

Abstract
Summary
We show that imatinib, nilotinib and dasatinib possess weak off-target activity against RAF and therefore drive paradoxical activation of BRAF and CRAF in a RAS-dependent manner. Critically, since RAS is activated by BCR-ABL, in drug-resistant chronic myeloid leukemia (CML) cells RAS activity persists in the presence of these drugs, driving paradoxical activation of BRAF, CRAF, MEK and ERK, and leading to an unexpected dependency on the pathway. Consequently, nilotinib synergizes with MEK inhibitors to kill drug-resistant CML cells and block tumor growth in mice. Thus, we show that imatinib, nilotinib and dasatinib drive paradoxical RAF/MEK/ERK pathway activation and have uncovered a synthetic lethal interaction that can be used to kill drug-resistant CML cell in vitro and in vivo.
Keywords: Chronic myeloid leukemia, BCR-ABLT315I, BRAF, CRAF, synthetic lethality
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disease characterised by myeloid cell expansion in the bone marrow and blood (O'Dwyer et al., 2001). CML accounts for about 15% of adult leukemias and there are about 5,000 cases each year in the US. The largely asymptomatic chronic phase of CML can last several years and is followed by an accelerated phase that indicates disease progression, leading eventually to a life-threatening acute phase called blast crisis. CML has complex pathophysiology, but its diagnosis depends on the presence of the Philadelphia chromosome, a chromosome 9/chromosome 22 translocation that fuses BCR (breakpoint cluster region) to the ABL (Abelson) tyrosine kinase. The normal function(s) of BCR are unclear, but ABL is a cytosolic/nuclear tyrosine kinase that regulates stress responses, cell growth and differentiation. Critically, fusion of ABL to BCR generates a constitutively active kinase that drives transformation and leukemogenesis by phosphorylating substrates such as CRKL and STAT5 and activating pathways such as NFkB and RAS/RAF/MEK/ERK (Deininger et al., 2000).
The clinical management of CML was revolutionized by imatinib, a small molecule ABL inhibitor (Druker et al., 2001). Imatinib mediates remission in the majority of CML patients, but patients can develop resistance through acquired point mutations that block imatinib binding to BCR-ABL. Fortunately, most imatinib-resistant BCR-ABL mutants are sensitive to nilotinib and dasatinib, next-generation drugs that provide vital second-line treatments (Kantarjian et al., 2010a). However, substitution of threonine 315 in ABL for isoleucine (BCR-ABLT315I) generates a protein that is resistant to all three drugs and this mutant remains a persistent clinical problem for the long-term CML management. Pan-ABL inhibitors effective against BCR-ABLT315I are undergoing clinical trials (reviewed in O'Hare et al., 2011), but compound mutants (two or more mutations in the same protein) are resistant to all current ABL inhibitors and may represent a future obstacle for CML management (O'Hare et al., 2009, Eide et al., 2011). Furthermore, patients can develop resistance that is mediated by BCR-ABL-independent mechanisms and for these patients, treatment options are limited (reviewed in Bixby and Talpaz, 2011).
The RAS/RAF/MEK/ERK pathway promotes CML cell survival (Goga et al., 1995). RAS is a small membrane bound G-protein and RAF, MEK and ERK are sequentially activated protein kinases. There are three RAS genes (HRAS, KRAS and NRAS) in humans and together they are mutated in about 30% of human cancers. There are also three RAF genes (ARAF, BRAF and CRAF) and BRAF is mutated in about half of melanomas and at a lower frequency in several other cancers (Wellbrock et al., 2004). BRAF inhibitors such as vemurafenib (PLX4032, RG7204) mediate dramatic responses in BRAF mutant melanoma patients, but not in BRAF wild-type patients (Flaherty et al., 2010), validating mutant BRAF as a therapeutic target in melanoma. However these drugs also reveal an unexpected paradox, because while they inhibit MEK and ERK in cells expressing oncogenic BRAF, they activate MEK and ERK in cells expressing oncogenic RAS (Halaban et al., 2010, Hatzivassiliou et al., 2010, Heidorn et al., 2010, Poulikakos et al., 2010). This is because in the presence of oncogenic RAS BRAF inhibition drives BRAF binding to CRAF, resulting in BRAF acting as a scaffold to facilitate CRAF hyper-activation by stimulating critical events such as serine 338 (S338) phosphorylation (Hatzivassiliou et al., 2010, Heidorn et al., 2010). Paradoxical activation of the pathway can also be achieved by CRAF inhibition, which drives CRAF homodimerization consisting of drug-bound monomers that facilitate the activation of drug-free monomer through scaffold functions or conformational changes (Poulikakos et al., 2010). Thus, under some circumstances RAF inhibitors drive paradoxical activation of BRAF and CRAF to accelerate tumorigenesis by hyper-activating MEK and ERK (Hatzivassiliou et al., 2010, Heidorn et al., 2010).
Here we investigated if other kinase inhibitors can also drive paradoxical activation of RAF, MEK and ERK. Surprisingly, we found that imatinib, nilotinib and dasatinib hyper-activated BRAF, CRAF, MEK and ERK in cells expressing oncogenic RAS or BCR-ABLT315I. We therefore investigated the underlying mechanisms and examined how this affected the growth of leukemia cells.
Results
Imatinib, nilotinib and dasatinib activate RAF, MEK and ERK in RAS mutant cells
To initiate our study we treated D04 cells, a melanoma line that expresses NRASQ61L, with a variety of protein kinase inhibitors and investigated their effects on the MEK/ERK pathway by measuring MEK and ERK phosphorylation by western blot. The majority of compounds tested did not affect MEK or ERK phosphorylation (Fig S1A), but surprisingly imatinib, nilotinib and dasatinib stimulated robust MEK and ERK phosphorylation at concentrations as low as 100nM (Fig 1A). Since the peak plasma/serum concentrations of imatinib, nilotinib and dasatinib are ∼5μM, 4μM and 90nM respectively (Weisberg et al., 2007, Demetri et al., 2002), these data show that the drugs activate this pathway at physiologically relevant concentrations.
Figure 1. Imatinib, nilotinib and dasatinib activate RAF, MEK and ERK in cells harbouring RAS mutations.
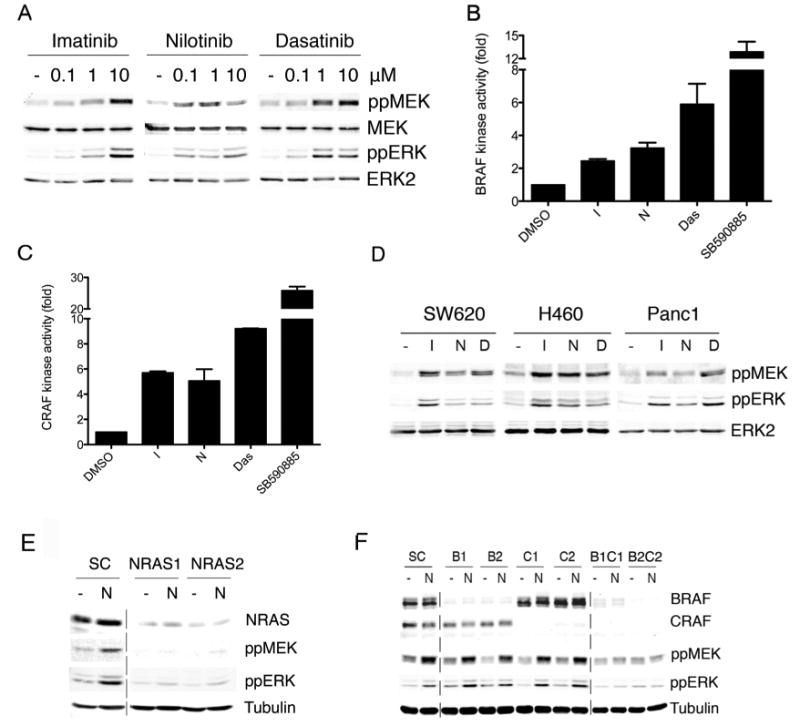
A. Western blot for phospho-MEK (ppMEK), MEK, phospho-ERK (ppERK) and ERK2 (loading control) in D04 cells treated with DMSO (-), imatinib, nilotinib or dasatinib at the indicated concentrations (μM).
B. Endogenous BRAF kinase activity in D04 cells treated with imatinib (10μM), nilotinib (1μM), dasatinib (5μM) or SB590885 (0.1μM) for 3 hr. Data represent means of triplicate measurements with error bars to represent standard deviation (SD).
C. Endogenous CRAF kinase activity in D04 cells treated with imatinib (10μM), nilotinib (1μM), dasatinib (5μM) or SB590885 (0.1μM) for 3 hr. Data represent means of triplicate measurements with error bars to represent standard deviation (SD).
