

Abstract
Neurons, like all cells, face the problem that tubulin forms microtubules with too many or too few protofilaments (pfs). Cells overcome this heterogeneity with the γ-tubulin ring complex, which provides a nucleation template for 13-pf microtubules. Doublecortin (DCX), a protein that stabilizes microtubules in developing neurons, also nucleates 13-pf microtubules in vitro. Using fluorescence microscopy assays, we show that the binding of DCX to microtubules is optimized for the lateral curvature of the 13-pf lattice. This sensitivity depends on a cooperative interaction wherein DCX molecules decrease the dissociation rate of their neighbors. Mutations in DCX found in patients with subcortical band heterotopia weaken these cooperative interactions. Using assays with dynamic microtubules, we discovered that DCX binds to polymerization intermediates at growing microtubule ends. These results support a mechanism for stabilizing 13-pf microtubules that allows DCX to template new 13-pf microtubules through associations with the sides of the microtubule lattice.
INTRODUCTION
Microtubules are long polymers of the protein tubulin that are essential to brain development and function. Neuronal migration, differentiation, and maturation each require the active remodeling of the neuron’s microtubule cytoskeleton (Conde and Caceres, 2009). Neurons and other cells must (1) nucleate new microtubules, (2) efficiently elongate these nascent microtubules, (3) selectively stabilize the microtubules they need, and (4) break down those they do not. A central question is how cells control the architecture of their microtubules, both in terms of the length of individual microtubules but also their thickness, meaning the specification of a 13-protofilament (pf) lattice.
Microtubules are “plastic” polymers (Kueh and Mitchison, 2009), which is to say that tubulin can form structures that differ in their lateral and longitudinal curvature. This plasticity is made possible by flexibility in the links between tubulin dimers and may give microtubules their unique mechanical properties (Sui and Downing, 2010), such as a length-dependent persistence length (Pampaloni et al., 2006). Cells use different microtubule curvatures to their advantage; for example, outwardly-curved pfs may enable cells to couple kinetochores to shrinking microtubule ends during mitosis (Koshland et al., 1988; Mandelkow et al., 1991). Structural plasticity also creates a challenge, however, in terms of controlling the number of pfs in the lattice. Microtubules form in a range of thicknesses when grown in cell extracts (Chretien et al., 1992) or in vitro (Wade and Chretien, 1993). Some have fewer than 13 pfs, whereas some have more. This heterogeneity is at odds with the uniform 13-pf microtubules observed in the majority of cell types (Tilney et al., 1973). Microtubules with too many or too few pfs have a supertwist in their pfs that causes motor proteins to spiral around the microtubule during intracellular transport (Ray et al., 1993). The disadvantages of such spiraling may explain the preference of most cells for 13-pf microtubules.
Cells overcome the intrinsic heterogeneity in pf-number via the centrosome (Evans et al., 1985), where the γ-tubulin ring-complex (γ-TuRC) (Zheng et al., 1995) provides a 13-pf nucleation template to ensure a uniform microtubule thickness (Moritz et al., 2000). Microtubules elongate from the γ-TuRC, which can remain attached as a minus-end cap (Keating and Borisy, 2000). What we do not understand is how microtubule thickness is controlled in regions devoid of γ-tubulin, such as neuronal processes (Baas and Joshi, 1992), or how the 13-pf lattice is maintained against defects that distort it during growth (Chretien et al., 1992). Doublecortin (DCX), a microtubule-associated protein (MAP) expressed in developing neurons (Gleeson et al., 1999), also nucleates uniform 13-pf microtubules in vitro (Moores et al., 2004), making it the first MAP after γ-tubulin to influence microtubule thickness. The γ-TuRC’s template mechanism was recently shown to originate from an intrinsic 13-fold symmetry of a minimal component set of the γ-TuRC (Kollman et al., 2010). DCX must specify 13-pf microtubules through a different mechanism, however, as DCX binds to the sides, not the base, of the lattice (Fourniol et al., 2010).
DCX was discovered in 1998 in a screen of patients with subcortical band heterotopia (“double cortex syndrome”) and X-linked lissencephaly (des Portes et al., 1998b; Gleeson et al., 1998), a malformation of the cerebral cortex caused by a failure in neuronal migration. The spectrum of this disease ranges from refractory epilepsy to severe mental retardation. DCX is enriched along the lengths of microtubules in the leading process of migrating neurons (Gleeson et al., 1999; Francis et al., 1999), extending immature neurites, and growth cones (Tint et al., 2009). RNAi of DCX caused a failure in radial migration of cortical neurons in rats (Bai et al., 2003) consistent with the disease phenotype; in cultured neurons, RNAi caused reductions in collateral branching (Tint et al., 2009), axon elongation, and dendrite arborization (Deuel et al., 2006). A prominent model is that DCX contributes to brain development through the stabilization of microtubules in the leading process of migrating neurons and other neuronal processes (Horesh et al., 1999), although the molecular mechanism of stabilization remains undefined.
Analysis of patient mutations identified tandem domains (Taylor et al., 2000), the N-DC and C-DC domains (Fig. 1A). The DC domains have a common ubiquitin-like fold (Kim et al., 2003) but are divergent in their primary sequence (Reiner et al., 2006). In vitro experiments showed that DCX stimulates the bulk rate of microtubule formation (Taylor et al., 2000), a result interpretable in light of the subsequent observations that DCX increases the number of nuclei formed in vitro (Moores et al., 2004) and also decreases the frequency of catastrophe and the rate of post-catastrophe shrinkage (Moores et al., 2006). Recently, electron microscopy has shown that a single DC domain binds to microtubules at the vertex of four tubulin dimers in the groove between pfs (Fourniol et al., 2010), a binding site that DCX shares with the microtubule end-tracking protein EB1 (Maurer et al., 2012). The microtubule saturates at one full-length DCX molecule per vertex (Moores et al., 2006), and the binding site is most-likely occupied by the N-DC domain (Fourniol et al., 2010). A large portion (~75%) of the molecule was not resolved in the EM reconstruction, however. Importantly, single DC domains alone are not sufficient to stimulate microtubule formation (Taylor et al., 2000) and thus the core molecular mechanism of DCX remains unsolved. We wanted to know the nature of this core mechanism and whether it is important to the interaction of DCX with microtubules during brain development. We also wanted to know how DCX might specify 13-pf microtubules in vitro.
Figure 1. DCX preferentially binds 13-pf microtubules.

(A) Schematic of DCX-GFP. The two DC-domains (labeled) are joined by a linker (labeled) and flanked by polypeptides. The C-terminal polypeptide is enriched in S/P residues (labeled). The GFP-tag is C-terminal. (B) Plot of absorbance at 350 nm versus time for tubulin alone (red), DCX-GFP alone (blue), and DCX-GFP with tubulin (green). DCX-GFP with tubulin produced a significant increase in absorbance, indicating microtubule formation. (C) Schematic of the single-molecule assay. (D) Schematic drawing of an axoneme (labeled), which nucleates axoneme-microtubules (MTs) with >90% 13-pf. (E) Chemical drawing of GMPCPP, which nucleates GMPCPP-MTs with >96% 14-pf. (F) Image of axoneme-nucleated microtubules (white arrow) and GMPCPP microtubules in the same microscope chamber (MTs); image of DCX-GFP exposed to these microtubules (DCX-GFP); color-combined image of axoneme-MTs, GMPCPP-MTs, and DCX-GFP. The DCX-GFP preferentially binds to the 13-pf axoneme MTs. (G) Schematic of a mixed population of MTs nucleated from purified tubulin. (H) Image of the mixed population of rhodamine-labeled microtubules (MTs); image of DCX-GFP exposed to this mixed population (DCX-GFP); color-combined image of MTs and DCX-GFP. Note that DCX-GFP binds preferentially to a subset of the mixed population, corresponding to the 13-pf subset, and that DCX-GFP binds to segments within individual microtubules (white arrow). See also Figure S1.
We chose to address these questions using a single-molecule fluorescence microscopy assay. We discovered that DCX recognizes 13-pf microtubules through a cooperative interaction between adjacent DCX molecules that reduces their dissociation rate. Mutations in DCX found in patients with double cortex syndrome disrupt this mechanism, demonstrating a link between the biophysical parameters of the molecule and the disease phenotype. Finally, we discovered that DCX binds to polymerization intermediates at growing microtubule ends, providing a mechanism by which DCX can specify 13-pf microtubules in vitro.
