

Abstract
Neuro-cognitive disabilities are a well-recognized complication of hypothermic circulatory arrest. We and others have reported that prolonged cardiac arrest (CA) produces neuronal death and microglial proliferation and activation that are only partially mitigated by hypothermia. Microglia, and possibly other cells, are suggested to elaborate tumor necrosis factor alpha (TNF-α) which can trigger neuronal death cascades and exacerbate edema after CNS insults. Minocycline is neuroprotective in some brain ischemia models in part by blunting the microglial response. We tested the hypothesis that minocycline would attenuate neuroinflammation as reflected by brain tissue levels of TNF-α after hypothermic CA in rats. Rats were subjected to rapid exsanguination, followed by a 6 min normothermic CA. Hypothermia (30 °C) was then induced by an aortic saline flush. After a total of 20 min CA, resuscitation was achieved via cardiopulmonary bypass (CPB). After 5 min reperfusion, minocycline (90 mg/kg; n=6) or vehicle (PBS; n=6) were given. Hypothermia (34 °C) was maintained for 6 h. Rats were sacrificed at 6 or 24 h. TNF-α was quantified (ELISA) in four brain regions (cerebellum, CEREB; cortex, CTX; hippocampus, HIP; striatum, STRI). Naïve rats (n=6) and rats subjected to the same anesthesia and CPB but no CA served as controls (n=6). Immunocytochemistry was used to localize TNF-α. Naïve rats and CPB controls had no detectable TNF-α in any brain region. CA markedly increased brain TNF-α. Regional differences were seen, with the highest TNF-α levels in striatum in CA groups (10-fold higher, P<0.05 vs. all other brain regions). TNF-α was undetectable at 24 h. Minocycline attenuated TNF-α levels in CTX, HIP and STRI (P<0.05). TNF-α showed unique co-localization with neurons. In conclusion, we report region-dependent early increases in brain TNF-α levels after prolonged hypothermic CA, with maximal increases in striatum. Surprisingly, TNF-α co-localized in neurons and not microglia. Minocycline attenuated TNF-α by approximately 50% but did not totally ablate its production. That minocycline decreased brain TNF-α levels suggests that it may represent a therapeutic adjunct to hypothermia in CA neuroprotection.
University of Pittsburgh IACUC 0809278B-3.
Introduction
Deep hypothermic circulatory arrest (DHCA) has been used to create a bloodless field in cardiac surgery, enabling repair of congenital anomalies of the heart or acquired diseases of the aorta. Deep hypothermia is also used experimentally as an emergency treatment for prolonged cardiac arrest (CA) with delayed resuscitation via cardiopulmonary bypass (CPB) in both small and large animal models.1–4 The underlying mechanisms of hypothermic preservation are not yet fully understood.
Neurocognitive disabilities are a well-recognized complication of hypothermic circulatory arrest. We and others have reported that prolonged CA produces neuronal death and microglial proliferation and activation that are only partially mitigated by hypothermia.5 The exposure to the artificial materials of the CPB circuit has also been reported to induce neuroinflammation.6,7
Cytokines are pleiotropic proteins produced by multiple central nervous system (CNS) cells that participate in an orchestrated reaction to an insult. Traditionally, they have been considered pro- or anti-inflammatory, depending on their main course of action. However, individual cytokines including tumor necrosis factor alpha (TNF-α), a principal mediator of neuroinflammation in the brain, could play a dual role depending on the intensity of the insult or the period of recovery.8–10
The source of cytokines, including TNF-α, in CNS injuries remains controversial. Most studies suggest that microglia are the major source of cytokines.11–13 Other glial cells14–17 and neurons18 are also capable of cytokine production, depending on the type of insult.19 The timing of the cytokine assessment could also be an important factor, since the sources of cytokines can vary over time.
TNF-α is one of the major cytokines released by microglia. TNF-α can induce apoptosis and necroptosis in neural tissue and increase inflammation. TNF-α mRNA expression is up-regulated in cerebral ischemia,20 with microglia purportedly acting as its major source.21 It has previously been shown that TNF-α expression in the hippocampus is attenuated by mild hypothermia following hypoxic insult.22 In addition, we recently reported a decrease in microglial activation in the hippocampus following prolonged CA when treated with deep hypothermia compared to moderate hypothermia, with improved neurologic outcome.5
There are well known regional differences in damage after CA or DHCA. The role of TNF-α or other cytokines in various brain regions has not yet been fully explored. Traditionally, studies focused on selectively vulnerable regions, namely hippocampal CA1 region or cerebellar Purkinje neurons. There is an emerging interest in striatum that also shows selective vulnerability in global brain ischemia.23
Therapies to attenuate TNF-α production – antibody based therapies may be promising in the setting of conditions such as traumatic brain injury (TBI) where blood-brain barrier (BBB) is injured, but there are challenges in the setting of CA and DHCA, where BBB is often intact or minimally affected.