D. Western blot for phospho-MEK (ppMEK), phospho-ERK (ppERK) and ERK2 (loading control) in SW620, H460 and Panc-1 cells treated with DMSO (-), imatinib (I; 10μM), nilotinib (N; 1μM) or dasatinib (D; 5μM).
E. Western blot for NRAS, phospho-MEK (ppMEK), phospho-ERK (ppERK) and tubulin (loading control) in D04 cells transfected with non-specific control (SC) or two NRAS (NRAS1, NRAS2) siRNAs. The cells were treated with DMSO (-) or nilotinib (N; 1μM) after 48 hours. The dotted line shows where discontinuous sections of the same blot were joined.
F. Western blot for BRAF, CRAF, phospho-MEK (ppMEK), phospho-ERK (ppERK) and tubulin (loading control) in D04 cells transfected with non-specific control (SC), or two different BRAF (B1, B2) or two different CRAF (C1, C2) siRNAs, or combinations thereof. After 72 hours the cells were treated with DMSO (-) or nilotinib (N; 1μM) as indicated.
Imatinib, nilotinib and dasatinib also activated BRAF and CRAF in D04 cells, albeit much less efficiently than SB590885 (Fig 1B, 1C), a BRAF selective inhibitor (Takle et al., 2006). We show that imatinib, nilotinib and dasatinib also activated MEK and ERK in SW620 (KRASG12V) colorectal carcinoma cells, Panc1 (KRASG12D) pancreatic carcinoma cells, and H460 (KRASQ61H) lung cancer cells (Fig 1D), but not in BRAFV600E expressing A2058 or A375P melanoma cells (Fig S1B). We used RNA interference (RNAi) to show that NRAS depletion blocked MEK and ERK activation in D04 cells (Fig 1E), whereas BRAF or CRAF depletion did not (Fig 1F). However, when BRAF and CRAF were both depleted, MEK and ERK activation was blocked (Fig 1F).
Imatinib, nilotinib and dasatinib induce paradoxical activation of the MEK/ERK pathway by inhibiting BRAF and CRAF
The data above show that imatinib, nilotinib and dasatinib activate BRAF, CRAF, MEK and ERK in RAS mutant, but not BRAF mutant cells. We therefore examined directly if this was driven by the paradoxical mechanism(s) previously described. First we show that although imatinib, nilotinib and dasatinib activated BRAF and CRAF in cells (Fig 1B, 1C), they inhibited BRAF and CRAF in vitro (Fig 2A), determining their IC50 values to be 1,630, 1,700 and 119nM respectively for BRAF and 515, 745 and 61nM respectively for CRAF.
Figure 2. Imatinib, nilotinib and dasatinib drive paradoxical activation of BRAF and CRAF in RAS mutant cells.
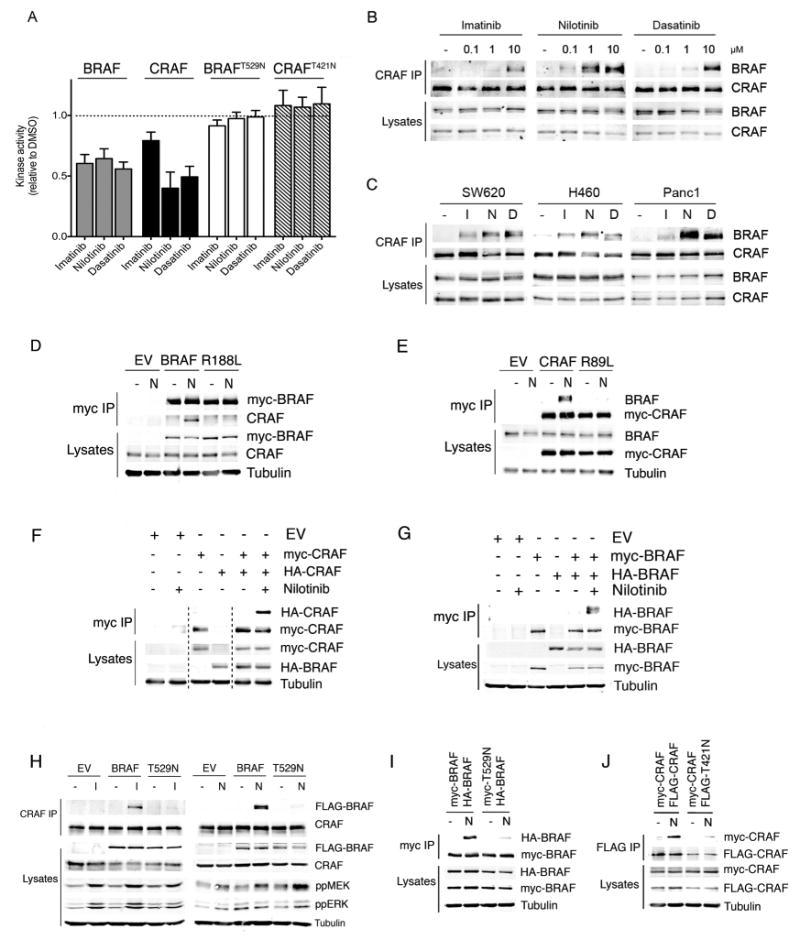
A. RAF kinase assays. Myc-epitope tagged BRAF, CRAF, BRAFT529N or CRAFT421N were transiently expressed in COS cells. BRAF and BRAFT529N were co-expressed with HRASG12V, and CRAF and CRAFT421N were co-expressed with HRASG12V and SRCY527F. The myc-tagged proteins were captured and their kinase activity was determined in the presence of imatinib (10μM), nilotinib (1μM) or dasatinib (5μM). The results are the mean of triplicate determinations and error bars to represent SD and are presented relative to DMSO-treated controls (dotted line).
B. Western blots for endogenous BRAF and CRAF in CRAF immunoprecipitates (CRAF IP) or cell lysates from D04 cells treated with DMSO (-), imatinib, nilotinib or dasatinib at the indicated concentrations.
C. Western blots for endogenous BRAF and CRAF in CRAF immunoprecipitates (CRAF IP) or cell lysates from SW620, H460 and Panc1 cells treated with DMSO (-), imatinib (10μM), nilotinib (1μM) or dasatinib (5μM).
D. Western blots for myc-tagged BRAF, endogenous CRAF or tubulin (loading control) in myc immunoprecipitates (myc IP) or cell lysates from D04 cells expressing empty vector control (EV), myc-BRAF (BRAF) or myc-BRAFR188L (R188L) and treated with DMSO (-) or nilotinib (N; 1μM).
E. Western blots for myc-tagged CRAF, endogenous BRAF or tubulin (loading control) in myc immunoprecipitates (myc IP) or cell lysates from D04 cells expressing empty vector control (EV), myc-CRAF (CRAF) or myc-CRAFR89L (R89L) and treated with DMSO (-) or nilotinib (N; 1μM).
F. Western blots for myc-tagged CRAF, HA-tagged CRAF or tubulin (loading control) in myc immunoprecipitates (myc IP) or cell lysates from D04 cells expressing empty vector control (EV), myc-CRAF or HA-CRAF and treated with DMSO (-) or nilotinib (+; 1μM).
G. Western blots for myc-tagged BRAF, HA-tagged BRAF or tubulin (loading control) in myc immunoprecipitates (myc IP) or cell lysates from D04 cells expressing empty vector control (EV), myc-BRAF or HA-BRAF and treated with DMSO (-) or nilotinib (+; 1μM).
H. Western blots for FLAG-tagged BRAF or FLAG-tagged BRAFT529N (FLAG-BRAF), endogenous CRAF, phospho-MEK (ppMEK), phospho-ERK (ppERK) and tubulin (loading control) in CRAF immunoprecipitates (CRAF IP) or cell lysates from D04 cells expressing empty vector control (EV), FLAG-tagged BRAF (BRAF) or FLAG-tagged BRAFT529N (T529N) and treated with DMSO (-), imatinib (I; 10μM), or nilotinib (N; 1μM).
I. Western blots for HA-tagged BRAF (HA-BRAF), myc-tagged BRAF (myc-BRAF) or tubulin (loading control) in myc immunoprecipitates (myc IP) or cell lysates from D04 cells expressing myc-BRAF, myc-BRAFT529N (myc-T529N) or HA-BRAF and treated with DMSO (-), or nilotinib (N; 1μM).
J. Western blots for myc-tagged CRAF (myc-CRAF), FLAG-tagged CRAF (FLAG-CRAF) or tubulin (loading control) in FLAG immunoprecipitates (FLAG IP) or cell lysates from D04 cells expressing myc-CRAF, FLAG-CRAF or FLAG-CRAFT421N (FLAG-T421N) and treated with DMSO (-), or nilotinib (N; 1μM).