RESULTS
DCX measures microtubule thickness
Previous work showed that DCX, although difficult to see directly by electron microscopy, caused 80 Å diffraction signals to appear preferentially on 13-pf microtubules (Moores et al., 2004). This observation indicated that DCX can “measure” microtubule thickness: in other words, the binding of DCX to pre-existing microtubules appeared to be sensitive to the number of pfs in the lattice. In order to study how DCX could measure microtubule thickness, we expressed and purified human DCX and a GFP-tagged version of DCX from E. coli. (Fig. S1A). We confirmed that our recombinant DCX and DCX-GFP were active using a standard bulk-biochemistry assay that monitors the absorbance at 350 nm (A350), also called turbidity. Large polymers, such as microtubules, cause scattering at this wavelength. DCX and DCX-GFP were added to purified brain tubulin, which caused a significant increase in A350 (Fig. 1B and S1B), as previously observed (Taylor et al., 2000). This experiment served as a positive control for our protein expression and purification protocols and confirmed that the GFP-tag on our DCX-GFP does not affect the protein’s function in this assay.
In order to test whether the binding of DCX-GFP to microtubules is sensitive to their thickness, we extended the single-molecule fluorescence microscopy assay for microtubule-associated proteins and kinesins (Gell et al., 2010) (Fig. 1C). We used controlled growth conditions to generate stable microtubules of different thickness. As a positive control for 13-pf microtubules, we used sea-urchin axonemes as a nucleation template (Fig. 1D, a generous gift of Dr. E.D. Salmon). Axonemes extend >90% 13-pf microtubules from the A-tubule and the central pair (Ray et al., 1993). We also generated microtubules with >96% 14-pf by polymerization with GMPCPP, a slowly-hydrolyzable analog of GTP (Meurer-Grob et al., 2001) (Fig. 1E). Both microtubule types have a 3-start helix and one “seam.” We introduced both types into the same microscope chamber sequentially and distinguished them by their order of introduction into the chamber (Fig. 1E, white arrow). Figure 1E shows an image taken when we exposed these different microtubules to 0.5 μM DCX-GFP. The 13-pf axoneme-nucleated microtubules showed a bright DCX-GFP signal (I = 7462 ± 655 a.u. for regions corresponding to single MTs, mean ± SD, n = 7 MTs), while the GFP signal for 14-pf GMPCPP microtubules was dimmer (I = 1794 ± 83 a.u., n = 7 MTs), indicating a significant preference (p ≪0.0001 by Welch’s t-test). These results confirm that DCX binds preferentially to 13-pf microtubules.
We wondered if DCX-GFP could distinguish the 13-pf microtubules from others in mixed populations. We polymerized tubulin into microtubules with GTP and then stabilized them with paclitaxel. These microtubules will be present in a range of different thicknesses, with an expected distribution ranging between ~35% 13-pf (Ray et al., 1993) and ~75% 13-pf microtubules (Vitre et al., 2008) and the remainder consisting primarily of 14-pf microtubules (Fig. 1G). We adhered microtubules from this mixed population to a cover glass surface and introduced 0.5 μM DCX-GFP. Figure 1H shows a representative field of view of paclitaxel microtubules. We observed that the DCX-GFP signal could be divided into two subsets, one bright (I = 15716 ± 337 a.u., n = 9 MTs) and one dim (I = 1783 ± 217 a.u., n = 9 MTs). We interpret the bright subset of microtubules as having 13-pf. In support of this interpretation, the proportion of bright paclitaxel-stabilized microtubules is in agreement with the expected proportion of 13-pf microtubules in the population (55%, based on n = 1.12 mm of microtubule length analyzed). Furthermore, the 14-pf GMPCPP microtubules could be included in the experiment as an internal control and distinguished by fluorescent labeling (see Fig. S1C). The DCX-GFP signal on the GMPCPP microtubules was equivalent to the signal on the dim subset of the paclitaxel microtubules (Fig. S1D, p = 0.28, n = 9 MTs), supporting the interpretation that the dim subset has 14-pf. We also observed DCX-GFP binding to specific segments within a single microtubule (Fig. 1H, white arrow), indicating that the microtubule changed thickness along its length. Similar transitions in pf-number were observed by electron microscopy (Chretien et al., 1992) and may arise during growth or from end-to-end annealing of microtubules of different thickness (Rothwell et al., 1986). Our mixed population of microtubules is therefore a useful tool for studying DCX-GFP, as we can exclude variations in DCX-GFP binding based on microtubule nucleation conditions, the fluorescent dye on tubulin, nucleotide state, and stabilizing agents. Thickness measurement is robust; 13-pf preference did not depend on ionic strength, the source of purified tubulin (porcine vs. bovine), or the presence of a poly-histidine purification tag (data not shown). In contrast to the 13-pf preference we observed for DCX, a control protein, kinesin-1-GFP, did not show a preference and bound equally to all microtubule types tested (Fig. S1E–G).
We have now established a fluorescence microscopy assay that allows us to directly visualize different microtubule architectures in vitro, something previously observable primarily in the Moiré patterns of electron microscopy images. These results confirm that DCX can “measure” microtubule thickness: the binding of DCX to microtubules is sensitive to their lateral curvature, as specified by their pf number. The differences between 13-pf and 14-pf microtubules are subtle. Their diameters differ by only 1 nm, while the arc-length between adjacent protofilament grooves differs by less than 3 Å, or the approximate diameter of a water molecule (see Fig. S1H & I for schematic). Despite these small differences, DCX binds specifically to 13-pf over 14-pf microtubules.
Thickness measurement is cooperative
We were curious whether sensitivity to thickness is an intrinsic property of individual DCX-GFP molecules. Put simply: could a single molecule of DCX measure thickness? This might occur if the binding site for DCX, which lies in the groove between pfs (Fourniol et al., 2010), changes shape with microtubule thickness and the affinity of DCX for the site (KA) changes with it. To test for an intrinsic preference, we exposed a mixed population of microtubules, shown in Figure 2A, to 10 nM DCX-GFP, a concentration at which single DCX-GFP molecules could be detected as diffraction-limited signals (Fig. 2B, right, white arrow). As shown in Movie S1, DCX-GFP bound transiently to the microtubule lattice, and we observed many association and dissociation events. We could not, however, detect any preference for microtubules of different thickness; the number of binding events and their duration appeared similar for all microtubules within the chamber. We confirmed this observation by summing the frames in Movie S1, which is similar to increasing the exposure time of the image. Longer camera exposures and/or summation integrate over many binding and unbinding events and will exaggerate any differences between microtubule types. Even with a summation over 10 seconds (100 frames), shown in Figure 2B, there was no detectable preference for microtubules of different thickness. Increasing the summation further still did not reveal a preference, nor could we force a preference to arise by varying buffer conditions such as ionic strength. This result indicates that 13-pf specificity is not an intrinsic property of a single DCX molecule and that the affinities are similar. When we increased the concentration to 0.5 μM DCX-GFP, however, and imaged the same population of microtubules, a distinct preference became evident (Fig. 2C). We then used these distinctions to confirm that there was no statistically-significant difference between microtubules in our 10-sec summation (I = 33699 ± 2749 a.u. for 13-pf vs. I = 32278 a.u. ± 1098 for non-13-pf, n = 7 MTs, p = 0.23). We interpret these results as evidence for a cooperative transition in binding; in other words, single DCX molecules do not bind specifically to 13-pf microtubules, but groups of DCX molecules do.
Figure 2. DCX undergoes a cooperative transition in microtubule binding.

(A) Image of a mixed population of rhodamine-labeled microtubules. (B) Left, Image of 10 nM DCX-GFP exposed to a mixed population of microtubules taken with a 0.1 s camera exposure. Single DCX-GFP molecules were observed as diffraction-limited signals (white arrow). Right, Image of a 10 s summation (100 × 0.1 s frames) of DCX-GFP. No preference for 13-pf microtubules could be measured. (C) Image of 0.5 μM DCX-GFP exposed to the same microtubules as in (A), taken with a 0.1 s camera exposure. A clear preference for 13-pf microtubules is evident. (D) Plot of DCX-GFP intensity on the microtubules against the protein concentration in solution during titration of DCX-GFP into the microscope chamber. The bright, 13-pf microtubules (blue squares) were distinguished from the dimmer, non-13-pf microtubules (red circles). Error bars represent the SEM (n = 10). For curve-fitting, the data were fitted to the Hill equation, y = y0 + (ymax − y0) · xnH=(KnH + xnH) (lines plotted). See also Figure S2.