Minocycline is a widely used antibiotic that readily passes BBB.24 Minocycline demonstrated anti-inflammatory and anti-apoptotic properties in several models of neurologic injury.25–30 The primary effect of minocycline is probably inhibition of activation of microglia30 although a number of direct neuroprotective effects have also been shown in neurons.31,32 Minocycline thus could have potential value in CA — for example — as an adjunct to hypothermia.33,34
In this study, we hypothesized that after rapid lethal hemorrhage and prolonged hypothermic CA 1) TNF-α will be increased in brain, 2) minocycline will attenuate TNF-α production, and 3) microglia will be the major source of TNF-α. Naïve rats and rats subjected to CPB were used as controls. In order to assess possible regional differences, four distinct brain regions were studied.
Methods
The study was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. We used our previously established model of rapid hemorrhage followed by prolonged hypothermic CA resuscitated with CPB5 (Figure 1) that has been described in detail before.
Figure 1.

Schematic representation of the experiment. Dotted line represents mean arterial pressure (left axis) and the dashedline represents tympanic temperature of the rats subjected to cardiac arrest.
In brief, adult male Sprague-Dawley rats were anesthetized with isoflurane, intubated and mechanically ventilated with FiO2 0.5. The left femoral artery and vein were cannulated for blood pressure monitoring and blood sampling. The right femoral artery and right jugular vein were cannulated for CPB resuscitation. Rectal and tympanic temperatures were monitored. Baseline blood samples were obtained, and hemodynamic values were recorded. Heparin sodium (200 units) was administered to prevent clotting.
After instrumentation, intubated rats were weaned to spontaneous ventilation with FiO2 0.3 and isoflurane 2%. Rapid exsanguination (12.5 ml of blood over 5 min) was performed via the internal jugular catheter. The shed blood was collected at room-temperature and used immediately for priming of the CPB circuit. After the rapid exsanguination phase, CA was ensured with intravenous administration of 9 mg of esmolol (0.9 ml) and 0.2 mEq of potassium chloride (0.1 ml). After 5 min of CA, hypothermia was induced with 270 ml of room-temperature flush solution (Plasma-Lyte A, Baxter) administered via the right femoral artery catheter at 50 ml/min. The flush was drained from the jugular vein catheter.
After 20 min of CA, resuscitation was started with CPB primed with the shed blood. After 5 min reperfusion, minocycline (90 mg/kg) or same volume of vehicle (PBS) were given according to randomization. Heating and cooling were achieved by warming the returned blood from CPB, heated blanket and overhead lamp. Blood samples for biochemistry and hematology were obtained every 15 min during CPB resuscitation and then every hour to guide treatment during the intensive care unit (ICU) phase. The samples were processed immediately using a point-of-care blood analyzer (Stat Profile, Nova Biomedical; Waltham, MA).
Arterial blood gas management followed alpha-stat principles. pH and electrolyte values outside of the normal range were corrected during CPB and ICU phases by adjustments in ventilation and/or administration of sodium bicarbonate, calcium chloride. Additional blood obtained from an isoflurane-anesthetized donor rat was used to maintain hematocrit > 25%. The blood was withdrawn immediately prior to the experiment, stored in a syringe at room temperature and transfused over 1 h during the CPB-resuscitation phase. CPB support was gradually discontinued after 60 min. Mechanical ventilation with an FiO2 of 1.0 was continued while maintaining normocapnia. Rats scheduled for sacrifice at 24 h were weaned from mechanical ventilation, decannulated, extubated and placed in a cage with supplemental O2 at 1 L/min, resulting in FiO2 ~ 0.3. At 6 h or 24 h after resuscitation, rats were deeply anesthetized with isoflurane, intubated, mechanically ventilated and perfused with heparinized ice-cold normal saline. The rats were then decapitated, brains removed and dissected into four regions of interest: cortex (CTX), striatum (STRI), hippocampus (HIP) and cerebellum (CEREB). Each of these regions is well known to develop selectively vulnerable neuronal death. The samples were snap-frozen in liquid nitrogen and then stored at −70 °C freezer until further processing.
Since others indicated that exposure to artificial materials of an experimental CPB circuit used in our study induces systemic and cerebral inflammation,6,7 we included a control group that was subjected to the same duration (60 min) of CPB but without preceding hemorrhage or CA. The temperature profile of this group was designed to mirror the CA group. We have also studied sham animals subjected to the same cannulation and anesthesia, without ischemic insult or CPB. This group was kept normothermic.
Rats were randomly assigned to four groups, n=6 per group: 1) sham rats (sham); 2) CPB controls (CPB); 3) CA group (CA); 4) CA treated with minocycline (CA+M).
Additional rats in group CA sacrificed at 6 h were used for immunocytochemistry (n=6) or at 24 h for ELISA TNF-α (n=6).
In additional experiments, we have screened the possibility of invasion of macrophages into the brain after hypothermic CA. Micrometer sized paramagnetic iron oxide particles (MPIO, 9 mg Fe/kg) were injected intravenously in shams (negative control), hypothermic CA or traumatic brain injury (TBI, positive control). Migration of these macrophages into the brain after 24 h was tracked ex-vivo using 11.7 T magnetic resonance imaging.