We next examined if these drugs drove RAF dimerization. Endogenous CRAF was immunoprecipitated and western blotted for endogenous BRAF. Imatinib, nilotinib and dasatinib all induced robust BRAF binding to CRAF in cells expressing oncogenic RAS (D04, SW620, H460 and Panc1 cells; Fig 2B, 2C), but not in cells expressing oncogenic BRAF (A2058 or A375 cells; Fig S2A). Mutations that prevented BRAF (BRAFR188L) or CRAF (CRAFR89L) binding to RAS (Fabian et al., 1994) blocked BRAF binding to CRAF (Fig 2D, 2E), confirming BRAF and CRAF must bind to RAS in order to dimerize. We also examined if these BRAF and CRAF formed homodimers. We expressed myc-epitope or HA-epitope tagged versions BRAF or CRAF in D04 cells, immunoprecipitated the myc-tagged proteins and western blotted for the HA-tagged proteins and show that both BRAF and CRAF homodimers were formed in D04 cells (Fig 2F, 2G).
To test directly if dimer formation was driven by drug binding to BRAF or CRAF, we used mutant versions of BRAF and CRAF in which the so-called gatekeeper residues were substituted with asparagine (BRAFT529N and CRAFT421N respectively). We have previously shown that this mutation blocks drug binding to BRAF (Whittaker et al., 2010) and confirm here that both BRAFT529N and CRAFT421N were resistant to imatinib, nilotinib and dasatinib (Fig 2A). Critically, BRAFT529N and CRAFT421N were severely impaired in their ability to form BRAF:CRAF heterodimers and BRAF:BRAF or CRAF:CRAF homodimers (Fig 2H, 2I, 2J, S2B).
Imatinib, nilotinib and dasatinib induce paradoxical MEK/ERK pathway activation in leukemia cells expressing BCR-ABLT315I
The data above show that imatinib, nilotinib and dasatinib are weak RAF inhibitors that drive formation of RAF hetero and homodimers, and stimulate paradoxical activation of BRAF and CRAF in the presence of activated RAS. Previous studies have shown that imatinib activates ERK in leukemia cells expressing imatinib-resistant BCR-ABL (Yu et al., 2002, Suzuki et al., 2010, Mohi et al., 2004, Chu et al., 2004), so we tested if this was also driven through paradoxical activation of RAF. For this we used isogenic clones of murine Ba/F3 pro-B cells whose growth was driven by either BCR-ABL or BCR-ABLT315I (Golub TR et al., 1996). We confirmed that imatinib, nilotinib and dasatinib blocked BCR-ABL phosphorylation on tyrosine 245 (Y245) and CRKL phosphorylation on tyrosine 207 (Y207) in BCR-ABL Ba/F3 cells (Fig 3A). Furthermore, imatinib, nilotinib and dasatinib blocked CRAF activity in these cells (Fig 3B) and consistent with this, they suppressed CRAF phosphorylation on serine 338 (S338) and blocked MEK and ERK activity (Fig 3A). In contrast, in BCR-ABLT315I Ba/F3 cells imatinib, nilotinib and dasatinib did not inhibit BCR-ABL or CRKL phosphorylation (Fig 3C). More importantly, in these cells all three drugs induced CRAF phosphorylation on S338 (Fig 3C) and activated CRAF (Fig 3D), MEK and ERK (Fig 3C). Critically, we show that whereas imatinib, nilotinib and dasatinib did not affect BRAF binding to CRAF in the BCR-ABL cells, they enhanced BRAF binding to CRAF in BCR-ABLT315I Ba/F3 cells (Fig 3A, 3C).
Figure 3. Imatinib, nilotinib and dasatinib induce paradoxical RAF, MEK and ERK activation in cells expressing BCR-ABLT315I.
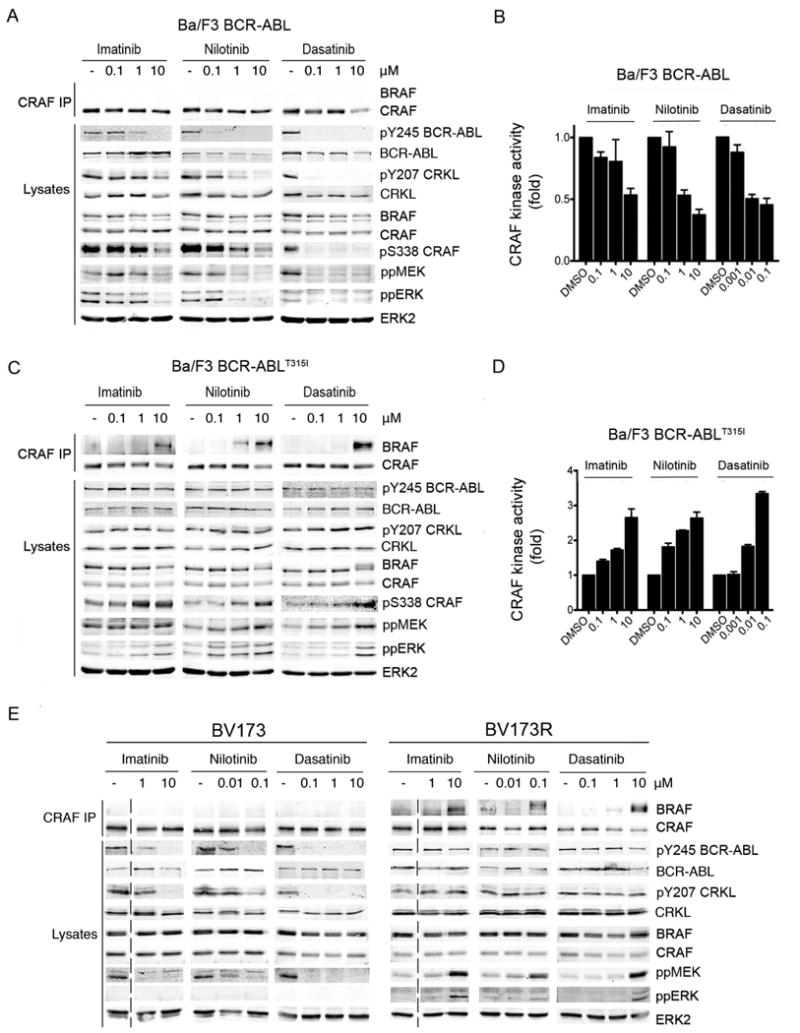
A. Western blots for endogenous BRAF, CRAF, pY245 BCR-ABL, BCR-ABL, pY207 CRKL, CRKL, pS338 CRAF, ppMEK, ppERK and ERK2 (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from Ba/F3 cells expressing BCR-ABL treated with DMSO (-), or the indicated concentrations of imatinib, nilotinib and dasatinib.
B. RAF kinase assays. Endogenous CRAF kinase activity in Ba/F3 cells expressing BCR-ABL treated with imatinib, nilotinib or dasatinib at the indicated concentrations (μM). Data represent means of triplicate measurements with error bars to represent standard deviation (SD).
C. Same as in A but with Ba/F3 cells expressing BCR-ABLT315I.
D. Same as in B but with BCR-ABLT315I Ba/F3 cells.
E. Western blots for endogenous BRAF, CRAF, pY245 BCR-ABL, BCR-ABL, pY207 CRKL, CRKL, ppMEK, ppERK and ERK2 (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from BV173 and BV173R cells treated with DMSO (-) or the indicated concentrations of imatinib, nilotinib and dasatinib. The dotted line indicates the joining of two discontinuous sections of the same blot.
We also compared responses in BV173 and BV173R cells. BV173 cells were derived from a blast crisis CML patient and express BCR-ABL endogenously, whereas BV173R cells were selected for imatinib-resistance and express BCR-ABLT315I (Pegoraro et al., 1983). Imatinib, nilotinib and dasatinib inhibited BCR-ABL and CRKL phosphorylation in BV173, but not BV173R cells (Fig 3E). Furthermore, whereas imatinib, nilotinib and dasatinib did not induce BRAF binding to CRAF and inhibited MEK and ERK in BV173 cells, they induced BRAF binding to CRAF and activated MEK and ERK in BV173R cells (Fig 3E).
RAS signaling is critical to paradoxical activation of the RAF–ERK pathway in CML cells
The results above show that imatinib, nilotinib and dasatinib block RAF/MEK/ERK signaling in BCR-ABL cells, but induce unexpected paradoxical activation of this pathway in BCR-ABLT315I cells. To investigate the mechanism(s) underlying this difference, we first examined RAS because of its critical role in RAF activation. Dominant-negative HRAS (HRASS17N) blocked ERK activation by nilotinib in BCR-ABLT315I Ba/F3 cells (Fig 4A) and nilotinib blocked RAS activity in BCR-ABL, but not BCR-ABLT315I cells (Fig 4B). We also show that imatinib, nilotinib and dasatinib did not induce BRAF binding to CRAF in K562 cells (which express BCR-ABL), but when these cells expressed HRASG12V, all three drugs induced BRAF binding to CRAF (Fig 4C). Note that imatinib, nilotinib and dasatinib did not increase MEK and ERK phosphorylation in K562 cells expressing HRASG12V, because the pathway is already saturated by the expression of HRASG12V (Fig 4C). Taken together, we conclude that RAS plays a critical role in paradoxical MEK/ERK pathway activation in BCR-ABLT315I expressing cells.