To test this interpretation, we developed an assay for cooperative binding using a continuous titration of DCX-GFP, in which we measure the intensity of DCX-GFP along a microtubule as a function of the DCX-GFP background intensity, which serves as a readout for DCX-GFP concentration in solution. Figure 2D shows a representative trace. Bright and dim subsets of microtubules were readily observable. For bright, 13-pf microtubules, we observed a sigmoid response, indicative of a cooperative transition in DCX binding (Fig. 2D, blue squares). For dim, non-13-pf microtubules in the same experiment, a sigmoid response in DCX binding was also apparent (Fig. 2D, red circles), although the trace was shifted rightward and the slope of the linear portion of the response was decreased. As a curve-fitting model, we used the Hill equation, which, for 13-pf microtubules, yielded a Hill coefficient of nH = 2.87 ± 0.01 and K = 0.57 ± 0.01 μM (n = 30) (Fig. 2C, blue line). Although the non-13-pf data did not saturate, we can extrapolate and fit the data (Fig. 2C, red line), with nH = 2.19 ± 0.02 and K = 1.23 ± 0.01 μM (n = 30). The change in both parameters is statistically-significant (p ≪ 0.0001 by Welch’s t-test). The Hill equation is phenomenological, however, so we cannot “base any direct physical meaning” on the parameters (Hill, 1910). As a non-cooperative, negative control for our assay, we used kinesin-1-GFP. We observed a linear increase in kinesin-1-GFP intensity with increasing concentration in solution (Fig. S2). This control gave us confidence that our assay is not confounded by, for example, detection thresholds at low protein concentrations that would flatten the left-most portion of the curve.
The sigmoid response seen on non-13-pf microtubules shows that cooperative binding can occur on all microtubules, but less favorably than on 13-pf, both in terms of the increased point of half-maximal saturation and the decreased slope of the response. These results explain the fundamental mechanism by which DCX binds specifically to 13-pf microtubules: DCX undergoes a cooperative transition in its binding to microtubules that is sensitive to the number of pfs. On 13-pf microtubules, cooperativity is optimized; on non-13-pf microtubules, cooperativity is below par.
Cooperativity decreases the dissociation rate of DCX
What is the mechanism that causes cooperative binding to microtubules? Cooperativity is commonly observed in receptor-ligand binding studies (Lauffenburger and Linderman, 1993), for example in the binding of acetylcholine to its receptor. In these cases, the binding of additional ligands to a multivalent receptor increases the affinity (KA) of every ligand for the receptor. Often, the affinity increase is caused by a change in the dissociation rate constant, kd, of the ligand. Specifically, kd falls, and each additional ligand increases the lifetime of the other ligands bound to the receptor. By analogy, we can consider DCX-GFP as a “ligand” and the microtubule lattice as a “multivalent receptor.” The prediction of this line of reasoning is that the lifetime of an individual DCX on the microtubule lattice will increase with DCX concentration due to the presence of neighboring DCX molecules. We tested this prediction by examining the residence time of single DCX-GFP molecules on the microtubule lattice. Under single-molecule conditions (Fig. 3A), where DCX-GFP molecules do not cooperate or measure thickness, DCX-GFP dissociated from the microtubule rapidly. Figure 3B shows a kymograph of these short-lived interactions. The interaction lifetimes of 10 nM DCX-GFP were distributed exponentially (Fig. 3C). We calculated an average lifetime of 〈τ〉 = 0.9 s (n = 115 events) corresponding to kd = 1.1 ± 0.1 s−1 when corrected for photobleaching. We then performed “spiking” experiments (Brouhard et al., 2008), where 10 nM DCX-GFP was added to a much higher concentration of unlabeled DCX (2 μM) interacting with the microtubule lattice (Fig. 3D). Figure 3E shows a kymograph under these conditions, and it is clear that single DCX-GFP molecules no longer dissociated rapidly. Rather, we measured an average lifetime of 〈τ〉 = 7.7 s (n = 301 events) (Fig. 3F) corresponding to kd = 0.13 ± 0.02 s−1 when corrected for photobleaching, a nearly 10-fold change. Note the different timescales of the two experiments (Fig. 3E, red arrow), which were chosen to minimize photobleaching in the spiking experiments. From these observations, we conclude that the cooperativity of DCX binding is caused by a decrease in kd with increasing occupancy on the microtubule lattice. In other words, the presence of neighboring molecules causes DCX to remain bound to the microtubule longer.
Figure 3. DCX decreases the dissociation rate of its neighbors.

(A) Schematic of the single-molecule experiment. (B) Image from the single-molecule experiment (top) showing a single DCX-GFP (green) bound to a microtubule (red). Kymograph (bottom) depicting the association and dissociation of DCX-GFP to/from the microtubule. (C) Histogram of durations of DCX-GFP microtubule interactions. An exponential curve fit, corrected for photobleaching, yields a mean lifetime of interaction, 〈τ〉 = 0.9 s. (D) Schematic of the “spiking” experiment, in which unlabeled DCX is added to a low concentration of DCXGFP. (E) Image from the spiking experiment (top) showing a single DCX-GFP (green) bound to a microtubule (red). Kymograph (bottom) depicting the association and dissociation of DCX-GFP to/from the microtubule in the presence of unlabeled DCX. Note the difference in timescale (red arrow) compared to (B). (F) Histogram of durations of DCX-GFP microtubule interactions. An exponential curve fit, corrected for photobleaching, yields a mean lifetime of interaction, 〈τ〉 = 7.7 s.
Patient mutations in DCX disrupt cooperative interactions
We wondered whether the cooperative interactions we observed might be important for the interaction of DCX with microtubules during neuronal migration. If so, we might expect that mutations in DCX known to impair neuronal migration, namely those found in patients with double cortex syndrome, would show, for example, a decreased sigmoid response. We tested this hypothesis by examining the effects of patient mutations on DCX in vitro. Missense mutations in the DC domains fall into two categories: (1) substitutions found at surface residues and (2) substitutions for hydrophobic residues embedded in the core ubiquitin fold. This latter category is expected to cause misfolding of DCX. We investigated the effect of 15 surface-residue mutations, 8 in the N-DC domain (see Fig. 4A) and 7 in the C-DC domain (see Fig. 4B), to determine if these mutations affect the cooperativity of DCX. We selected mutations from familial as well as sporadic cases of double cortex syndrome, covering >75% of affected residues distributed across the surfaces of the DC domains (Kim et al., 2003).
Figure 4. Missense mutations found in human patients disrupt cooperative interactions.

(A) Crystal structure of the N-DC domain (PDB: 2BQQ) showing the patient mutations used in this study. (B) Homology model of the C-DC domain based on the N-DC structure (Fourniol et al., 2010) showing the patient mutations used in this study. (C) Plot of T203R-DCX-GFP intensity on the microtubules against protein concentration in solution during titration of T203R-DCX-GFP into the microtubule chamber. The paclitaxel-MTs (blue) were plotted separately from the GMPCPP MTs (red). Error bars represent the SEM (n = 10). (D) Plot of R89G-DCX-GFP intensity on the microtubules against protein concentration in solution during titration of R89G-DCX-GFP into the microtubule chamber. The paclitaxel-MTs (blue) were plotted separately from the GMPCPP MTs (red). Error bars represent the SEM (n = 10). Data were fitted to the Hill equation (lines plotted). (E) Left, Image of two types of rhodamine-labeled MTs, dim paclitaxel-microtubules (labeled, mixed-pf number) and bright GMPCPP-MTs (labeled, 14-pf). Right, Image of T203R-DCX-GFP exposed to the two microtubule types. No preference for microtubule types was measured. (F) Plot of absorbance at 350 nm against time in the turbidity assay for wild-type DCX-GFP and T222I-DCX-GFP. See also Figure S3.
We introduced each mutation into our protein expression constructs using site-directed mutagenesis and expressed and purified mutant-DCX-GFP (see Fig. S3). We tested each mutant in cooperativity assays with both paclitaxel-microtubules (mixed-pf number) and GMPCPP-microtubules (14-pf), which we distinguished by fluorescence intensity. We also tested each mutant in the turbidity assay in order to determine if the mutation affected the bulk rate of microtubule formation. Our assays allowed us to document three classes of biochemical defect in mutant DCX: (a) disrupted cooperative interactions and decreased microtubule binding, (b) loss of 13-pf specificity, and (c) reduced performance in the turbidity assay. The results are surveyed below.
The most prominent phenotype was a decreased sigmoid response in the cooperativity assay. An example of loss of cooperative binding is shown in Figure 4C, which shows the results for the switch of an threonine for an arginine at position 203 (T203R, see Fig. 4B), a mutation found in both familial and sporadic cases. The increase in signal appeared much less sigmoid than wild-type DCX (Fig. 4C). To confirm that the reduced sigmoid response corresponded to weakened nearest-neighbor interactions, we performed “spiking experiments” for T203R-DCX (see Fig. 3). Unlike wild-type, T203R-DCX-GFP dissociated rapidly from the microtubule even with a high concentration of unlabeled T203R-DCX in the background (Fig. 4D), indicating that cooperative interactions are impaired. Because the lack of clear saturation makes fitting the Hill equation unreliable, we compared the slope of the linearly-increasing portion of the T203R response to wild-type and noted a significant decrease (p ≪ 0.0001, n ≥ 10 MTs). All 15 mutants tested showed a reduced slope (see Fig. S4 and Table 1). Both N-DC mutations (e.g., R78H) and C-DC mutations (e.g., T222I) showed a severely impaired response, indicating that both domains play a role in cooperative interactions. Although single-molecule affinities may also be affected, the consistency of the decreased sigmoid response as a phenotype indicates that cooperative interactions are at the core of the DCX mechanism.
