22 X-rays of the two siblings (aged 16 and 18 years) do not show monkey wrench appearance of the femoral neck or advanced carpal bone age, but these features disappear in the course of DBQD. However, among features listed in the two siblings, coxa valga, broad thumbs, abnormal feet, and obesity are reminiscent of DBQD spectrum. The milder phenotype observed in the two siblings might be due to a partial loss of function of XT-I.22
PG formation begins with the synthesis of a common linker chain of four sugar residues. The very first step of this process can be catalyzed by the two paralogs XT-I and XT-II14 leading to the initiation of the two types of GAG chains (Figure 3).23 Our finding of decreased XYLT2 cDNA level in two individual fibroblasts suggests that XYLT1 loss of function may affect XYLT2 gene expression. However, because of the variability observed in cultured fibroblasts (even in control samples), this observation will need to be confirmed in additional individual samples compared to age- and gender-matched control samples.
The two paralogs XT-I and XT-II exhibit a high degree of homology within their catalytic domain24 and are ubiquitously expressed in human tissues (data not shown).25,26 Despite the apparent redundancy in catalytic activity as demonstrated by in vitro enzymatic assays25,26 and in gene expression, the identification of XYLT1 mutations in a severe chondrodysplasia supports a pivotal role of XYLT1 during the ossification process. The specific implication of XT-I in skeletal development and during cartilage repair has been previously reported.27,28 Moreover, a recent study describing the phenotype of the pug mouse mutant carrying a homozygous missense mutation in Xylt1 and characterized by a disproportionate dwarfism, premature chondrocyte maturation, and increased Indian Hedgehog signaling further supports a key role of XT-I in early chondrocyte maturation and skeletal length.29
The presence of intellectual disability was observed in at least five subjects and highlights also a possible role of XYLT1 in brain development. Interestingly, common CSPGs such as aggrecan, perlecan, and biglycan are both found in cartilage and expressed during brain development.30,31 Our findings that mainly CSPGs were secreted into the cell media and that synthesis of large CSPGs was clearly reduced for the p.Arg147∗ cell line suggest that both PG-specific expression pattern and impaired synthesis of large CSPGs may contribute to the tissue specificity of the disease. Interestingly, a strong decrease of sulfate incorporation in the high-density PG fraction (mainly CSPGs) was also observed in the pug mutant.29 Finally, the accumulation of PGs in the range of 90 kDa observed for the p.Arg147∗ cell line could compensate the important loss of high-molecular-weight CSPGs, whereas for the p.Pro93Alafs∗69 cell line, the loss of large CSPGs was less pronounced.
The consequences of XYLT1 mutations on PG synthesis were also clearly illustrated by a significant reduction of cellular PG synthesis in the two cultured individual fibroblasts. This reduction could not be rescued by the addition of 4-MUX, indicating that most of the 4-MUX-primed GAGs were presumably secreted into the cell media. Because XT-I catalyzes the first step of PG synthesis, an altered expression of this enzyme is likely to impact PG synthesis either by limiting the initiation of the GAG chains or (less probably) by leading to an abnormal decrease of the PG core protein level.
Interestingly, trials based on the use of either heparin-like molecules in animals32 to repair skull defects or HS supplementation33 in cultures of human mesenchymal stem cells have already been tested. Although protein xylosylation is known to be the rate-limiting step in PG biosynthesis, this can be bypassed by the addition of exogenous xylosides (such as 4-MUX) into cell media owing to the GalT-I activity. Our findings of identical B4GALT7 cDNA level in controls and individual fibroblasts but increased GAG production in response to 4-MUX treatment in individual fibroblasts may support the development of new approaches based on the use of GAGs.