Figure 4. Active RAS is required for paradoxical activation of RAF/MEK/ERK signalling.
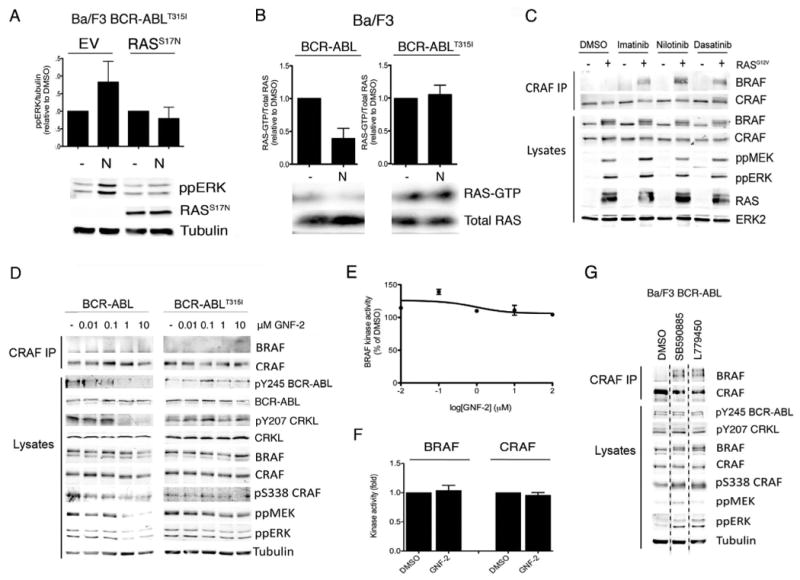
A. Graph showing phospho-ERK quantification in BCR-ABLT315I Ba/F3 cells expressing empty vector control (EV) or RASS17N and treated with DMSO (-) or nilotinib (N; 1μM). The data were quantified by western blotting, a representative of which is below the graph and shows phospho-ERK (ppERK), myc-RASS17N and tubulin (loading control). The ppERK levels are presented relative to tubulin and were calculated from triplicate determinations with error bars to represent the SD.
B. Graph showing RAS-GTP quantification in BCR-ABL and BCR-ABLT315I Ba/F3 cells treated with DMSO (-) or nilotinib (N; 1μM, 60 min). The data were quantified by western blotting, a representative of which is below the graph and shows RAS-GTP and total RAS levels in the extracts. The RAS-GTP levels are shown relative to total RAS levels and are from triplicate experiments with error bars to represent the SD.
C. Western blots for endogenous BRAF, CRAF, phospho-MEK (ppMEK), phospho-ERK (ppERK), RAS and ERK2 (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from K562 cells expressing an empty vector control (-) or HRASG12V (+) treated with DMSO, imatinib (10μM), nilotinib (1μM) or dasatinib (5μM).
D. Western blots for BRAF, CRAF, pY245 BCR-ABL, BCR-ABL, pY207 CRKL, CRKL, pS338 CRAF, ppMEK and ppERK and tubulin (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from BCR-ABL and BCR-ABLT315I Ba/F3 cells treated with GNF-2 at the indicated concentrations.
E. BRAF kinase assay. Myc-epitope tagged BRAF was transiently expressed in COS cells with HRASG12V. Myc-BRAF was captured and its kinase activity was determined in the presence of various GNF-2 concentrations for 30 min. The results are the mean of triplicate determinations and error bars to represent SD and are presented relative to DMSO-treated (100%) controls.
F. BRAF and CRAF kinase assays. Endogenous BRAF and CRAF kinase activities were measured in D04 cells treated with DMSO or GNF-2 (1μM). The data are presented relative to DMSO and represent triplicate measurements with error bars to represent standard deviation (SD).
G. Western blot for endogenous BRAF, CRAF, pY245 BCR-ABL, pY207 CRKL, pS338 CRAF, ppMEK and ppERK and tubulin (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from BCR-ABL Ba/F3 cells treated with DMSO (-), SB590885 (0.3μM) or L779450 (0.3μM). The dotted line indicates where discontinuous sections of the same blot were joined.
We next examined cell responses to GNF-2, an allosteric inhibitor of BCR-ABL. As a control we show that GNF-2 blocked BCR-ABL, CRKL, CRAF, MEK and ERK phosphorylation in BCR-ABL Ba/F3 cells and confirmed that BCR-ABLT315I was resistant to GNF-2 by showing that it did not block BCR-ABL or CRKL phosphorylation in cells expressing this mutant (Fig 4D). Critically, GNF-2 did not inhibit BRAF activity in vitro (Fig 4E), and in BCR-ABLT315I Ba/F3 cells it did not induce BRAF binding to CRAF, did not increase CRAF, MEK or ERK phosphorylation (Fig 4D), and did not activate BRAF or CRAF (Fig 4F). We also performed apposite experiments with the BRAF selective inhibitors SB590885 and L779450. Neither agent inhibited BCR-ABL or CRKL phosphorylation in BCR-ABL Ba/F3 cells and accordingly, they both stimulated BRAF binding to CRAF and CRAF, MEK and ERK phosphorylation in these cells (Fig 4G). Thus, BCR-ABL inhibitors that do not inhibit BRAF do not activate the pathway in BCR-ABLT315I cells, whereas BRAF inhibitors activate the pathway in BCR-ABL cells.
Taking these data together, we propose the following model. We posit that imatinib, nilotinib and dasatinib are weak RAF inhibitors that drive paradoxical activation of BRAF and CRAF in the presence of activated RAS. Since RAS is activated downstream of BCR-ABL (Goga et al., 1995, Suzuki et al., 2010), when BCR-ABL is inhibited, so is RAS (Fig 4B) and although BRAF and CRAF are also inhibited, the lack of RAS activity means that they are not paradoxically activated. In contrast, since BCR-ABLT315I is resistant to these three inhibitors, RAS activity persists in the presence of the drugs and consequently they are able to drive paradoxically activation of BRAF and CRAF.
Nilotinib synergizes with MEK inhibition to induce synthetic lethality in drug-resistant CML cells in vitro
We next investigated how paradoxical MEK/ERK pathway activation affected the growth of leukemia cells expressing BCR-ABLT315I. As mentioned, imatinib, nilotinib and dasatinib reach concentrations of ∼5μM, 4μM and 90nM respectively in patient plasma (Weisberg et al., 2007, Demetri et al., 2002). We therefore the effects of imatinib and nilotinib at 3μM and 1μM respectively, but since dasatinib only activated the RAF/MEK/ERK pathway at concentrations above 1μM, we did not further examine the effects of this drug. As expected, BCR-ABL Ba/F3 cells were sensitive to imatinib and nilotinib, whereas BCR-ABLT315I Ba/F3 cells were resistant (Fig 5A). The MEK inhibitor PD184352 did not inhibit the growth of BCR-ABL or BCR-ABLT315I Ba/F3 cells (Fig 5A) and PD184352 did not synergize with imatinib, to inhibit the growth of BCR-ABLT315I Ba/F3 cells (Fig 5A). Importantly, whereas PD184352 and nilotinib did not synergize to inhibit the growth of the BCR-ABL Ba/F3 cells, they synergized to inhibit the growth of BCR-ABLT315I Ba/F3 cells.
Figure 5. PD184352 synergizes with nilotinib to induce synthetic lethality in cells expressing BCR-ABLT315I in vitro.
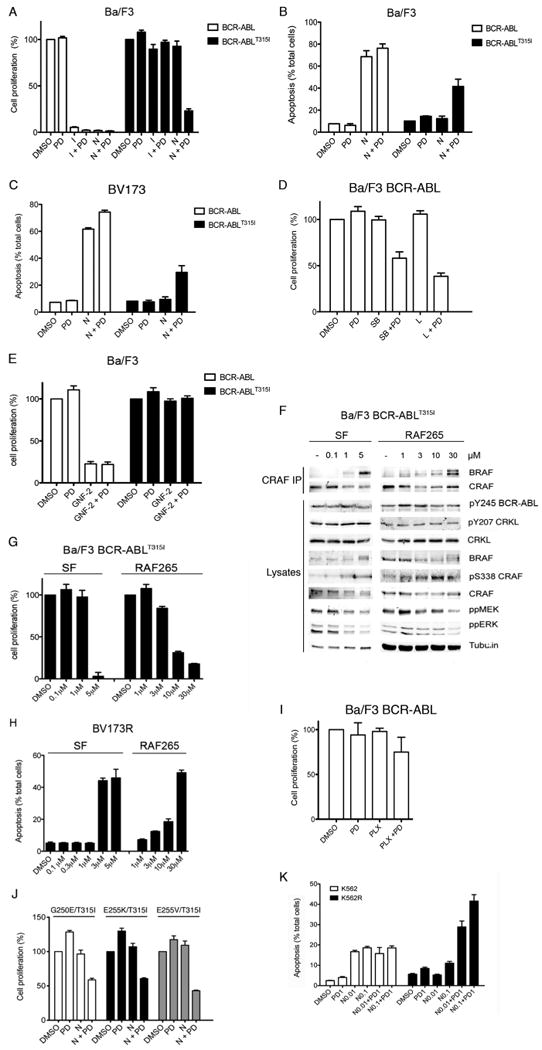
A. Cell proliferation was measured in BCR-ABL and BCR-ABLT315I Ba/F3 cells treated with DMSO, PD184352 (PD; 2μM), imatinib (I; 3μM), nilotinib (N; 1μM) or the indicated combinations for 4 days. Growth, determined in triplicate, is expressed as a percentage of the DMSO controls with error bars to represent SD from the mean.