Assessment of TNF-α levels
Each tissue region was homogenized in 5× volume of 1X PBS. Homogenates were centrifuged at 14000g for 30 min and supernatant was retained. Protein concentration was measured using the BCA assay (Pierce). TNF-α was measured by enzyme-linked immunosorbent assay (ELISA TNF-α kit, R&D Systems) according to manufacturer’s instructions. The final concentrations were then adjusted for the protein content in the sample.
Immunocytochemistry
Samples were mounted in OTC media and 10 μm sections were cut and mounted on Superfrost slides (Fisher). Tissue sections were fixed for 10 min in acetone, rinsed with 1× TBST and blocked in 3% Normal Horse Serum for 30 min at room temperature. Slides were washed in 1× TBST and incubated overnight at 4 °C in primary antibody (TNF-α 1:40, R&D Systems, NeuN 1:100, Millipore). The next day, sections were rinsed 3 times with 1× TBST and incubated for 1 hour at room temperature in secondary antibody (donkey anti-goat Alexa 488 for TNF-α, horse anti-rabbit Alex 594 for NeuN, both at 1:400 dilution). Sections were rinsed, coverslipped with aqueous mounting media and visualized for co-localization. For Iba-1 staining, the adjacent 10um sections were used, but fixed in 4% paraformaldehyde for 10 minutes. The same procedure was used above with a 1:250 dilution for Iba-1 (Wako Chemicals) and a goat anti-rabbit Alexa 594 as the secondary antibody.
Statistical analysis
Repeated measures analysis of variance (ANOVA) was used to compare heart rate, mean arterial pressure and tympanic and rectal temperatures between groups. One-way ANOVA was used to compare physiologic and biochemical data and TNF-α levels between groups or regions. Post-hoc Tukey’s test was used. A p value < 0.05 was considered statistically significant.
Results
All rats survived the insult until the scheduled timepoint of sacrifice.
There were no differences in physiologic or biochemical data between groups at baseline. Physiologic parameters (heart rate, blood pressure) and temperature profiles during resuscitation are shown in Figs. 2 and 3. Heart rate was significantly higher in shams vs. other groups (p<0.01) (Fig. 2, top panel). Mean arterial pressure profiles were similar in CA and CA+M groups, with significant differences vs. sham (p<0.01) or CPB (p<0.05) groups, respectively (Fig. 2, bottom panel). Tympanic and rectal temperature profiles were also similar in both CA groups, but differed vs. shams or CPB control group (Fig. 3).
Figure 2.

Heart rate (top panel) and mean arterial pressure (bottom panel) after prolonged hypothermic CA. Heart rate: p<0.01sham vs. other groups. Mean arterial pressure: p<0.01 sham vs. CA or CA+M groups; p<0.05 CPB vs. CA or CA+M groups. BL, baseline; HS, end of hemorrhagic shock; CA, cardiac arrest; CPB, cardiopulmonary bypass; ICU, intensive care.
Figure 3.

Tympanic (Tty, top) and rectal (Trec, bottom) temperatures during CA. Tty: p<0.01 sham vs. other groups; CA vs. CPB. p<0.05 CPB vs. CA+M. Trec: p<0.01 CA or CA+M vs. other groups, and sham vs. CPB. CA, cardiac arrest.
Prolonged CA resulted in marked physiologic and biochemical disturbances, including extremely low pH <7.0, BE exceeding −20, and increased lactate up to 6 mmol/L. These changes were gradually improved during resuscitation and were largely ameliorated by the end of the ICU phase. Minor changes were also observed in the CPB group. (Table 1)
Table 1.
Biochemical and hematological values after prolonged cardiac arrest.