To date, mutations in genes involved in the synthesis of the common linker region have been identified in β1,4-galactosyltransferase 7 (GalT-I = B4GALT7 [MIM 604327]),34–36 β1,3-galactosyltransferase 6 (GalT-II = B3GALT6 [MIM 615291]),37,38 and β1,3-glucuronosyltransferase I (GlcAT-I = B3GAT3 [MIM 606374]),39 and the human phenotypes associated included Ehlers-Danlos progeroid type (type 1 MIM 130070, type 2 MIM 615349), spondylo-epiphyseal-meta-dysplasia with joint laxity type 1 (SEMD-JL1 [MIM 271640]), and a Larsen-like phenotype with heart defect (MIM 245600). Perturbations in GAG modifications (mainly sulfation) have been also reported in (1) spondyloepiphyseal dysplasia (SED) with dislocations resulting from CHST3 mutations (C6ST-1) responsible for altered sulfation of CSPGs,40 (2) a chondrodysplasia with joint dislocations resulting from mutations in IMPAD1 that encodes for the golgi-resident nucleotide phosphatase gPAPP and resulting in an undersulfation of PGs,11 and (3) DBQD resulting from CANT1 mutations accounting for shorter GAG chains.7
Our findings of XYLT1 mutations in DBQD type 2 further confirm a common physiological basis in the group of multiple dislocation disorders involving PG synthesis and also shed light on the pivotal and specific role of XT-I during the ossification process.
Acknowledgments
This work has been supported by FRM (Fondation pour la Recherche Médicale) funding (DEQ20120323703).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Alamut Interpretation Software 2.0 (gateway for PolyPhen-2, SIFT, SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and Human Splicing Finder), http://www.interactive-biosoftware.com
Ensembl Genome Browser, http://www.ensembl.org/index.html
GeneCards, http://www.genecards.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faivre L., Cormier-Daire V., Eliott A.M., Field F., Munnich A., Maroteaux P., Le Merrer M., Lachman R. Desbuquois dysplasia, a reevaluation with abnormal and “normal” hands: radiographic manifestations. Am. J. Med. Genet. A. 2004;124A:48–53. doi: 10.1002/ajmg.a.20440. [DOI] [PubMed] [Google Scholar]
- 3.Faivre L., Le Merrer M., Al-Gazali L.I., Ausems M.G., Bitoun P., Bacq D., Maroteaux P., Munnich A., Cormier-Daire V. Homozygosity mapping of a Desbuquois dysplasia locus to chromosome 17q25.3. J. Med. Genet. 2003;40:282–284. doi: 10.1136/jmg.40.4.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huber C., Oulès B., Bertoli M., Chami M., Fradin M., Alanay Y., Al-Gazali L.I., Ausems M.G., Bitoun P., Cavalcanti D.P. Identification of CANT1 mutations in Desbuquois dysplasia. Am. J. Hum. Genet. 2009;85:706–710. doi: 10.1016/j.ajhg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim O.H., Nishimura G., Song H.R., Matsui Y., Sakazume S., Yamada M., Narumi Y., Alanay Y., Unger S., Cho T.J. A variant of Desbuquois dysplasia characterized by advanced carpal bone age, short metacarpals, and elongated phalanges: report of seven cases. Am. J. Med. Genet. A. 2010;152A:875–885. doi: 10.1002/ajmg.a.33347. [DOI] [PubMed] [Google Scholar]
- 6.Furuichi T., Dai J., Cho T.J., Sakazume S., Ikema M., Matsui Y., Baynam G., Nagai T., Miyake N., Matsumoto N. CANT1 mutation is also responsible for Desbuquois dysplasia, type 2 and Kim variant. J. Med. Genet. 2011;48:32–37. doi: 10.1136/jmg.2010.080226. [DOI] [PubMed] [Google Scholar]