B. Percentage of apoptotic cells in BCR-ABL and BCR-ABLT315I Ba/F3 cells treated with DMSO, PD184352 (PD; 2μM), nilotinib (N; 1μM) or the indicated combinations for 4 days. Apoptosis was detected by staining cells with annexin V and propidium iodide. The mean percentage of apoptotic cells is shown, as determined by triplicate samples, along with the SD.
C. Percentage of apoptosis in BV173 and BV173R cells treated with DMSO, PD184352 (PD; 0.2μM), nilotinib (N; 0.1μM) or the indicated combinations for 6 days. The cells were fixed and stained with propidium iodide and sub-G1 cells were measured by flow cytometry. The mean of triplicate samples is shown with the SD.
D. Cell proliferation was measured in BCR-ABL Ba/F3 cells treated with PD184352 (PD; 2μM), SB590885 (SB; 0.3μM), L779450 (L; 0.3μM), or the indicated combinations for 72 hr. Cell growth determinations in triplicate are expressed relative to the DMSO control along with the SD.
E. Cell proliferation was measured in BCR-ABL and BCR-ABLT315I Ba/F3 cells treated with PD184352 (PD; 2μM), GNF-2 (1μM), or the indicated combination for 4 days. Cell growth determinations in triplicate are expressed relative to the DMSO control along with the SD.
F. Western blots for endogenous BRAF, CRAF, pY245 BCR-ABL, pY207 CRKL, CRKL, pS338 CRAF, ppMEK and ppERK and tubulin (loading control) in CRAF immunoprecipitates (CRAF IP) and cell lysates from BCR-ABLT315I Ba/F3 cells treated with the indicated concentrations of sorafenib (SF) and RAF265.
G. Cell proliferation was measured in BCR-ABLT315I Ba/F3 cells treated with the indicated concentrations of sorafenib (SF) and RAF265 for 72 hr. Growth, determined in triplicate, is expressed as a percentage of the DMSO controls with the SD.
H. Percentage of apoptotic BV173R cells treated with the indicated concentrations of sorafenib (SF) and RAF265 for 72 hr. Cells were fixed and stained with propidium iodide and sub-G1 cells were measured by flow cytometry. The mean of triplicate samples is shown with the SD.
I. Cell proliferation was measured in BCR-ABL Ba/F3 cells treated with PD184352 (PD; 2μM), PLX4720 (PLX; 1μM) or the indicated combinations for 72 hr. Growth, determined in triplicate, is expressed as a percentage of the DMSO controls and error bars represent SD.
J. Cell proliferation was measured in BCR-ABLG250E/T315I, BCR-ABLE255K/T315I and BCR-ABLE255V/T315I Ba/F3 cells treated with DMSO, PD184352 (PD; 2μM), nilotinib (N; 1μM) or the indicated combinations for 4 days. Growth, determined in triplicate, is expressed as a percentage of the DMSO controls and error bars represent SD.
K. Percentage of apoptotic K562 and K562R cells treated with PD184352 (PD; 1μM), the indicated concentrations of nilotinib (N) or combinations of both for 4 days. The cells were fixed and stained with propidium iodide and sub-G1 cells were measured by flow cytometry. The mean percentage of apoptotic cells is shown, as determined by triplicate samples, along with the SD.
These responses were accompanied by apposite responses in apoptosis. Thus, imatinib and nilotinib induced apoptosis in BCR-ABL, but not in BCR-ABLT315I Ba/F3 cells (Fig 5B and Fig S3A). PD184352 did not induce apoptosis in either line (Fig 5B and Fig S3A) and whereas it did not synergize with imatinib, it did synergise with nilotinib to induce apoptosis in BCR-ABLT315I cells (Fig 5B and Fig S3A). We observed similar responses in BV173 and BV173R cells. Imatinib and nilotinib inhibited cell proliferation and induced apoptosis in BV173 cells, but not BV173R cells (Fig 5C and Fig S3B). PD184352 did not inhibit cell proliferation or induce apoptosis in either line and while it synergized with nilotinib to inhibit cell proliferation and induce apoptosis in BV173R cells, we saw no such synergy with imatinib (Fig 5C and Fig S3B).
These data show that paradoxical activation of RAF leads CML cells to develop an unexpected dependence on MEK/ERK signaling, such that if MEK is inhibited, proliferation is inhibited and apoptosis induced. We support this model by showing that PD184352 synergized with the BRAF inhibitors SB590885 and L779450 to inhibit the growth of BCR-ABL Ba/F3 cells (Fig 5D), whereas GNF-2 did not synergize with PD184352 to inhibit the growth of BCR-ABLT315I Ba/F3 cells (Fig 5E). Thus BRAF inhibitors that did not inhibit BCR-ABL were able to drive paradoxical activation of RAF and synergy with MEK inhibitors to kill cells expressing BCR-ABL. Furthermore, GNF-2, which did not drive paradoxical activation of RAF did not synergize with MEK to kill BCR-ABLT315I Ba/F3 cells.
We further show that the pan-RAF inhibitors sorafenib and RAF265 did not inhibit BCR-ABL or CRKL phosphorylation in BCR-ABLT315I Ba/F3 cells and although they induced BRAF binding to CRAF, they inhibited, rather than activated MEK and ERK (Fig 5F). Critically, even in the absence of PD184352, these agents inhibited proliferation and induced cell death in cells expressing BCR-ABLT315I (Fig 5G, 5H). In line with our previous conclusions (Heidorn et al., 2010) we posit that because sorafenib and RAF265 are relatively potent pan-RAF inhibitors, they drive RAF dimerization but also inhibit the RAF proteins in the complexes that are formed. By simultaneously driving the paradoxical activation of RAF and inhibiting MEK/ERK signaling, they therefore inhibit proliferation and induce death in CML cells even in the absence of MEK inhibitors. Note also that the BRAF inhibitor PLX4720, which did not induce strong binding of BRAF to CRAF (Fig 3SC, Heidorn et al., 2010), only produced weak synergy with PD184352 to inhibit cell proliferation of these cells (Fig 5I). These data suggest that the formation of RAF dimers in the presence of RAF inhibitors is critical to the ability of these agents to synergize with PD184352 and kill the cells.
Nilotinib synergizes with MEK inhibition to induce synthetic lethality in cells expressing compound BCR-ABL mutants
Next we tested if similar responses occurred in cells expressing compound BCR-ABL mutants, because clinical resistance to ABL inhibitors is mediated largely by T315I or compound mutants that emerge following sequential treatment with imatinib and then nilotinib or dasatinib (Shah et al., 2007). We show that in Ba/F3 cells expressing BCR-ABLG250E/T315I, BCR-ABLE255K/T315I and BCR-ABLE255V/T315I, nilotinib did not inhibit BCR-ABL or CRKL phosphorylation, and induced BRAF binding to CRAF as well as MEK and ERK activation (Fig S3D). Further, whereas nilotinib and PD184352 by themselves did not affect proliferation of cells expressing these compound BCR-ABL mutants, they synergized to induce synthetic lethality in these cells (Fig 5J).
Nilotinib synergizes with MEK inhibition to induce synthetic lethality in cells whose resistance is BCR-ABL-independent
We also tested if similar responses occurred in CML cells whose resistance was mediated by non BCR-ABL mechanisms. K562 cells were derived from a patient in terminal blast crisis and K562R cells are a clone that is resistant due to overexpression of the SRC family kinase LYN (Donato et al., 2003). In K562 cells nilotinib inhibited BCR-ABL and CRKL phosphorylation, suppressed RAS activity and inhibited CRAF, MEK and ERK phosphorylation (Fig S3E, S3F). Nilotinib also blocked BCR-ABL and CRKL phosphorylation in K562R cells (Fig S3E), but nevertheless did not inhibit RAS (Fig S3F) and did not block CRAF, MEK or ERK phosphorylation (Fig S3E). Nilotinib induced apoptosis in K562 cells, but PD184352 did not kill these cells and did not enhance nilotinib-induced cell death (Fig 5K). In contrast, nilotinib and PD184352 alone did not affect the growth of K562R cells, but together they synergized to induce death in these cells (Fig 5K).