| BL | CPB5 | ICU15 | ICU120 | ICU300 | ||
|---|---|---|---|---|---|---|
| pHa | sham | 7.39±0.03 | 7.39±0.05abc | 7.39±0.01f | 7.40±0.04 | 7.37±0.03 |
| CPB | 7.41±0.06 | 7.19±0.17bcd | 7.26±0.06eg | 7.32±0.06g | 7.33±0.06 | |
| CA | 7.41±0.02 | 6.95±0.09ef | 7.36±0.06a | 7.41±0.06a | 7.37±0.06 | |
| CA+M | 7.37±0.05 | 6.93±0.10ef | 7.34±0.07 | 7.41±0.07 | 7.32±0.07 | |
| paO2 | sham | 259±65 | 384±69 | 403±25 | 364±105 | 420±61 |
| CPB | 304±64 | 460±77 | 338±64 | 396±130 | 434±44 | |
| CA | 224±44 | 451±73 | 323±74 | 394±62 | 414±70 | |
| CA+M | 256±80 | 482±55 | 370±63 | 356±78 | 414±53 | |
| paCO2 | sham | 39±5 | 41±2bc | 40±5 | 37±5 | 41±3 |
| CPB | 37±7 | 48±8bc | 44±9 | 38±2 | 38±4 | |
| CA | 39±5 | 31±5ef | 43±7 | 40±7 | 42±9 | |
| CA+M | 46±9 | 28±4ef | 43±5 | 38±7 | 36±8 | |
| BE | sham | −0.5±1.7 | 0.1±1.4bcf | −0.2±2.3f | −1.6±1.6 | −1.3±1.7 |
| CPB | −0.8±1.0 | −5.9±1.3bce | −7.0±2.5ebh | −6.0±3.0gh | −5.0±2.8 | |
| CA | −0.3±1.8 | −22.9±2.7ef | −2.0±2.4f | 0.9±5.3a | −1.0±4.6 | |
| CA+M | 1.8±1.0 | −24.2±2.3ef | −2.6±2.4 | 0.5±3.1a | −5.6±6.1 | |
| Lactate | sham | 1.2±0.5 | 1.4±0.6bc | 1.5±0.6bc | 1.8±0.3gh | 2.2±0.4c |
| CPB | 1.9±0.1 | 2.5±0.6bc | 4.8±2.4h | 4.7±1.7 | 3.1±0.8h | |
| CA | 1.2±0.6 | 6.6±1.0ef | 6.9±2.9e | 5.3±3.2d | 4.6±2.4 | |
| CA+M | 1.0±0.4 | 5.4±1.6ef | 8.6±2.0ae | 5.6±1.2d | 7.3±3.4ae | |
| Hct | sham | 40±4 | 42±3bcf | 40±4bcf | 37±3 | 40±5 |
| CPB | 41±2 | 26±3eh | 27±3e | 32±2 | 33±2 | |
| CA | 40±5 | 25±3e | 28±3e | 36±5 | 36±4 | |
| CA+M | 37±3 | 20±4ae | 30±3e | 37±4 | 35±7 | |
| Glucose | sham | 256±62 | 266±53 | 273±61 | 243±77bc | 230±78agh |
| CPB | 340±63h | 292±52 | 358±55b | 167±48 | 128±26d | |
| CA | 233±61 | 223±59 | 200±74f | 117±29e | 121±48d | |
| CA+M | 212±51a | 302±59 | 265±53 | 96±36e | 134±59d |
BL = baseline, CPB5 = 5 min after start of CPB, ICU15 = 15 min after weaning from CPB, ICU120= 2 h after weaning from CPB, ICU300 = at 6 h resuscitation time.
p<0.05 vs. CPB;
p<0.01 vs. CA;
p<0.01 vs. CA+M;
p<0.05 vs. sham;
p<0.01 vs. sham;
p<0.01 vs. CPB;
p<0.05 vs. CA;
p<0.05 vs. CA+M.
Shams and CPB controls had no detectable TNF-α in any brain region. CA markedly increased brain TNF-α in all regions compared to shams or CPB controls. Striatum showed an early specific increase in TNF-α levels after CA. The levels measured in the striatum were 10-fold higher than in the hippocampus or other brain regions. Minocycline decreased TNF-α levels in CTX, HIP and STRI by ~ 50% but did not totally ablate TNF-α increase. (Fig. 4) At 24 h, the levels of TNF-α were undetectable across all regions (data not shown).
Figure 4.
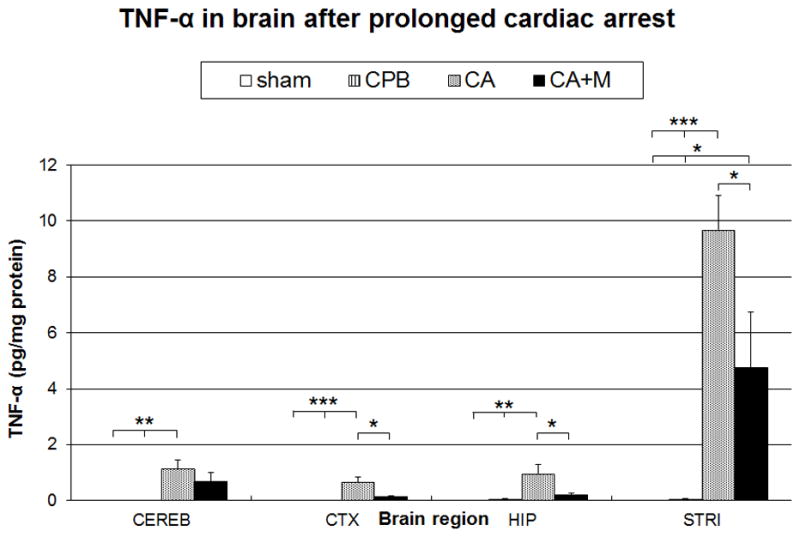
TNF-α protein levels in individual brain regions at 6 hours after prolonged cardiac arrest. A unique increase of TNF-α is observed in the striatum. Minocycline attenuated the TNF-α increase in selected brain regions.* p<0.05; ** p<0.01; *** p<0.001
TNF-α showed selective co-localization with NeuN, identifying neurons. (Fig. 5) The morphological type and cell density of TNF-α positive cells did not correspond with microglia (Figure 6).
Figure 5.