- 7.Nizon M., Huber C., De Leonardis F., Merrina R., Forlino A., Fradin M., Tuysuz B., Abu-Libdeh B.Y., Alanay Y., Albrecht B. Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 2012;33:1261–1266. doi: 10.1002/humu.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang D., Herring J.A., Swaney S.S., McClendon T.B., Gao X., Browne R.H., Rathjen K.E., Johnston C.E., Harris S., Cain N.M., Wise C.A. Mutations responsible for Larsen syndrome cluster in the FLNB protein. J. Med. Genet. 2006;43:e24. doi: 10.1136/jmg.2005.038695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unger S., Lausch E., Rossi A., Mégarbané A., Sillence D., Alcausin M., Aytes A., Mendoza-Londono R., Nampoothiri S., Afroze B. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A. 2010;152A:2543–2549. doi: 10.1002/ajmg.a.33641. [DOI] [PubMed] [Google Scholar]
- 10.Hästbacka J., de la Chapelle A., Mahtani M.M., Clines G., Reeve-Daly M.P., Daly M., Hamilton B.A., Kusumi K., Trivedi B., Weaver A. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78:1073–1087. doi: 10.1016/0092-8674(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 11.Vissers L.E., Lausch E., Unger S., Campos-Xavier A.B., Gilissen C., Rossi A., Del Rosario M., Venselaar H., Knoll U., Nampoothiri S. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am. J. Hum. Genet. 2011;88:608–615. doi: 10.1016/j.ajhg.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prydz K., Dalen K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000;113:193–205. doi: 10.1242/jcs.113.2.193. [DOI] [PubMed] [Google Scholar]
- 13.Nizon M., Alanay Y., Tuysuz B., Kiper P.O., Geneviève D., Sillence D., Huber C., Munnich A., Cormier-Daire V. IMPAD1 mutations in two Catel-Manzke like patients. Am. J. Med. Genet. A. 2012;158A:2183–2187. doi: 10.1002/ajmg.a.35504. [DOI] [PubMed] [Google Scholar]
- 14.Voglmeir J., Voglauer R., Wilson I.B. XT-II, the second isoform of human peptide-O-xylosyltransferase, displays enzymatic activity. J. Biol. Chem. 2007;282:5984–5990. doi: 10.1074/jbc.M608087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bui C., Ouzzine M., Talhaoui I., Sharp S., Prydz K., Coughtrie M.W., Fournel-Gigleux S. Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J. 2010;24:436–450. doi: 10.1096/fj.09-136291. [DOI] [PubMed] [Google Scholar]
- 20.Revenkova E., Eijpe M., Heyting C., Gross B., Jessberger R. Novel meiosis-specific isoform of mammalian SMC1. Mol. Cell. Biol. 2001;21:6984–6998. doi: 10.1128/MCB.21.20.6984-6998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Götting C., Müller S., Schöttler M., Schön S., Prante C., Brinkmann T., Kuhn J., Kleesiek K. Analysis of the DXD motifs in human xylosyltransferase I required for enzyme activity. J. Biol. Chem. 2004;279:42566–42573. doi: 10.1074/jbc.M401340200. [DOI] [PubMed] [Google Scholar]
- 22.Schreml J., Durmaz B., Cogulu O., Keupp K., Beleggia F., Pohl E., Milz E., Coker M., Ucar S.K., Nürnberg G. The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 2014;133:29–39. doi: 10.1007/s00439-013-1351-y. [DOI] [PubMed] [Google Scholar]
- 23.Pönighaus C., Ambrosius M., Casanova J.C., Prante C., Kuhn J., Esko J.D., Kleesiek K., Götting C. Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem. 2007;282:5201–5206. doi: 10.1074/jbc.M611665200. [DOI] [PubMed] [Google Scholar]
- 24.Götting C., Kuhn J., Kleesiek K. Human xylosyltransferases in health and disease. Cell. Mol. Life Sci. 2007;64:1498–1517. doi: 10.1007/s00018-007-7069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Götting C., Kuhn J., Zahn R., Brinkmann T., Kleesiek K. Molecular cloning and expression of human UDP-d-Xylose:proteoglycan core protein beta-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000;304:517–528. doi: 10.1006/jmbi.2000.4261. [DOI] [PubMed] [Google Scholar]
- 26.Roch C., Kuhn J., Kleesiek K., Götting C. Differences in gene expression of human xylosyltransferases and determination of acceptor specificities for various proteoglycans. Biochem. Biophys. Res. Commun. 2010;391:685–691. doi: 10.1016/j.bbrc.2009.11.121. [DOI] [PubMed] [Google Scholar]