Nilotinib synergizes with MEK inhibition to induce synthetic lethality in primary drug-resistant CML cells
We next determined if nilotinib and PD184352 also inhibited the growth of primary cells from patients with BCR-ABL-driven CML. Mononuclear cells derived from blood or bone marrow of CML patients harbouring native BCR-ABL or BCR-ABLT315I and from healthy individuals were treated with nilotinib, PD184352 or both for 96 hours and cell viability was measured. Consistent with the cell lines, nilotinib inhibited the proliferation of cells expressing BCR-ABL from newly diagnosed CML patients and PD184352 did not substantially enhance this effect (Fig 6A). In contrast, nilotinib and PD184352 alone did not affect the growth of BCR-ABLT315I cells, but synergized to inhibit growth of these cells (Fig 6B). As a control, we show that CD34+ hematopoietic cells from healthy patients were resistant to all combinations of nilotinib and PD184352 (Fig 6C).
Figure 6. PD184352 synergizes with nilotinib to inhibit cell proliferation of CML primary cells and induce synthetic lethality in cells expressing BCR-ABLT315I in vivo.
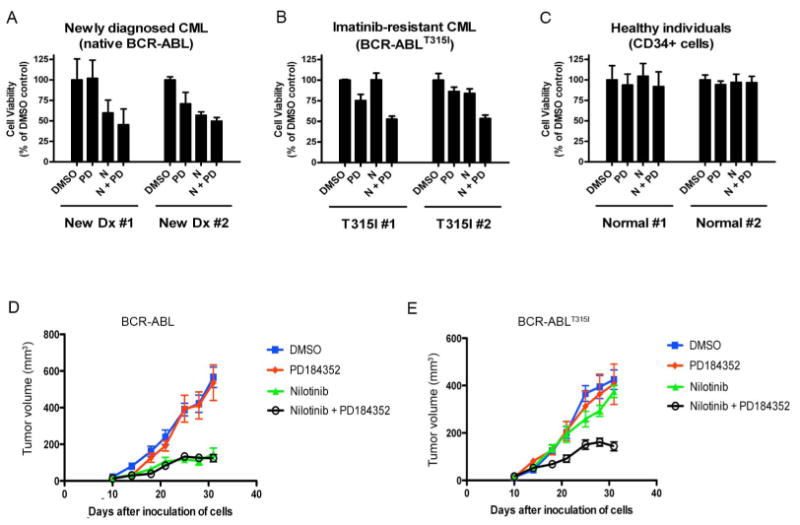
A. Cell proliferation was measured in mononuclear cells harbouring native BCR-ABL isolated from newly diagnosed CML patients (n=2) treated ex vivo with nilotinib (1μM), PD184352 (0.3μM) or both for 96 hr.
B. As in A, but with BCR-ABLT315I expressing mononuclear cells isolated from imatinibresistant CML patients (n=2).
C. As in A, but with CD34+ cells from healthy individuals (n=2).
D and E. The growth of tumor allografts formed by Ba/F3 cells expressing BCR-ABL (D) or BCR-ABLT315I (E) grown in nude mice is shown. The mice were treated with vehicle, PD184352 (25mg/kg), nilotinib (25mg/kg), or both. The results show mean tumor volumes for groups of 6 animals with error bars to represent the standard error.
Nilotinib and PD184352 induce synthetic lethality in drug-resistant CML cells in vivo
Finally, we tested the implications of our findings in vivo by examining how the drugs affected the growth of subcutaneously implanted Ba/F3 allografts expressing BCR-ABL or BCR-ABLT315I. The growth of BCR-ABL tumors was strongly suppressed by nilotinib, but not by PD184352, and PD184352 did not enhance the growth-inhibitory activity of nilotinib (Fig 6D). In contrast, BCR-ABLT315I tumors were insensitive to both nilotinib and PD184352, but together these drugs synergized to inhibit the growth of these tumors (Fig 6E).
Taking all of these data together, we conclude that nilotinib and PD184352 induced synthetic lethality in drug-resistant CML cells both in vitro and in vivo.
Discussion
Building on our previous studies, we tested a panel of drugs for their ability to activate MEK and ERK in cells expressing oncogenic RAS. Most of the drugs were ineffective, but imatinib, nilotinib and dasatinib activated MEK and ERK in a variety of lines. Critically, we show that these drugs are weak RAF inhibitors whose binding to BRAF and CRAF drives BRAF:CRAF heterodimer and BRAF and CRAF homodimer formation, leading to paradoxical activation of both BRAF and CRAF. We established an essential role for RAS in these responses by showing that its depletion blocked MEK/ERK activation and if BRAF or CRAF were unable to bind to RAS, they did not form dimers. We also established a critical role for BRAF and CRAF by showing that depletion of both was necessary to block MEK and ERK activation by these drugs. Thus, although they only inhibit RAF weakly, imatinib, nilotinib and dasatinib possess sufficient off-target activity to drive the formation of BRAF:CRAF dimers and stimulate paradoxical activation of the pathway. It has previously been shown that RAF inhibitors also drive paradoxical activation of BRAF and CRAF (Halaban et al., 2010, Hatzivassiliou et al., 2010, Heidorn et al., 2010, Poulikakos et al., 2010) and our data show that imatinib, nilotinib and dasatinib appear to mimic these effects. We therefore posit that like RAF inhibitors (Downward, 2011), imatinib, nilotinib and dasatinib bind to monomeric RAF and induce RAF dimerization in which one partner is bound to drug and the other is not. The drug-bound partner then acts as a scaffold, or induces a conformational change to facilitate activation of the drug-free partner.
We extended these observations to show that imatinib, nilotinib and dasatinib drove paradoxical activation of the RAF/MEK/ERK pathway in drug-resistant leukemia cells. Critically, we showed that inhibition of BCR-ABL causes RAS inactivation in BCR-ABL expressing, but not BCR-ABLT315I expressing cells. We further showed that dominant negative RAS blocked MEK/ERK activation in BCR-ABLT315I cells and that imatinib, nilotinib and dasatinib drove RAF dimerization in BCR-ABL cells when oncogenic RAS was ectopically introduced. These data establish that RAS plays a key role in these responses and accordingly, we propose the following model. We posit that BCR-ABL inhibition leads to RAS inhibition and so although RAF is also inhibited, it is not paradoxically activated (Fig 7A). In contrast, because BCR-ABLT315I is resistant to imatinib, nilotinib and dasatinib, RAS activity persists in the presence of these drugs and hence the off-target inhibition of RAF leads to its paradoxical activation (Fig 7B). This model also explains why BRAF inhibitors drive paradoxical activation of the pathway in BCR-ABL cells: they do not inhibit BCR-ABL, so do not inhibit RAS and hence can drive paradoxically activation of RAF. It also explains why BCR-ABL inhibitors such as GNF-2 do not drive paradoxical activation of the pathway: although they do not inhibit BCR-ABLT315I and therefore do not inhibit RAS, they are not BRAF/CRAF inhibitors and so cannot drive their paradoxical activation.
Figure 7. Model of paradoxical RAF/MEK/ERK activation in drug-resistant CML cells by nilotinib.
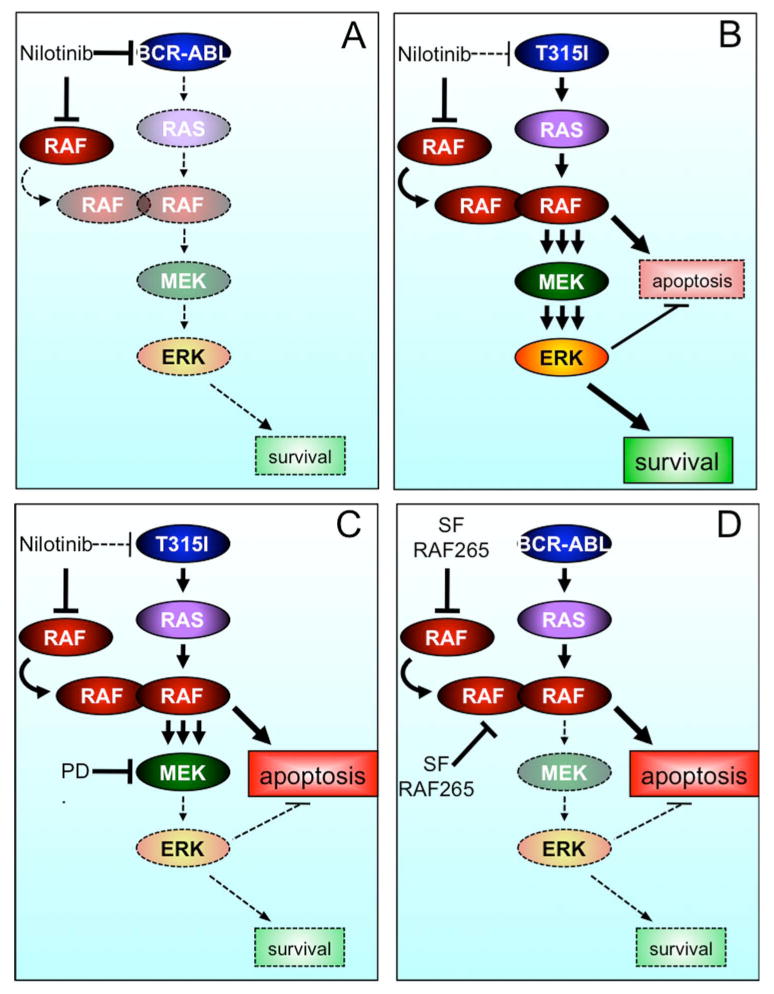
A. Nilotinib binds to and inhibits BCR-ABL, which inhibits downstream signaling, including RAS. Thus, the inhibition of RAF is inconsequential.