Table 1. Compiled Data for Patient Mutations.
Left, Mutations in the N-terminal DC domains are listed in the first column, followed by a column reporting their origin (familial, sporadic, or both), a column reporting the relative slope of the linearly-increasing portion of the response curve (normalized to the slope of the 13-pf wild-type curve, Fig. 2D, blue), and a column reporting whether they bind preferentially to 13-pf microtubules (+ = yes, − = no). Right, Mutations in the C-terminal DC domains are listed, followed by the same columns described above. See also Figure S4.
| Mutation N-DC | Origin | Relative Slope | 13-pf? | Mutation C-DC | Origin | Relative Slope | 13-pf? |
|---|---|---|---|---|---|---|---|
| S47R | Familial | 0.43 | + | R178L | Familial | 0.22 | − |
| Y64N | Sporadic | 0.12 | + | P191R | Sporadic | 0.46 | − |
| A71S | Familial | 0.14 | − | R192W | Familial | 0.33 | + |
| R76S | Familial | 0.38 | − | N200K | Sporadic | 0.58 | − |
| R78H | Sporadic | 0.11 | − | T203R | Both | 0.18 | − |
| D86H | Familial | 0.37 | + | T222I | Sporadic | 0.21 | − |
| R89G | Familial | 0.33 | − | G223E | Sporadic | 0.03 | − |
| R102S | Unknown | 0.23 | − |
The majority of mutants showed a severely disrupted response (e.g., R78H), with slope parameters <25% of wild-type. Other mutants retained signs of a cooperative transition in binding, albeit an impaired one. For example, we examined the switch of an arginine to a glycine at position 89 in the N-DC domain (R89G). This mutation causes a mild form of double cortex syndrome (Gleeson et al., 1998). Figure 4E shows the results from the cooperativity assay, and the response appears sigmoid. Nevertheless, R89G-DCX-GFP also dissociated rapidly from the microtubule in the “spiking experiment” (Fig. 4F). In addition, the slope of the linearly-increasing portion of the response was significantly decreased and the point of half-maximal saturation was shifted rightward. In other words, a weakened cooperative transition occurred at a higher concentration in solution.
In Figures 4C and 4E, the data for paclitaxel-microtubules (mixed-pf number) and GMPCPP-microtubules (14-pf) were plotted separately. To our surprise, T203R-DCX and R89G-DCX bound to the paclitaxel- and GMPCPP-microtubules equivalently (Fig. 4F), indicating that these patient mutations disrupt the specificity for 13-pf microtubules. For 11 of 15 mutations that we tested, the data for paclitaxel-microtubules were statistically-equivalent to the data for GMPCPP-microtubules. We interpret this result as evidence that 13-pf specificity is finely tuned and easily disrupted.
Finally, we tested our mutants in the turbidity assay in order to determine if the mutation affected the bulk rate of microtubule formation. For all mutations, we observed a significant increase in A350, demonstrating that mutated DCX can stimulate microtubule formation, although never to peak wild-type levels (e.g., N200K, Fig. 4G). Therefore, the disease phenotype is not explainable by a complete loss of function in this bulk assay, in contrast to previous findings (Taylor et al., 2000).
Table 1 summarizes the data for all 15 mutations (for individual curves, see Fig. S4). These biochemical defects are not the only possible explanation for double-cortex syndrome in these patients. Other issues such as localization defects, premature degradation, and/or aberrant post-translational modifications could also contribute to the disease phenotype, but our results document a specific loss of function in terms of a decreased sigmoid response and impaired cooperative binding to microtubules. We attempted to correlate the severity of the biochemical defects with the severity of the disease phenotype. The clinical literature on double cortex syndrome patients is heterogeneous, so it is not possible to rank the severity of mutations. For 4 of the mutations we tested, the relative thickness of the subcortical heterotopic band was reported. The “thick” band mutant (G223E) (des Portes et al., 1998a) had a low slope parameter; mutations that produced “thin” bands (Matsumoto et al., 2001) were better (R89G & R192W); the slope parameter for a “moderate” band (T203R) (Matsumoto et al., 2001) lay in between. We expect, however, that the diverse genetic background of patients, as well as the mosaicism due to X-inactivation (Matsumoto et al., 2001), will contribute significantly to the penetrance of individual mutations.
DCX tracks microtubule ends in vitro
Although DCX is often considered a microtubule stabilizing protein, we remained curious as to the mechanism underlying the specification of 13-pf microtubules in vitro. Starting from first principles, there are two ways to specify 13-pf microtubules without a γ-TuRC template. In the first, a nucleation reaction produces an initially heterogeneous population of microtubules. DCX stabilizes the 13-pf fraction over the others; the non-13-pf fraction undergo catastrophes and, over time, the population evolves toward homogeneity (Fig. S5A). In the second, DCX acts earlier by binding to intermediates that occur during the polymerization process (Fig. S5B), such as small oligomeric species (Fig. S5B, i) or microtubule end structures (Fig. S5B, ii). These polymerization intermediates are made of GTP- or GDP·Pitubulin and may also have a sheet-like structure (Erickson, 1974; Chretien et al., 1995).
Our data show that preferential binding to 13-pf microtubules occurs only within a certain concentration range, above which all microtubules saturate (see Fig. 2D), and that DCX-GFP binds to 14-pf microtubules within this range, albeit at lower levels. These observations made us skeptical that DCX could stabilize only 13-pf microtubules in the conditions used by Moores et al. (2004). Therefore, we looked for evidence that DCX interacts with polymerization intermediates by visualizing the interaction of DCX with dynamic microtubules. We prepared a flow chamber in which antibodies specific to rhodamine were used to adhere GMPCPP-stabilized, rhodamine-labeled microtubule seeds to the cover glass surface (Fig. 5A). We introduced 10 μM tubulin and 5 nM DCX into the flow chamber. Microtubules grew by extension from the microtubule seeds and, remarkably, a bright DCX-GFP signal was observed at the growing microtubule end (see Fig. 5B and Movies S2 & S3). In other words, DCX is an end-tracking protein; Figure 5B shows a kymograph of DCX end-tracking, which appears similar to data for the canonical end-tracking protein EB1 (Bieling et al., 2007). The bright DCX-GFP signal at growing microtubule ends was well-described by a Gaussian distribution (data not shown), indicating that the structural or biochemical feature that DCX-GFP recognizes is within the diffraction limit under these conditions. We repeated the experiment with a higher concentration of DCX-GFP (50 nM). In this case, the entire microtubule extension showed a bright DCX-GFP signal, although the 14-pf GMPCPP seed remained dimmer (see Fig. 5C and Movie S4). Figure 5C shows a kymograph of DCX binding preferentially to the microtubule extension. This result suggests that a transition occurred in the number of pfs as the 14-pf GMPCPP seed was elongated. DCX was not responsible for this pf-number transition, however, as we could detect it on microtubules elongated from GMPCPP seeds in the absence of DCX. These results demonstrate that the affinity of DCX-GFP is highest for growing microtubule ends, intermediate for the 13-pf lattice, and lower for the 14-pf lattice.
Figure 5. DCX tracks microtubule ends.
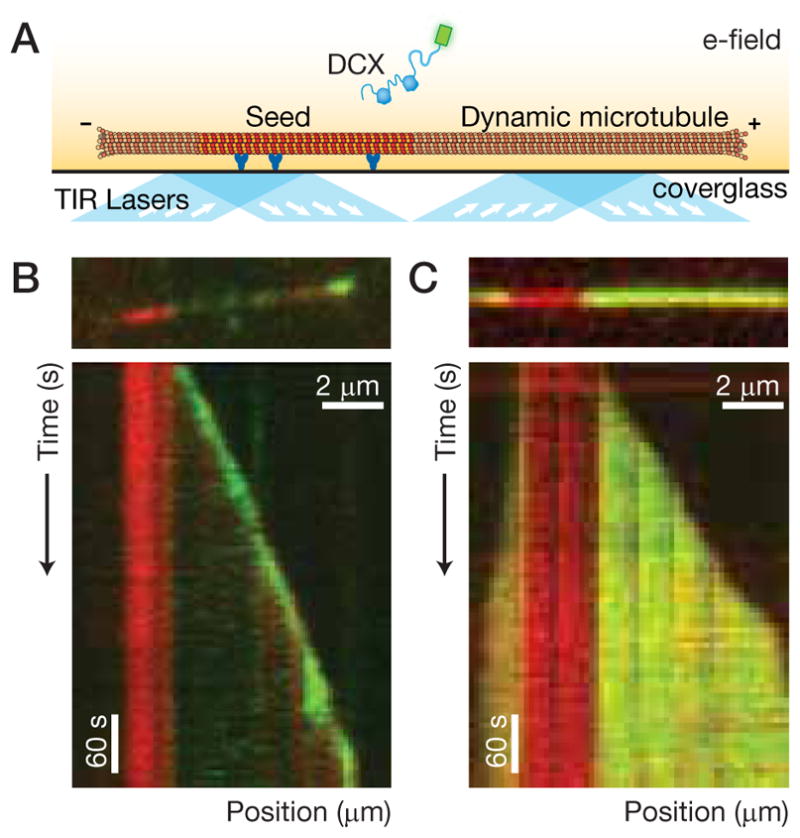
(A) Schematic of the single-molecule dynamic assay. A dynamic microtubule (labeled) grows by extension of a GMPCPP-seed microtubule (labeled) adhered to a coverglass surface. (B) Image (top) and kymograph (bottom) depicting the interaction of 10 nM DCX-GFP with a dynamic microtubule. A bright DCX-GFP signal is observed to track the growing microtubule end. (C) Image (top) and kymograph (bottom) depicting the interaction of 100 nM DCX-GFP with a dynamic microtubule. A bright DCX-GFP signal is observed along the entire length of the microtubule extensions but the GMPCPP-seed microtubule is dimmer. See also Figure S5.