- 27.Venkatesan N., Barré L., Bourhim M., Magdalou J., Mainard D., Netter P., Fournel-Gigleux S., Ouzzine M. Xylosyltransferase-I regulates glycosaminoglycan synthesis during the pathogenic process of human osteoarthritis. PLoS ONE. 2012;7:e34020. doi: 10.1371/journal.pone.0034020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eames B.F., Yan Y.L., Swartz M.E., Levic D.S., Knapik E.W., Postlethwait J.H., Kimmel C.B. Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 2011;7:e1002246. doi: 10.1371/journal.pgen.1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mis E.K., Liem K.F., Jr., Kong Y., Schwartz N.B., Domowicz M., Weatherbee S.D. Forward genetics defines Xylt1 as a key, conserved regulator of early chondrocyte maturation and skeletal length. Dev. Biol. 2014;385:67–82. doi: 10.1016/j.ydbio.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz N.B., Domowicz M. Proteoglycans in brain development. Glycoconj. J. 2004;21:329–341. doi: 10.1023/B:GLYC.0000046278.34016.36. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz N.B., Domowicz M. Chondrodysplasias due to proteoglycan defects. Glycobiology. 2002;12:57R–68R. doi: 10.1093/glycob/12.4.57r. [DOI] [PubMed] [Google Scholar]
- 32.Blanquaert F., Saffar J.L., Colombier M.L., Carpentier G., Barritault D., Caruelle J.P. Heparan-like molecules induce the repair of skull defects. Bone. 1995;17:499–506. doi: 10.1016/8756-3282(95)00402-5. [DOI] [PubMed] [Google Scholar]
- 33.Helledie T., Dombrowski C., Rai B., Lim Z.X., Hin I.L., Rider D.A., Stein G.S., Hong W., van Wijnen A.J., Hui J.H. Heparan sulfate enhances the self-renewal and therapeutic potential of mesenchymal stem cells from human adult bone marrow. Stem Cells Dev. 2012;21:1897–1910. doi: 10.1089/scd.2011.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidler D.G., Faiyaz-Ul-Haque M., Hansen U., Yip G.W., Zaidi S.H., Teebi A.S., Kiesel L., Götte M. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7) J. Mol. Med. 2006;84:583–594. doi: 10.1007/s00109-006-0046-4. [DOI] [PubMed] [Google Scholar]
- 35.Guo M.H., Stoler J., Lui J., Nilsson O., Bianchi D.W., Hirschhorn J.N., Dauber A. Redefining the progeroid form of Ehlers-Danlos syndrome: report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. A. 2013;161:2519–2527. doi: 10.1002/ajmg.a.36128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okajima T., Fukumoto S., Furukawa K., Urano T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999;274:28841–28844. doi: 10.1074/jbc.274.41.28841. [DOI] [PubMed] [Google Scholar]
- 37.Malfait F., Kariminejad A., Van Damme T., Gauche C., Syx D., Merhi-Soussi F., Gulberti S., Symoens S., Vanhauwaert S., Willaert A. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 2013;92:935–945. doi: 10.1016/j.ajhg.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakajima M., Mizumoto S., Miyake N., Kogawa R., Iida A., Ito H., Kitoh H., Hirayama A., Mitsubuchi H., Miyazaki O. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 2013;92:927–934. doi: 10.1016/j.ajhg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baasanjav S., Al-Gazali L., Hashiguchi T., Mizumoto S., Fischer B., Horn D., Seelow D., Ali B.R., Aziz S.A., Langer R. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermanns P., Unger S., Rossi A., Perez-Aytes A., Cortina H., Bonafé L., Boccone L., Setzu V., Dutoit M., Sangiorgi L. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 2008;82:1368–1374. doi: 10.1016/j.ajhg.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.