B. In the presence of BCR-ABLT315I, nilotinib inhibits BRAF and CRAF and due to the presence of active RAS, induces the formation of RAF dimers and activation of the RAF/MEK/ERK survival signal. We posit that these RAF complexes also activate a MEK/ERK-independent apoptotic signal, but this is overridden by the dominant survival signal.
C. Nilotinib inhibits RAF in the presence of BCR-ABLT315I, leading to paradoxical activation of RAF/MEK/ERK. MEK inhibition by PD184352 blocks this survival signal, allowing apoptosis to predominate.
D. Pan-RAF drugs such as sorafenib (SF) and RAF265 inhibit both BRAF and CRAF with high potency. So although they induce RAF dimers, they simultaneously inhibit these dimers, blocking MEK/ERK signaling, thereby favoring apoptosis.
It has been reported that imatinib activates MEK and ERK in cells expressing imatinib-resistant BCR-ABL (Yu et al., 2002, Suzuki et al., 2010, Mohi et al., 2004, Chu et al., 2004) and our studies now provide a mechanistic explanation for those observations. More importantly, we show that whereas the growth of the drug-resistant cells was unaffected by nilotinib and PD184352 in vitro and in vivo, these drugs synergized to inhibit cell growth and induce apoptosis in vitro, and to suppress tumor growth in mice. Thus, we show that drug-resistant cells develop an unexpected dependency on MEK/ERK signaling when the pathway is paradoxically activated. We therefore posit that in these cells paradoxical activation of this pathway drives both a MEK/ERK-dependent anti-apoptotic signal and a MEK/ERK-independent pro-apoptotic signal (Fig 7B). Under normal conditions the anti-apoptotic signal overcomes the pro-apoptotic signal (Fig 7B), but when MEK is inhibited, the pro-apoptosis signal predominates (Fig 7C).
It is unclear how MEK inhibition induces apoptosis under these conditions, but one possibility is that it is driven by the formation of the RAF dimers. Previous studies have shown that CRAF opposes cell death in a MEK/ERK independent manner by sequestering the pro-apoptotic kinases ASK1, MST2, ROCK1 and RIP2 (O'Neill et al., 2004, Navas et al., 1999, Chen et al., 2001, Piazzolla et al., 2005). We posit that the recruitment of CRAF into homo and heterodimers releases these binding partners, allowing them to induce apoptosis. Our preliminary experiments failed to establish a clear role for ASK1 and MST2 in the death of BCR-ABLT315I cells, but the response of these cells to RAF inhibitors supports our model. We show that SB590885 and L779450 induced robust BRAF binding to CRAF and synergized with PD184352 to induce synthetic lethality (Fig 4G, 5D). In contrast, PLX4720, which induced weak BRAF binding to CRAF (Fig S3C) only weakly synergized with the MEK inhibitor to inhibit cell proliferation (Fig 5I). Furthermore, although sorafenib and RAF265 induced strong BRAF binding to CRAF, they simultaneously inhibited MEK signaling and were thus able to induce cell death without the need of a MEK inhibitor. It has been proposed that sorafenib induces apoptosis in imatinib-resistant leukemia cells by targeting multiple kinases (Rahmani et al., 2007, Kurosu et al., 2009), but our data suggest that pan-RAF inhibitors such as sorafenib induce apoptosis because they induce paradoxical activation of RAF and simultaneously inhibit MEK/ERK, thereby favoring the pro-apoptotic signal (Fig 7D).
Imatinib was approved for first-line treatment of CML over a decade ago and is generally well tolerated, but 20-30% of patients do not achieve complete responses and acquired resistance is a persistent clinical problem (Quintás-Cardama et al., 2009). Most imatinib-resistant BCR-ABL mutants remain sensitive to nilotinib and dasatinib providing vital second-line treatments (Saglio et al., 2010, Kantarjian et al., 2010b), and both were recently approved as first-line CML drugs. However, BCR-ABLT315I and the compound mutants that arise following long-term or sequential drug treatment are resistant to all three drugs (Shah et al., 2007) and some patients develop resistance that is mediated by BCR-ABL-independent mechanisms. Thus, new treatments are still required for relapsed patients and agents active against BCR-ABLT315I are undergoing clinical trials (O'Hare et al., 2011).
We propose that the synthetic lethality we describe could provide an approach to block the emergence of drug-resistance in patients. This is based on the observation that BCR-ABL cells are sensitive to imatinib and nilotinib alone, whereas the resistant cells are sensitive to nilotinib plus the MEK inhibitor. Thus, if these drugs were to be combined the primary disease would be treated by nilotinib and the resistant clones by nilotinib plus a MEK inhibitor. Thus, this combination has the potential to treat both the bulk disease and prevent the emergence of resistance. Critically, this synthetic lethality also occurred in K562R cells, where resistance was mediated by BCR-ABL-independent mechanisms, suggesting that our findings could have wide utility. In this context, it is intriguing to note a recent report where acute lymphoblastic leukemia resistance was shown to be mediated by EphB4 receptor tyrosine kinase overexpression that led to constitutive RAS activation and ERK hyper-activation following imatinib treatment (Suzuki et al., 2010). Importantly, the MEK inhibitor U0126 synergized with imatinib to inhibit proliferation of these cells, corroborating our model. Clearly, not all BCR-ABL drugs will mediate these responses. GNF-2 lacks off-target RAF activity, and dasatinib, which only inhibits RAF at levels above those that can be achieved in patients' blood would not be suitable. We wish also to be clear that we are not proposing BRAF inhibitors for this approach and indeed, we show that PLX4720 did not induce robust RAF dimerization or efficient synthetic lethality.
In summary, CML is a heterogeneous disease characterized by the evolution of drug-resistance. We have elaborated an unexpected synthetic lethality mediated by paradoxical activation of RAF in drug-resistant cells. Importantly this response could provide approaches to extend clinical responses to nilotinib by preventing the emergence of the drug resistant clones.
Experimental Procedures
See Supplemental Experimental Procedures for detailed protocols.
Reagents
Expression vectors for epitope-tagged BRAF, BRAFT529N, BRAFR188L, CRAF, CRAFT421N, CRAFR89L, HRASS17N and HRASG12V have been described (Marais et al., 1998, Marais et al., 1995). For western blotting the antibodies used were: rabbit anti-ppMEK1/2 and rabbit anti-phospho-c-Abl (Tyr245), c-Abl, rabbit anti-phospho-CRKL (Tyr207), rabbit anti-phospho-CRAF (Ser338) (Cell Signaling Technology); mouse anti-NRAS (C-20), rabbit anti-ERK2 (C-14), mouse anti-BRAF (F-7), Crkl (Santa Cruz Biotechnology); mouse anti-FLAG, mouse anti-Tubulin, and mouse anti-ppERK1/2 (Sigma); mouse anti-CRAF, mouse-anti RAS and mouse anti-MEK1 (BD Transduction Laboratories). For immunoprecipitation, the antibodies used were: rabbit anti-myc (Abcam); rabbit anti-CRAF (C-20; Santa Cruz Biotechnology). Imatinib, nilotinib, dasatinib and sorafenib were from LC Laboratories (Woburn, MA, USA), GNF-2 from Sigma, SB590885 from Symansis (Timaru, New Zealand); L779450 from Tocris Bioscience (Ellisville, MO, USA); RAF265 from American Custom Chemicals (San Diego, CA, USA), and PD184352 was synthesized in-house. All drugs were prepared in DMSO.