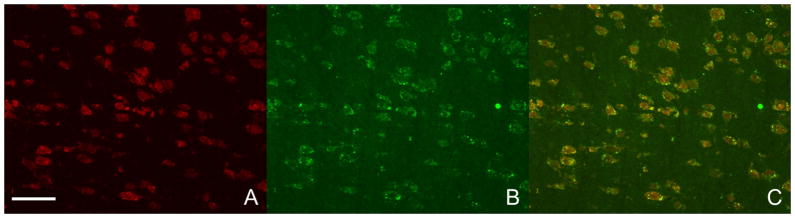
Prolonged 20 min cardiac arrest resulted in a massive TNF-α expression in the striatum (A, NeuN staining visualizing neurons; B, TNF-α staining; C, merge A&B). The TNF-α-expressing cells co-localize with neurons (striatum, 20x). The marker in panel A represents 40 μm.
Figure 6.
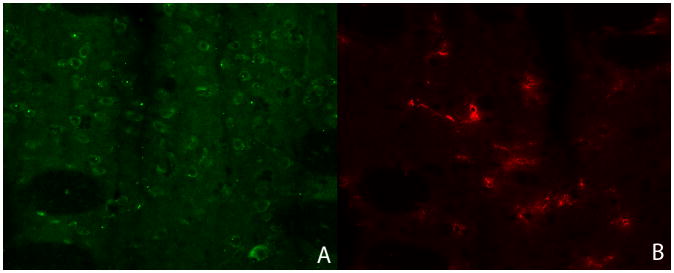
TNF-alpha positive cells (green, panel A) and microglia (red, panel B) in striatum show different morphology and cell density, further supporting our findings that microglia are an unlikely to be a major source of the TNF-alpha production at 6 h.
No difference in MPIO-labeled macrophage invasion between shams and rats subjected to hypothermic CA was appreciated, while rats subjected to TBI showed massive macrophage infiltration (Supplemental File 1).
Discussion
In our model of prolonged DHCA, we report 1) a marked increase of TNF-α across multiple brain regions, and 2) a unique finding of an early selective increase in TNF-α levels in the striatum, 10-fold higher than other brain regions studied. Using immunocytochemistry, we identified that the increased TNF-α colocalized with neurons. Minocycline was able to attenuate the increase in most regions, but did not fully mitigate TNF-α production.
The severe hemodynamic and physiologic derangements resulting from the insult are consistent with our previous reports, documenting a good reproducibility of the model. Importantly, there were no differences in any parameters between CA and CA+M groups, respectively, speaking against a major hemodynamic or physiologic effect of minocycline that could independently affect regional brain TNF-α production after prolonged hypothermic CA.
TNF-α
Multiple pro- and anti-inflammatory cytokines were identified and studied in experimental models of brain ischemia and in clinical settings.35 In our study, we focused on TNF-α as an important cytokine mediating neuronal injury. Our choice of TNF-α as a footprint for neurologic damage is supported by a body of evidence suggesting that TNF-α is linked to worse neurologic outcome, although some reports ascribed beneficial effects to TNF-α.36 Neutralization of TNF-α after focal brain ischemia models in rats led to decreased infarct volume and cerebral edema.37 In transgenic mice overexpressing TNF-α, ischemia led to a five-fold increase in TNF-α in the border zone approximating the ischemic penumbra and the infarct core, and to a worse neurologic outcome, including larger infarct volume and increased neuronal apoptosis.38 In contrast, others reported no differences between wild type TNF-α mice and TNF-α gene-deficient mice in hippocampal neuronal damage after global brain ischemia.39 This is in line with our findings that TNF-α may not be the key mediator in hippocampus but could play a major role in other brain regions.
The effect of TNF-α can be also dependent on the level of its expression. In the nigrostriatal dopaminergic circuit of adult mice, chronic low expression of TNF-α was neuroprotective, while high expression of TNF-α produced neurodegenerative effects associated with gliosis and inflammatory infiltration.40
The finding of TNF-α co-localization with neurons in striatum is surprising but does have support in the literature. Under physiological situations, neurons are the only CNS cells expressing TNF-α.41 After incomplete brain ischemia in rats, early increase of of TNF-α mRNA was observed in the cortex as early as at 3 h, peaking at 12 h. TNF-α protein was found associated with neurons in the ischemic cortex at 6 h, before neutrophil or macrophage infiltration. Additionally, scattered TNF-α immunoreactive neurofilaments were observed in the striatum at 12–24 h. No colocalization with the astrocytes was seen. Unfortunately, microglia were not studied.18 Early colocalization of TNF-α in neurons in that study agrees with our current work in striatum after DHCA.
After focal cerebral ischemia, TNF-α was found in the neural processes, and to a moderate extent also in neuronal soma and glia. Many of TNF-α expressing neurons were in close proximity to activated microglial cells, which exhibited a high level of TNF-α immunoreactivity.42
Yasuda et al. studied neuronal degeneration, gliosis and temporospatial cytokine profile in the hippocampus in a global brain ischemia model. Extensive hippocampal CA1 cell loss was accompanied by reactive gliosis. An early isolated peak of TNF-α at 6 h was seen. After a period of low TNF-α levels, a second period of four-fold higher levels stretched from day 7 to 21. The source of TNF-α were not identified.43 Given the similarity with our model, it is possible that the early release of TNF-α may have originated from a separate source, possibly neurons, while delayed TNF-α increase coincided with increased microglial proliferation and activation.