DISCUSSION
Flexibility in the links between pfs allows microtubules to form in a range of thicknesses. We established a fluorescence microscopy assay that showed the binding of DCX to be sensitive to microtubule thickness. It is our hope that this assay will serve as a general tool for interrogating microtubule structure by fluorescence. We discovered that DCX binds to microtubules cooperatively in a manner optimized for 13-pf microtubules. Based on our results, we propose the following mechanism (Figure 6A). When a single DCX binds to the microtubule lattice without other DCX molecules nearby, it dissociates rapidly (Fig. 6A, top). In the presence of adjacent DCX molecules, however, an interaction occurs that stabilizes each of the DCX molecules involved, decreasing their dissociation rate constant (Fig. 6A, bottom). Missense mutations found in patients with double cortex syndrome caused a failure in this mechanism, although single-molecule affinities may also be affected. Taylor et al. (2000) studied the effects of 4 patient mutations in the turbidity assay. They observed reduced absorbance for all 4 mutants but wrote that “The precise defect of mutant DCX in microtubule polymerization is still unclear.” We have defined this defect, namely that mutant DCX shows a loss of cooperative microtubule binding.
Figure 6. Model schematic for the mechanism of DCX.
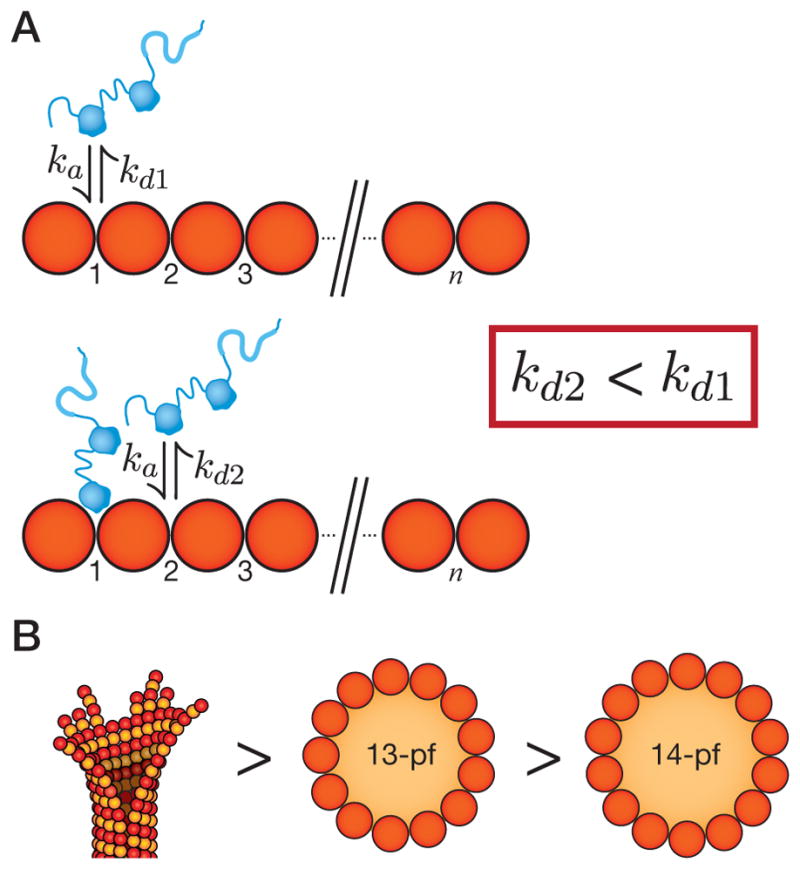
(A) The microtubule is depicted as an array of binding sites (numbered). DCX binds to microtubules in the groove between protofilaments, at the vertex of four tubulin dimers. In the absence of nearby molecules (top), DCX associates with an association rate constant, ka and dissociates with a dissociation rate constant, kd1. In the presence of nearby molecules (bottom), the dissociation rate constant falls (kd2 < kd1). (B) The affinity of DCX for microtubules is highest for microtubule end structures (left), intermediate for the 13-pf microtubule lattice (center), and lower for the 14-pf microtubule lattice (right).
We do not know the structural basis of the cooperative interaction. There are observations, however, that constrain any model for how DCX binds cooperatively to 13-pf microtubules. First, a single DC domain binds to microtubules at the vertex of four tubulin dimers (Fourniol et al., 2010) and the microtubule saturates at one full-length DCX molecule per vertex (Moores et al., 2006). This tells us that ~75% of the molecule, including a DC domain, probably extends into solution at saturation. The domain that fills the vertex is most likely the N-DC domain, based on the superior docking of the N-DC structure into the EM density map (Fourniol et al., 2010), leaving the C-DC domain and SP-rich tail domain to extend into solution. Additionally, the DC domains are divergent in their primary sequence (Reiner et al., 2006) and differ both in their distribution of surface charge (Fourniol et al., 2010) and the compactness of their fold (Kim et al., 2003).
The cooperative interaction is either direct or indirect (or both). In our reconstitution experiments, an indirect interaction implies an allosteric process wherein the binding of one DCX molecule changes the conformation of an adjacent binding site. The finding that some mutants can bind to the microtubules with weakened cooperative interactions suggests that microtubule binding alone is not sufficient to trigger allostery. Alternatively, adjacent DCX molecules may contact each other directly. There are two findings that temper this idea as well. First, EM density maps resolved only a single DC domain (Fourniol et al., 2010), so direct interactions between adjacent molecules must be sufficiently flexible that the image averaging employed in EM reconstruction has made them invisible. Flexibility may originate from heterogeneity in the orientation of the linker sequence as it emerges from the N-DC domain, which is observed in the available atomic structures (Cierpicki et al., 2006). Second, DCX was shown to be monomeric in the absence of microtubules by analytic ultracentrifugation (Moores et al., 2006). Therefore, any direct interactions may be microtubule-dependent. Microtubule-mediated oligomerization has been observed previously for the kinetochore protein Ndc80 (Ciferri et al., 2008) and for Ase1, a microtubule-crosslinking protein in S. pombe (Kapitein et al., 2008). We currently favor a model of direct interactions based on the results for the mutants, but our present knowledge is not sufficient to close the case.
DCX was the first protein after the γ-TuRC to specify 13-pf microtubules in nucleation reactions in vitro (Moores et al., 2004). We considered two models that could explain this phenomenon: selective stabilization and interaction with polymerization intermediates. By visualizing the interaction of DCX-GFP with dynamic microtubules, we made the discovery that DCX tracks microtubule ends like the canonical end-binding protein EB1, demonstrating that the affinity of DCX is highest for microtubule ends (Fig. 6B). This result provides direct evidence for the polymerization intermediates model. Polymerization intermediates are made of GTP- or GDP·Pi-tubulin. In addition to their nucleotide state, polymerization intermediates may differ structurally from the microtubule lattice. Electron microscopy of microtubule ends has shown elongated, curved protofilaments, often interpreted as sheet-like structures (Chretien et al., 1995), and early observations of spontaneous-nucleation intermediates also reported sheet-like fragments of the microtubule wall (Erickson, 1974). Indeed, tubulin polymerized with the GTP-analog GMPCPP can form sheet-like helical ribbons (Wang and Nogales, 2005), indicating that the nucleotide state of tubulin and the structure of the polymer are linked. EB1 has been shown to have bind preferentially to microtubules polymerized with GTP-analogs, first GMPCPP (Zanic et al., 2009) and then GTPγS (Maurer et al., 2011), indicating that EB1 recognizes the nucleotide state of the lattice. EB1, like DCX, binds to microtubules at the vertex of four tubulin dimers (Maurer et al., 2012), where it contacts the helices of tubulin that coordinate nucleotide hydrolysis. We note that EB1 has also been shown to promote the formation of 13-pf microtubules (Vitre et al., 2008), although EB1 does not bind well to the lattice like DCX. Nevertheless, the common binding site and the shared features of end-tracking and 13-pf promotion suggest that interaction with polymerization intermediates at the vertex of four tubulin dimers is a robust mechanism for influencing microtubule structure.