Cell culture techniques
Human cell lines were cultured in DMEM (A375, A2058, COS-7, Panc1, SW620, H460, K562) or RPMI (D04, BV173, K562R) supplemented with 10% fetal bovine serum. Drug treatments were performed for 3 hours, unless otherwise specified. For RNAi studies, D04 cells were transfected with lipofectamine (Invitrogen, Paisley, UK) and 10nM of double stranded siRNAs. See Supplemental Experimental Procedures for sequences. Transient and stable expression of proteins was as described (Wan et al., 2004). For immunoprecipitation, lysates were incubated with 5μg CRAF C-20 or 3μg myc antibody (Abcam), captured on Protein G sepharose 4B beads (Sigma) and western blotted using standard protocols. FLAG-tagged BRAF was captured using Anti-FLAG-M2 Agarose beads (Sigma). Coupled RAF assays were performed as described (Wan et al., 2004). Cell proliferation was determined using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) and apoptotic cells were detected using the FITC Annexin V Apoptosis Detection Kit (BD Pharmingen) or propidium iodide staining of fixed cells followed by fluorescence-activated cell sorting (FACS) analysis. RAS capture assays were performed as described (Marais et al., 1998). BRAF and CRAF IC50 determinations were performed using Z-lyte technology (Invitrogen) at 100μM ATP. Mononuclear cells from newly diagnosed and imatinib-resistant CML patients harboring native BCR-ABL or BCR-ABLT315I were isolated by Ficoll. Bone marrow CD34+ cells from healthy individuals were purchased from Lonza. Cells were distributed in 96-well plates (4 × 104 cells/well) in the presence of PD184352 (0.3 μM) and/or nilotinib (1 μM), and cell proliferation at 96 hours was measured using MTS assay.
In vivo approaches
All procedures involving animals were approved by the Animal Ethics Committees of the Institute of Cancer Research in accordance with National Home Office regulations under the Animals (Scientific Procedures) Act 1986 and according to the guidelines of the Committee of the National Cancer Research Institute (Workman et al., 2010). Nude mice were injected subcutaneously with 5 × 106 BCR-ABLT315I Ba/F3 or 2 × 107 BCR-ABL Ba/F3 cells. Tumors were allowed to establish for seven days, size matched and allocated to groups of 6 animals. Treatment was by oral gavage daily with vehicle (5% DMSO, 95% water), 25 mg/kg nilotinib, 25 mg/kg PD184352 or both. Tumor size was determined by calliper measurements of tumor length, width and depth and volume was calculated as Volume = 0.5236 × length × width × depth (mm).
Supplementary Material
SIGNIFICANCE.
Acquired drug resistance through BCR-ABL-dependent and BCR-ABL-independent mechanisms is a persistent problem for the treatment of chronic myeloid leukemia (CML). We show that some front-line CML drugs are RAF inhibitors and they therefore drive paradoxical activation of BRAF and CRAF in drug-resistant CML cells. This leads to an unexpected dependency on the pathway and accordingly, nilotinib and MEK inhibitors mediate synthetic lethal killing of these cells in vitro and in vivo. Thus, our study shows that paradoxical activation of BRAF and CRAF can drive unexpected biological responses in CML and provide an intriguing strategy that may prevent the emergence of drug resistance in CML patients.
Highlights.
Imatinib, nilotinib and dasatinib are RAF inhibitors
Nilotinib drives paradoxically activation of RAF in drug-resistant CML cells
Nilotinib and MEK inhibitors synergize to kill drug-resistant CML cells in vitro
Nilotinib and MEK inhibitors synergize to block drug-resistant CML tumor growth
Acknowledgments
This work was supported by a Training Fellowship from The Harry J Lloyd Charitable Trust (L.P.), Cancer Research UK (ref: C107/A10433 and C309/A2187) and The Institute of Cancer Research. We are grateful to Dr Nicholas Donato for providing the BV173, BV173R and K562R cells.
Footnotes
CONFLICT OF INTEREST: The authors do not have conflicts of interest relating to this work.
References
- Bixby D, Talpaz M. Seeking the causes and solutions to imatinib-resistance in chronic myeloid leukemia. Leukemia. 2011;25:7–22. doi: 10.1038/leu.2010.238. [DOI] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci U S A. 2001;98:7783–8. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Holtz M, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103:3167–74. doi: 10.1182/blood-2003-04-1271. [DOI] [PubMed] [Google Scholar]
- Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–56. [PubMed] [Google Scholar]
- Demetri G, Von Mehren M, Blanke C, Van Den Abbeele A, Eisenberg B, Roberts P, Heinrich M, Tuveson D, Singer S, Janicek M, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Donato N, Wu J, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–8. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAF: trials and tribulations. Nat Med. 2011;17:286–8. doi: 10.1038/nm0311-286. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Eide CA, Adrian LT, Tyner JW, Mac Partlin M, Anderson DJ, Wise SC, Smith BD, Petillo PA, Flynn DL, Deininger MW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71:3189–95. doi: 10.1158/0008-5472.CAN-10-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, Mcarthur GA, Sosman JA, O'dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goga A, Mclaughlin J, Afar D, Saffran D, Witte O. Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell. 1995;82:981–8. doi: 10.1016/0092-8674(95)90277-5. [DOI] [PubMed] [Google Scholar]
- Golub Tr, Goga A, Barker Gf, Afar De, Mclaughlin J, Bohlander Sk, Rowley Jd, Witte On, Dg G. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol. 1996;16:4107–16. doi: 10.1128/mcb.16.8.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, Krauthammer M, Mccusker Jp, Kluger Y, S M. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H, Cortes J, La Rosée P, Hochhaus A. Optimizing therapy for patients with chronic myelogenous leukemia in chronic phase. Cancer. 2010a;116:1419–30. doi: 10.1002/cncr.24928. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz JA, Larson RA, Gattermann N, Ottmann OG, Hochhaus A, Radich JP, Saglio G, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase following imatinib resistance or intolerance: 24-month follow-up results. Blood. 2010b doi: 10.1182/blood-2010-03-277152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu T, Ohki M, Wu N, Kagechika H, Miura O. Sorafenib induces apoptosis specifically in cells expressing BCR/ABL by inhibiting its kinase activity to activate the intrinsic mitochondrial pathway. Cancer Res. 2009;69:3927–36. doi: 10.1158/0008-5472.CAN-08-2978. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109–12. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. Embo J. 1995;14:3136–45. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohi MG, Boulton C, Gu TL, Sternberg DW, Neuberg D, Griffin JD, Gilliland DG, Neel BG. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:3130–5. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas T, Baldwin D, Stewart T. RIP2 is a Raf1-activated mitogen-activated protein kinase kinase. J Biol Chem. 1999;274:33684–90. doi: 10.1074/jbc.274.47.33684. [DOI] [PubMed] [Google Scholar]
- O'dwyer ME, Druker BJ. Chronic myelogenous leukaemia--new therapeutic principles. J Intern Med. 2001;250:3–9. doi: 10.1046/j.1365-2796.2001.00823.x. [DOI] [PubMed] [Google Scholar]
- O'hare T, Deininger MW, Eide CA, Clarkson T, Druker BJ. Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin Cancer Res. 2011;17:212–21. doi: 10.1158/1078-0432.CCR-09-3314. [DOI] [PubMed] [Google Scholar]
- O'hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–70. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- Pegoraro L, Matera L, Ritz J, Levis A, Palumbo A, Biagini G. Establishment of a Ph1-positive human cell line (BV173) J Natl Cancer Inst. 1983;70:447–53. [PubMed] [Google Scholar]
- Piazzolla D, Meissl K, Kucerova L, Rubiolo C, Baccarini M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J Cell Biol. 2005;171:1013–22. doi: 10.1083/jcb.200504137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás-Cardama A, Kantarjian H, Cortes J. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009;16:122–31. doi: 10.1177/107327480901600204. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Nguyen TK, Dent P, Grant S. The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate-resistant bcr/abl+ human leukemia cells in association with signal transducer and activator of transcription 5 inhibition and myeloid cell leukemia-1 down-regulation. Mol Pharmacol. 2007;72:788–95. doi: 10.1124/mol.106.033308. [DOI] [PubMed] [Google Scholar]
- Saglio G, Kim DW, Issaragrisil S, Le Coutre P, Etienne G, Lobo C, Pasquini R, Clark RE, Hochhaus A, Hughes TP, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–9. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–9. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Abe A, Imagama S, Nomura Y, Tanizaki R, Minami Y, Hayakawa F, Ito Y, Katsumi A, Yamamoto K, et al. BCR-ABL-independent and RAS / MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur J Haematol. 2010;84:229–38. doi: 10.1111/j.1600-0609.2009.01387.x. [DOI] [PubMed] [Google Scholar]
- Takle AK, Brown MJ, Davies S, Dean DK, Francis G, Gaiba A, Hird AW, King FD, Lovell PJ, Naylor A, et al. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2006;16:378–81. doi: 10.1016/j.bmcl.2005.09.072. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–56. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nature Reviews. Molecular Cell Biology. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, Affolter A, Nourry A, Niculescu-Duvaz D, Springer C, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2:35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Krystal G, Varticovksi L, Mckinstry R, Rahmani M, Dent P, Grant S. Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res. 2002;62:188–99. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.