Regionality
Hippocampus and especially its CA1 area are considered the most vulnerable region to ischemic insults. Moreover, recent findings showed that important regional differences in response of individual brain regions exist. This may require individual, region-specific treatment. Our model is characterized with extensive neuronal degeneration associated with microglial activation and proliferation. At one week after the insult, there were ~ 30% Fluoro-Jade B positive neurons in the hippocampus, and ~38% in striatum, respectively. CA triggered an increase of TNF-α in all regions that may have biological relevance, but they were dwarfed by the marked increase in striatum early after DHCA.
Saito et al. previously reported increased TNF-α levels in both hippocampus and striatum early after global ischemia model in gerbils, with levels in hippocampus higher than in striatum.44 Several prior reports identified striatum as a selectively vulnerable region. In a neonatal piglet model of DHCA, the ratio of pro- vs. anti-apoptotic proteins was the least favorable in striatum, compared to hippocampus or cortex.45 It has also been reported that striatum can react to remote injury, e.g. in hippocampus, by elaborating cytokines.46
Hypothermia
Our model used moderate intra-arrest hypothermia, followed by a prolonged mild hypothermia as an integral part of the current clinical post-resuscitative paradigm. Experimentally, hypothermia effectively ameliorated neuronal degeneration after global and focal brain ischemia in multiple models.47,48
The exact mechanisms by which hypothermia exerts its effects are not yet fully understood. Effects of hypothermia on brain cytokine production after ischemic insults have not been studied systematically. The effect of hypothermia itself was not explored in our study since both groups subjected to CA followed an identical hypothermic protocol.
Our current results indicate that minocycline attenuates TNF-α which is to the greatest extent increased in the striatum. This region was not previously studied in our model. We cannot rule out that benefits previously seen with minocycline or deeper hypothermia were stemming from effects exerted outside hippocampus.
Minocycline
We chose minocycline as a treatment based on previous reports of its beneficial effects in multiple models, including global and focal brain ischemia, TBI, spinal cord injury and intracerebral hemorrhage.25 Both motor and neurocognitive behavior were improved by treatment with minocycline, even when the initiation of treatment was delayed for several hours after the insult.49 We used high-dose intravenous administration of minocycline to ensure adequate brain tissue levels.50
Minocycline is considered to target primarily microglia, but effects on other CNS cells including oligodendrocytes,51 astrocytes52 or neurons31,32,49 have been described. Most importantly, minocycline attenuated cell death induced by oxygen-glucose deprivation in neuronal culture (without microglia) to the same extent as TNF-αantibody, supporting a possible direct effect of minocycline on neurons.53
Studies in brain ischemia models showed beneficial effects of minocycline on behavioral outcome in juvenile rats,26,54,55 but only a limited effect in adult rats.56 One study reported a positive effect of minocycline but not hypothermia after focal brain ischemia in adult rats.33 These effects were suggested to be mediated by microglia,26 or independent of microglia.55 Several cytokines were altered by minocycline treatment in the developing rats, but no effect on TNF-α was seen.54 This argues for a different pattern of neuroinflammatory response to CA, depending on the developmental stage of the animal. These findings along with our current results identifying neurons as the likely source of early TNF-α may explain the relative lack of a definitive effect of minocycline in our prior study using an identical model.
Sanchez Mejia et al. showed that minocycline was able to ameliorate activation of caspases and neuronal death early after TBI. Of note, caspase-1 and -3 were also localized specifically in neurons.30 It should be noted that the aforementioned studies usually used acute minocycline treatment, usually limited to three days. Prolonged administration of minocycline over 4 weeks resulted in improved neurobehavioral deficits and reduced microgliosis.57
Control groups
We included a control group subjected to CPB without CA to explore the effects of CPB in our model that incorporated CPB as an integral part of the resuscitation efforts. Jungwirth et al. previously described increased cerebral TNF-α mRNA expression 4 h after CPB vs. shams.58 In our study, we were unable to detect TNF-α protein 6 h after CPB in any of the regions. It is acknowledged that increased mRNA expression does not need to be translated into an increased protein production, or the production could be delayed. Our CPB control group used hypothermia (25–34 °C) to mirror the hypothermic phase of CPB-resuscitation in the CA groups, contrasting normothermia maintained in the study by Jungwirth et al. This could have ameliorated TNF-α induction in brain.
Our study has certain limitations. We were unable to include a normothermic control group that would eliminate the protective effects of hypothermia that could have blunted the effect of minocycline. We have demonstrated in the model development feasibility study that normothermic controls are unable to survive this insult.1 This is certainly not surprising given that we are modeling a 20 min CA insult that can only be survived with hypothermia. We explored two early timepoints after resuscitation. It is possible that earlier or later phases of reperfusion could produce different results, and other CNS cells may be involved at different stages.