Neurons and other cells must control the size of individual microtubules not only with respect to their length but also their thickness. The most conservative interpretation of our data is that DCX stabilizes microtubules in the leading process of a migrating neuron by cooperative interactions, a mechanism distinct from other known MAPs, and that DCX has evolved to perform this duty optimally on 13-pf microtubules, which are, after all, its native substrate. The observation that DCX increases the number of 13-pf nuclei formed in vitro has also led to the proposal that DCX nucleates new microtubules in neuronal processes, making it a microtubule nucleation factor distinct from γ-TuRC (Moores et al., 2004). At present, the data on microtubule dynamics in cultured neurons depleted of DCX or taken from knockout mice is not sufficient to distinguish between these models.
New microtubules are assembled locally in neuronal processes (Ma et al., 2004), which are devoid of γ-TuRC (Baas and Joshi, 1992). Furthermore, recent data showed that centrosomes are dispensable for axon elongation (Stiess et al., 2010), indicating that new microtubules come from either non-centrosomal γ-TuRC’s or from an independent nucleation pathway. It is now well-established that non-centrosomal γ-TuRC’s nucleate microtubules in many contexts (Luders and Stearns, 2007), but there is also evidence for a γ-TuRC-independent nucleation pathway in metazoans. In C. elegans, for example, RNAi of γ-tubulin did not eliminate the microtubule cytoskeleton (Hannak et al., 2002) and new microtubules continued to emanate from the centrosome (Srayko et al., 2005). Likewise, Drosophila S2 cells continued to nucleate large numbers of microtubules after RNAi of γ-tubulin (Mahoney et al., 2006). The molecules involved in these independent pathways remain uncharacterized.
At present, we can only say that DCX nucleates 13-pf microtubules in vitro. In this context, how would DCX function as a nucleation factor? Broadly, a nucleation factor either (a) directly provides or (b) helps monomers form a template for the elongation of a polymer. The γ-TuRC uses the first mechanism: it directly provides a pre-formed “lock-washer” template for 13-pf microtubules (Kollman et al., 2010). Similarly, the Arp2/3 complex nucleates actin filaments by providing a template that persists at the base of new daughter-filaments (Volkmann et al., 2001). The actin cytoskeleton, however, has other nucleation factors. One such nucleator, Drosophila Spire, binds several actin monomers in solution and arranges them into a stable nucleus (Quinlan et al., 2005). This mechanism is distinct from γ-TuRC and Arp2/3 in that Spire binds to the sides, not the base, of the new polymer and helps actin monomers form their own template, rather than directly providing one. DCX acts as a Spire-like nucleation factor for microtubules, where the interaction with polymerization intermediates assembles tubulin dimers into a 13-pf nucleus.
EXPERIMENTAL PROCEDURES
Expression and Purification of DCX, related constructs, and control proteins
The coding sequence for human Doublecortin (accession number NP_835365) was a gift from Dr. Christopher Walsh. The coding sequence for DCX was PCR amplified using PfuX7 polymerase (Norholm, 2010) and cloned into a pHAT protein expression vector as described (Bitinaite et al., 2007). The pHAT vector contained an N-terminal poly-His tag followed by a PreScission site and a C-terminal EGFP-tag followed by a Strep-tag II (Schmidt and Skerra, 2007) for affinity purification. We induced expression of DCX-GFP in the BL21(DE3) strain of E. coli at OD(600) = 0.6 with 0.5 mM IPTG for 14 h at 18 °C. Cells were harvested by centrifugation, resuspended in 50 mM Na2HPO4, 300 mM NaCl, 10 mM imidazole, 10% glycerol and lysed using a French press in the presence of a protease inhibitor cocktail. Bacterial lysates were clarified by centrifugation. The cleared lysate was loaded onto a Ni-NTA affinity column (1 ml His-Trap HP, GE Healthcare) and eluted using a continuous imidazole gradient from 0 to 250 mM on an FPLC system (Akta Purifier, GE Healthcare). Peak fractions were loaded onto a 1 ml Strep-Tactin Superflow gravity flow column (IBA, Germany), which bound the C-terminal Strep-tag II. Purified DCX was eluted with desthiobiotin following manufacturer’s protocols. Optionally, Strep-Tactin-bound protein was incubated overnight with PreScission protease before elution, which cleaved the poly-His tag. The protein was used fresh and/or aliquots were flash frozen in LN2 in the presence of 10% glycerol and stored in elution buffer. Protein concentration and purity were determined using SDS-PAGE and absorbance at 280 nm and 488 nm using a NanoDrop spectrophotometer (Thermo Scientific). The eluted DCX-GFP or other proteins were >99% pure (e.g., Fig. S1A). Missense mutations corresponding to those found in human patients were introduced using site-directed mutagenesis by the Kunkel method (Kunkel, 1985). The cDNA for a constitutively-active rat kinesin-1-GFP (rKin430-GFP) was a gift of Dr. Rob Cross to Dr. Jonathon Howard (Rogers et al., 2001). rKin430-GFP was expressed and purified as described (Varga et al., 2009).
Total-Internal-Reflection Fluorescence Microscopy and Preparation of Microscope Chambers
The single-molecule assay for kinesins and microtubule-associated proteins was performed as described (Gell et al., 2010), with specific modifications described below. The microscope setup uses a Zeiss Axiovert Z1 microscope chassis, 100x 1.45 NA Plan-apochromat objective lens, and the Zeiss TIRF III slider. Diode-pumped solid-state lasers (Cobolt Jive, Cobolt Calypso) were coupled to fiber-optic cables in free space and introduced into the Zeiss slider. Images were recorded using an Andor iXon+ DV-897 EMCCD camera. Microscope chambers were constructed using custom-machined mounts diagrammed in Gell et al. (2010). In brief, microscope cover glass were silanized as described (Helenius et al., 2006). A 22x22 mm glass and an 18x18 mm glass were separated by double-sided tape, such that a narrow channel was created for the exchange of solution. Images were acquired using Metamorph. Simple measurements of microtubule intensities were made using the Linescan feature in Metamorph, and all intensity values are reported as mean ± SD. The absolute values of intensity are a function of laser power, laser alignment, and camera gain. Camera gain settings and laser powers were chosen to maximize the sensitivity and dynamic range of the camera, which prevents direct comparisons in intensity values between experiments in some cases. All experiments reported were repeated in a minimum of three independent trials.
Bulk-phase Microtubule Polymerization Assay
The formation of microtubule polymer was followed as the absorbance of 350 nm light using a Cary 300 Bio UV-Visible spectrophotometer (Varian Inc., Agilent Technologies, Santa Clara, CA, USA). The tubulin concentration was 10 μM in BRB80 (80 mM PIPES, 1 mM EGTA, 1 mM MgCl2) + 1 mM GTP. The concentration of recombinant protein (DCX-GFP, etc.) was 1 μM.
Tubulin and Microtubule Preparations
Tubulin was purified from juvenile bovine brain homogenates as described previously (Ashford et al., 1998). Labeling of cycled tubulin with Alexa Fluor 546 or TAMRA (Invitrogen) was performed as described (Hyman et al., 1991); fluorescently-labeled tubulin was typically used at a labeling ratio of 1:4 labeled:unlabeled tubulin dimers. Tubulin was polymerized into microtubules of different thickness as follows: (1) Microtubule polymerization in the presence of GTP followed by stabilization with paclitaxel (paclitaxel-MTs, mixed-pf number): A polymerization mixture was prepared with BRB80 + 32 μM tubulin + 1 mM GTP + 4 mM MgCl2 + 5% DMSO. The mixture was incubated on ice for 5 min, followed by incubation at 37 °C for 30 min. The polymerized microtubules were diluted into pre-warmed BRB80 + 10 μM paclitaxel, centrifuged at maximum speed in a Beckman Airfuge, and resuspended in BRB80 + 10 μM paclitaxel. (2) Microtubule Polymerization in the presence of GMPCPP (14-pf): A polymerization mixture was prepared with BRB80 + 2 μM tubulin, 1 mM GMPCPP (Jena Biosciences), and 1 mM MgCl2. The mixture was incubated on ice for 5 min, followed by incubation at 37 °C for 2 hours. The polymerized GMPCPP microtubules were centrifuged at maximum speed in a Beckman Airfuge and resuspended in BRB80. (3) Comparison of Microtubule polymerized from Axonemes (13-pf) with GMPCPP microtubules (14-pf): Sea urchin sperm axonemes were a generous gift from Dr. Ted Salmon (University of North Carolina at Chapel Hill). The purified axoneme stock was diluted 1:4000 in BRB80 and introduced into a microscope chamber together with an anti-TAMRA antibody (1:200, Invitrogen). The axonemes adhered non-specifically to the cover glass surface. The chamber was rinsed with BRB80, followed by a 5 min incubation with BRB80 + 1% Pluronic F-127, followed by a rinse with BRB80. At this point, the chamber was placed onto the microscope with the objective heater set to 35 °C. Polymerization buffer was then introduced: BRB80 + 12 μM Alexa-546 tubulin, 1 mM GTP, 1 mM MgCl2, 0.1 mg/ml bovine serum albumin (BSA). Microtubules were grown from the axonemes for 10 min, at which point the microscope chamber was rinsed with BRB80 + 10 μM paclitaxel, which stabilized the axonemal microtubules, and an image was taken to record the location of the axonemal microtubules. TAMRA-labeled GMPCPP microtubules were diluted into BRB80 + paclitaxel and introduced into the flow chamber. The GMPCPP microtubules bound to the chamber surfaces via the anti-TAMRA antibody, creating a microscope chamber containing both axonemal microtubules and GMPCPP microtubules. A solution of 0.5 μM DCX-GFP was prepared in imaging buffer: BRB80 + 10 μM paclitaxel + 0.1 mg/ml BSA + antifade reagents (Gell et al., 2010); this solution was introduced into the microscope chamber and images were recorded. (4) Comparison of mixed-pf Microtubules and GMPCPP microtubules (14-pf): Microtubules were prepared as described in (1) and (2) above, with the modification that either the GTP + paclitaxel microtubules or the GMPCPP microtubules were prepared with a lower ratio of fluorescently-labeled tubulin, creating, for example, bright GMPCPP-MTs and dim paclitaxel-MTs. Both types of microtubules were adhered to the surface of the microscope chamber using anti-TAMRA antibodies as described (Gell et al., 2010). This microtubule preparation was the default condition for the majority of experiments described in this manuscript, as the GMPCPP microtubules served as a consistent internal control for the behavior of the paclitaxel-MT population.
Continuous Titration Assay
A microscope chamber was prepared with a mixture of paclitaxel-MTs and GMPCPP-MTs (see above). The chamber was rinsed with imaging buffer, placed on the microscope, and an appropriate field of view was identified near the center of the imaging chamber. We prepared a high-concentration mixture of the protein of interest in imaging buffer, e.g., 1.5 μM DCX-GFP. A large drop of this concentrated solution was placed on one side of the open microscope chamber. Diffusion of the concentrated solution into the microscope chamber creates a continuous gradient of protein concentration that spreads over time. At this point, a streaming-video acquisition of 10 fps was initiated. After video acquisition began, we used a piece of pre-wetted chromatography paper to slowly draw the protein-containing mixture through the microscope chamber. The intensities of n ≥ 10 MTs were measured using the Linescan feature of Metamorph (Universal Imaging). The mean of these intensities was plotted against the background intensity, which served as a readout for protein concentration in solution, of a region that did not contain microtubules. Regions corresponding to 13-pf and non-13-pf microtubules were identified in frames of the movie where 13-pf specificity was most pronounced; in the absence of 13-pf specificity (e.g., for rKin430-GFP), the paclitaxel-MTs and GMPCPP-MTs were binned separately. The data were fit to the Hill equation using OriginPro 8.5 (OriginLab, Northampton, MA, USA).
Single-Molecule Image Analysis
Single-molecule particle tracking was performed as described (Helenius et al., 2006) using the Kalaimo-scope software package (Transinsight GmbH, Dresden, Germany). In brief, single molecules were identified as diffraction-limited point-spread-functions. The pixel-intensity pattern was fit to a 2D Lorentzian function to provide the (x,y) coordinates of the signal. Signals from consecutive frames were linked into trajectories using a cost-minimization function. The data for these trajectories was exported to MATLAB (MathWorks, Natick, MA, USA) for further analysis. The data were fit to an exponential decay function using OriginPro 8.5. Corrections for photobleaching were performed as described (Brouhard et al., 2008).
Supplementary Material
Acknowledgments
We thank C. Moores for many thoughtful discussions and for sharing cryo-EM data prior to publication. We thank Abattoir Jacques Forget (Terrebonne, Québec) for source material for tubulin purification, C. Walsh for the gift of DCX cDNA, J. Howard for the gift of a kinesin-1-GFP expression vector and porcine tubulin, and E.D. Salmon for the gift of sea-urchin axonemes. We thank J. Howard, S. Michnick, C. Moores, J. Stear, J. Vogel, P. Widlund, and M. Zanic for feedback on this work. We thank S. Wolfson for editing. This work was supported by the Canadian Institutes of Health Research (MOP-104150 and MOP-111265 to G.B.) and McGill University.
References
- Ashford A, Andersen S, Hyman AA. Preparation of Tubulin from Bovine Brain. In: Celis J, editor. Cell Biology, a Laboratory Handbook. Vol. 2. Academic Press; New York: 1998. pp. 205–212. [Google Scholar]
- Baas PW, Joshi HC. Gamma-tubulin distribution in the neuron: implications for the origins of neuritic microtubules. J Cell Biol. 1992;119(1):171–8. doi: 10.1083/jcb.119.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6(12):1277–83. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bieling P, Laan L, Schek H, Munteanu EL, Sandblad L, Dogterom M, Brunner D, Surrey T. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 2007;450(7172):1100–5. doi: 10.1038/nature06386. [DOI] [PubMed] [Google Scholar]
- Bitinaite J, Rubino M, Varma KH, Schildkraut I, Vaisvila R, Vaiskunaite R. USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res. 2007;35(6):1992–2002. doi: 10.1093/nar/gkm041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, Stear JH, Noetzel TL, Al-Bassam J, Kinoshita K, Harrison SC, Howard J, Hyman AA. XMAP215 is a processive microtubule polymerase. Cell. 2008;132(1):79–88. doi: 10.1016/j.cell.2007.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chretien D, Fuller SD, Karsenti E. Structure of growing microtubule ends: two-dimensional sheets close into tubes at variable rates. J Cell Biol. 1995;129(5):1311–28. doi: 10.1083/jcb.129.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chretien D, Metoz F, Verde F, Karsenti E, Wade RH. Lattice defects in microtubules: protofilament numbers vary within individual microtubules. J Cell Biol. 1992;117(5):1031–40. doi: 10.1083/jcb.117.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cierpicki T, Kim MH, Cooper DR, Derewenda U, Bushweller JH, Derewenda ZS. The DC-module of doublecortin: dynamics, domain boundaries, and functional implications. Proteins. 2006;64(4):874–82. doi: 10.1002/prot.21068. [DOI] [PubMed] [Google Scholar]
- Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, Salek M, Rappsilber J, Moores CA, Salmon ED, Musacchio A. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133(3):427–39. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C, Caceres A. Microtubule assembly, organization and dynamics in axons and den-drites. Nat Rev Neurosci. 2009;10(5):319–32. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- des Portes V, Francis F, Pinard JM, Desguerre I, Moutard ML, Snoeck I, Meiners LC, Capron F, Cusmai R, Ricci S, Motte J, Echenne B, Ponsot G, Dulac O, Chelly J, Beldjord C. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH) Hum Mol Genet. 1998a;7(7):1063–70. doi: 10.1093/hmg/7.7.1063. [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998b;92(1):51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49(1):41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Erickson HP. Microtubule surface lattice and subunit structure and observations on reassembly. J Cell Biol. 1974;60(1):153–67. doi: 10.1083/jcb.60.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans L, Mitchison T, Kirschner M. Influence of the centrosome on the structure of nucleated microtubules. J Cell Biol. 1985;100(4):1185–91. doi: 10.1083/jcb.100.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourniol F, Sindelar C, Amigues B, Clare D, Thomas G, Perderiset M, Francis F, Houdusse A, Moores C. Template-free 13-protofilament microtubule-MAP assembly visualized at 8A resolution. J Cell Biol. 2010 doi: 10.1083/jcb.201007081. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23(2):247–56. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Gell C, Bormuth V, Brouhard GJ, Cohen DN, Diez S, Friel CT, Helenius J, Nitzsche B, Petzold H, Ribbe J, Schaffer E, Stear JH, Trushko A, Varga V, Widlund PO, Zanic M, Howard J. Microtubule dynamics reconstituted in vitro and imaged by single-molecule fluorescence microscopy. Methods Cell Biol. 2010;95:221–45. doi: 10.1016/S0091-679X(10)95013-9. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92(1):63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23(2):257–71. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Hannak E, Oegema K, Kirkham M, Gonczy P, Habermann B, Hyman AA. The kinetically dominant assembly pathway for centrosomal asters in Caenorhabditis elegans is gamma-tubulin dependent. J Cell Biol. 2002;157(4):591–602. doi: 10.1083/jcb.200202047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius J, Brouhard G, Kalaidzidis Y, Diez S, Howard J. The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature. 2006;441(7089):115–9. doi: 10.1038/nature04736. [DOI] [PubMed] [Google Scholar]