The use of FiO2 1.0 after resuscitation from CA is controversial but may aggravate injury.59–61 In addition, the impact of of hyperoxia post CA on cytokines in brain remains unclear. Systemic cytokine levels were increased with hyperoxia after experimental CPB in a rat62 but brain tissue levels were not studied.
The role of TNF-α identified early after CA in neurons in our study is not yet elucidated. Future studies aimed at selective attenuation of the TNF-α surge in the striatum may help to identify whether TNF-α is linked with cell death by apoptosis and necroptosis or cell survival cascades in a cause-and-effect relationship. Based on our findings its role in striatal neuronal death deserves to be explored.
In conclusion, we report unique regional differences in early TNF-α production after prolonged hypothermic CA in rats resuscitated with CPB, identifying striatum as a region with the highest TNF-α levels. Surprisingly, TNF-α co-localized with neurons. Minocycline attenuated the TNF-α increase in most brain regions. Our data suggest that if neuroinflammatory pathways contribute to neuronal death early after CA, region specific differences in these pathways could play important roles in mediating selective vulnerabilities.
Supplementary Material
Migration of macrophages labeled with micrometer sized paramagnetic iron oxide particles s into the brain at 24 h after the injury. No difference between shams (panel A) and rats subjected to hypothermic CA (panel B) was appreciated, while rats subjected to TBI showed massive macrophage infiltration (panel C). These results argue against a role for circulatory inflammatory cells in our model.
Acknowledgments
Dr. Drabek was supported by the American Heart Association Beginning Grant-in-Aid #09BGIA2310196. Dr. Janatawas supported by the Laerdal Foundation for Acute Medicine and the Erwin Schroedinger Stipend. Dr. Kochanek was supported by NS30318.
Footnotes
All other authors report no conflict of interest.
Conflict of Interest statement: Dr. Drabek was supported by the American Heart Association Beginning Grant-in-Aid #09BGIA2310196.
Dr. Janata was supported by the Laerdal Foundation for Acute Medicine and the Erwin Schroedinger Stipend. Dr. Kochanek was supported by NS30318.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drabek T, Stezoski J, Garman RH, Wu X, Tisherman SA, Stezoski SW, Fisk JA, Jenkins L, Kochanek PM. Emergency preservation and delayed resuscitation allows normal recovery after exsanguination cardiac arrest in rats: a feasibility trial. Crit Care Med. 2007;35:532–7. doi: 10.1097/01.CCM.0000253398.61666.0D. [DOI] [PubMed] [Google Scholar]
- 2.Wu X, Drabek T, Kochanek PM, Henchir J, Stezoski SW, Stezoski J, Cochran K, Garman R, Tisherman SA. Induction of profound hypothermia for emergency preservation and resuscitation allows intact survival after cardiac arrest resulting from prolonged lethal hemorrhage and trauma in dogs. Circulation. 2006;113:1974–82. doi: 10.1161/CIRCULATIONAHA.105.587204. [DOI] [PubMed] [Google Scholar]
- 3.Wu X, Drabek T, Tisherman SA, Henchir J, Stezoski SW, Culver S, Stezoski J, Jackson EK, Garman R, Kochanek PM. Emergency preservation and resuscitation with profound hypothermia, oxygen, and glucose allows reliable neurological recovery after 3 h of cardiac arrest from rapid exsanguination in dogs. J Cereb Blood Flow Metab. 2008;28:302–11. doi: 10.1038/sj.jcbfm.9600524. [DOI] [PubMed] [Google Scholar]
- 4.Sailhamer EA, Chen Z, Ahuja N, Velmahos GC, de Moya M, Rhee P, Shults C, Alam HB. Profound hypothermic cardiopulmonary bypass facilitates survival without a high complication rate in a swine model of complex vascular, splenic, and colon injuries. J Am Coll Surg. 2007;204:642–53. doi: 10.1016/j.jamcollsurg.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Drabek T, Tisherman SA, Beuke L, Stezoski J, Janesko-Feldman K, Lahoud-Rahme M, Kochanek PM. Deep hypothermia attenuates microglial proliferation independent of neuronal death after prolonged cardiac arrest in rats. Anesth Analg. 2009;109:914–23. doi: 10.1213/ane.0b013e3181b0511e. [DOI] [PubMed] [Google Scholar]
- 6.Jungwirth B, Eckel B, Blobner M, Kellermann K, Kochs EF, Mackensen GB. The impact of cardiopulmonary bypass on systemic interleukin-6 release, cerebral nuclear factor-kappa B expression, and neurocognitive outcome in rats. Anesth Analg. 2010;110:312–20. doi: 10.1213/ANE.0b013e3181bbc42e. [DOI] [PubMed] [Google Scholar]
- 7.Jungwirth B, Kellermann K, Qing M, Mackensen GB, Blobner M, Kochs EF. Cerebral tumor necrosis factor alpha expression and long-term neurocognitive performance after cardiopulmonary bypass in rats. J Thorac Cardiovasc Surg. 2009;138:1002–7. doi: 10.1016/j.jtcvs.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10:119–30. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 9.Sriram K, O’Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–53. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- 10.Pan W, Zadina JE, Harlan RE, Weber JT, Banks WA, Kastin AJ. Tumor necrosis factor-alpha: a neuromodulator in the CNS. Neurosci Biobehav Rev. 1997;21:603–13. doi: 10.1016/s0149-7634(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 11.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 12.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 13.Perry VH, Bell MD, Brown HC, Matyszak MK. Inflammation in the nervous system. Curr Opin Neurobiol. 1995;5:636–41. doi: 10.1016/0959-4388(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 14.Uno H, Matsuyama T, Akita H, Nishimura H, Sugita M. Induction of tumor necrosis factor-alpha in the mouse hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:491–9. doi: 10.1097/00004647-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol. 1990;144:2999–3007. [PubMed] [Google Scholar]