- Hill A. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol. 1910;40(Suppl):iv–vii. [Google Scholar]
- Horesh D, Sapir T, Francis F, Wolf SG, Caspi M, Elbaum M, Chelly J, Reiner O. Doublecortin, a stabilizer of microtubules. Hum Mol Genet. 1999;8(9):1599–610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison T. Preparation of modified tubulins. Methods Enzymol. 1991;196:478–85. doi: 10.1016/0076-6879(91)96041-o. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Janson ME, van den Wildenberg SM, Hoogenraad CC, Schmidt CF, Peterman EJ. Microtubule-driven multimerization recruits ase1p onto overlapping microtubules. Curr Biol. 2008;18(21):1713–7. doi: 10.1016/j.cub.2008.09.046. [DOI] [PubMed] [Google Scholar]
- Keating TJ, Borisy GG. Immunostructural evidence for the template mechanism of microtubule nucleation. Nat Cell Biol. 2000;2(6):352–7. doi: 10.1038/35014045. [DOI] [PubMed] [Google Scholar]
- Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, Walsh CA, Otlewski J, Bushweller JH, Derewenda ZS. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol. 2003;10(5):324–33. doi: 10.1038/nsb918. [DOI] [PubMed] [Google Scholar]
- Kollman JM, Polka JK, Zelter A, Davis TN, Agard DA. Microtubule nucleating gamma-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature. 2010;466(7308):879–82. doi: 10.1038/nature09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE, Mitchison TJ, Kirschner MW. Polewards chromosome movement driven by microtubule depolymerization in vitro. Nature. 1988;331(6156):499–504. doi: 10.1038/331499a0. [DOI] [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325(5943):960–3. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82(2):488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Linderman JJ. Receptors : models for binding, trafficking, and signaling. Oxford University Press; New York: 1993. [Google Scholar]
- Luders J, Stearns T. Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol. 2007;8(2):161–7. doi: 10.1038/nrm2100. [DOI] [PubMed] [Google Scholar]
- Ma Y, Shakiryanova D, Vardya I, Popov SV. Quantitative analysis of microtubule transport in growing nerve processes. Curr Biol. 2004;14(8):725–30. doi: 10.1016/j.cub.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Mahoney NM, Goshima G, Douglass AD, Vale RD. Making microtubules and mitotic spindles in cells without functional centrosomes. Curr Biol. 2006;16(6):564–9. doi: 10.1016/j.cub.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E, Milligan RA. Microtubule dynamics and microtubule caps: a time-resolved cryo-electron microscopy study. J Cell Biol. 1991;114(5):977–91. doi: 10.1083/jcb.114.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Leventer RJ, Kuc JA, Mewborn SK, Dudlicek LL, Ramocki MB, Pilz DT, Mills PL, Das S, Ross ME, Ledbetter DH, Dobyns WB. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001;9(1):5–12. doi: 10.1038/sj.ejhg.5200548. [DOI] [PubMed] [Google Scholar]
- Maurer SP, Bieling P, Cope J, Hoenger A, Surrey T. GTPgammaS microtubules mimic the growing microtubule end structure recognized by end-binding proteins (EBs) Proc Natl Acad Sci U S A. 2011;108(10):3988–93. doi: 10.1073/pnas.1014758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer SP, Fourniol FJ, Bohner G, Moores CA, Surrey T. EBs Recognize a Nucleotide-Dependent Structural Cap at Growing Microtubule Ends. Cell. 2012;149(2):371–82. doi: 10.1016/j.cell.2012.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurer-Grob P, Kasparian J, Wade RH. Microtubule structure at improved resolution. Biochemistry. 2001;40(27):8000–8. doi: 10.1021/bi010343p. [DOI] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Francis F, Chelly J, Houdusse A, Milligan RA. Mechanism of microtubule stabilization by doublecortin. Mol Cell. 2004;14(6):833–9. doi: 10.1016/j.molcel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Kappeler C, Kain S, Drummond D, Perkins SJ, Chelly J, Cross R, Houdusse A, Francis F. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006;25(19):4448–57. doi: 10.1038/sj.emboj.7601335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz M, Braunfeld MB, Guenebaut V, Heuser J, Agard DA. Structure of the gamma-tubulin ring complex: a template for microtubule nucleation. Nat Cell Biol. 2000;2(6):365–70. doi: 10.1038/35014058. [DOI] [PubMed] [Google Scholar]
- Norholm MH. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol. 2010;10:21. doi: 10.1186/1472-6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampaloni F, Lattanzi G, Jonas A, Surrey T, Frey E, Florin EL. Thermal fluctuations of grafted microtubules provide evidence of a length-dependent persistence length. Proc Natl Acad Sci U S A. 2006;103(27):10248–53. doi: 10.1073/pnas.0603931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Drosophila Spire is an actin nucleation factor. Nature. 2005;433(7024):382–8. doi: 10.1038/nature03241. [DOI] [PubMed] [Google Scholar]
- Ray S, Meyhofer E, Milligan RA, Howard J. Kinesin follows the microtubule’s protofilament axis. J Cell Biol. 1993;121(5):1083–93. doi: 10.1083/jcb.121.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O, Coquelle FM, Peter B, Levy T, Kaplan A, Sapir T, Orr I, Barkai N, Eichele G, Bergmann S. The evolving doublecortin (DCX) superfamily. BMC Genomics. 2006;7:188. doi: 10.1186/1471-2164-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KR, Weiss S, Crevel I, Brophy PJ, Geeves M, Cross R. KIF1D is a fast non-processive kinesin that demonstrates novel K-loop-dependent mechanochemistry. EMBO J. 2001;20(18):5101–13. doi: 10.1093/emboj/20.18.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell SW, Grasser WA, Murphy DB. End-to-end annealing of microtubules in vitro. J Cell Biol. 1986;102(2):619–27. doi: 10.1083/jcb.102.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TG, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc. 2007;2(6):1528–35. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Srayko M, Kaya A, Stamford J, Hyman AA. Identification and characterization of factors required for microtubule growth and nucleation in the early C. elegans embryo. Dev Cell. 2005;9(2):223–36. doi: 10.1016/j.devcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Stiess M, Maghelli N, Kapitein LC, Gomis-Ruth S, Wilsch-Brauninger M, Hoogenraad CC, Tolic-Norrelykke IM, Bradke F. Axon extension occurs independently of centrosomal microtubule nucleation. Science. 2010;327(5966):704–7. doi: 10.1126/science.1182179. [DOI] [PubMed] [Google Scholar]
- Sui H, Downing KH. Structural basis of interprotofilament interaction and lateral deformation of microtubules. Structure. 2010;18(8):1022–31. doi: 10.1016/j.str.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275(44):34442–50. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Bryan J, Bush DJ, Fujiwara K, Mooseker MS, Murphy DB, Snyder DH. Microtubules: evidence for 13 protofilaments. J Cell Biol. 1973;59(2 Pt 1):267–75. doi: 10.1083/jcb.59.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint I, Jean D, Baas PW, Black MM. Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J Neurosci. 2009;29(35):10995–1010. doi: 10.1523/JNEUROSCI.3399-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga V, Leduc C, Bormuth V, Diez S, Howard J. Kinesin-8 motors act cooperatively to mediate length-dependent microtubule depolymerization. Cell. 2009;138(6):1174–83. doi: 10.1016/j.cell.2009.07.032. [DOI] [PubMed] [Google Scholar]
- Vitre B, Coquelle FM, Heichette C, Garnier C, Chretien D, Arnal I. EB1 regulates microtubule dynamics and tubulin sheet closure in vitro. Nat Cell Biol. 2008;10(4):415–21. doi: 10.1038/ncb1703. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Amann KJ, Stoilova-McPhie S, Egile C, Winter DC, Hazelwood L, Heuser JE, Li R, Pollard TD, Hanein D. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 2001;293(5539):2456–9. doi: 10.1126/science.1063025. [DOI] [PubMed] [Google Scholar]
- Wade RH, Chretien D. Cryoelectron microscopy of microtubules. J Struct Biol. 1993;110(1):1–27. doi: 10.1006/jsbi.1993.1001. [DOI] [PubMed] [Google Scholar]
- Wang HW, Nogales E. Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature. 2005;435(7044):911–5. doi: 10.1038/nature03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanic M, Stear JH, Hyman AA, Howard J. EB1 recognizes the nucleotide state of tubulin in the microtubule lattice. PLoS One. 2009;4(10):e7585. doi: 10.1371/journal.pone.0007585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wong ML, Alberts B, Mitchison T. Nucleation of microtubule assembly by a gamma-tubulin-containing ring complex. Nature. 1995;378(6557):578–83. doi: 10.1038/378578a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.