- 16.Orzylowska O, Oderfeld-Nowak B, Zaremba M, Januszewski S, Mossakowski M. Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci Lett. 1999;263:72–6. doi: 10.1016/s0304-3940(99)00043-9. [DOI] [PubMed] [Google Scholar]
- 17.Sairanen TR, Lindsberg PJ, Brenner M, Siren AL. Global forebrain ischemia results in differential cellular expression of interleukin-1beta (IL-1beta) and its receptor at mRNA and protein level. J Cereb Blood Flow Metab. 1997;17:1107–20. doi: 10.1097/00004647-199710000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–8. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 19.Munoz-Fernandez MA, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–40. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 20.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–44. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 21.Dziewulska D, Mossakowski MJ. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin Neuropathol. 2003;22:35–40. [PubMed] [Google Scholar]
- 22.Xiong M, Yang Y, Chen GQ, Zhou WH. Post-ischemic hypothermia for 24h in P7 rats rescues hippocampal neuron: association with decreased astrocyte activation and inflammatory cytokine expression. Brain Res Bull. 2009;79:351–7. doi: 10.1016/j.brainresbull.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Yang ZJ, Carter EL, Kibler KK, Kwansa H, Crafa DA, Martin LJ, Roman RJ, Harder DR, Koehler RC. Attenuation of neonatal ischemic brain damage using a 20-HETE synthesis inhibitor. J Neurochem. 2012;121:168–79. doi: 10.1111/j.1471-4159.2012.07666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saivin S, Houin G. Clinical pharmacokinetics of doxycycline and minocycline. Clin Pharmacokinet. 1988;15:355–66. doi: 10.2165/00003088-198815060-00001. [DOI] [PubMed] [Google Scholar]
- 25.Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist. 2005;11:308–22. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- 26.Fan LW, Lin S, Pang Y, Rhodes PG, Cai Z. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur J Neurosci. 2006;24:341–50. doi: 10.1111/j.1460-9568.2006.04918.x. [DOI] [PubMed] [Google Scholar]
- 27.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–74. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52:54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 29.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–9. doi: 10.1097/00006123-200106000-00051. discussion 1399–401. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez JC, Egea J, Del Carmen Godino M, Fernandez-Gomez FJ, Sanchez-Prieto J, Gandia L, Garcia AG, Jordan J, Hernandez-Guijo JM. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca(2+) signalling in hippocampal neurons. Eur J Neurosci. 2007;26:2481–95. doi: 10.1111/j.1460-9568.2007.05873.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhong J, Lee WH. Hydrogen peroxide attenuates insulin-like growth factor-1 neuroprotective effect, prevented by minocycline. Neurochem Int. 2007;51:398–404. doi: 10.1016/j.neuint.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Wang CX, Yang T, Noor R, Shuaib A. Delayed minocycline but not delayed mild hypothermia protects against embolic stroke. BMC Neurol. 2002;2:2. doi: 10.1186/1471-2377-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang CX, Yang T, Shuaib A. Effects of minocycline alone and in combination with mild hypothermia in embolic stroke. Brain Res. 2003;963:327–9. doi: 10.1016/s0006-8993(02)04045-3. [DOI] [PubMed] [Google Scholar]
- 35.Szelenyi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–38. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 36.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–94. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 37.Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, Kohno M. Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:959–67. doi: 10.1038/sj.jcbfm.9600086. [DOI] [PubMed] [Google Scholar]
- 38.Pettigrew LC, Kindy MS, Scheff S, Springer JE, Kryscio RJ, Li Y, Grass DS. Focal cerebral ischemia in the TNFalpha-transgenic rat. J Neuroinflammation. 2008;5:47. doi: 10.1186/1742-2094-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakami Y, Saito K, Hara A, Zhu Y, Sudo K, Niwa M, Fujii H, Wada H, Ishiguro H, Mori H, Seishima M. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J Neurochem. 2005;93:1616–22. doi: 10.1111/j.1471-4159.2005.03163.x. [DOI] [PubMed] [Google Scholar]
- 40.Chertoff M, Di Paolo N, Schoeneberg A, Depino A, Ferrari C, Wurst W, Pfizenmaier K, Eisel U, Pitossi F. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor alpha in the nigrostriatal dopaminergic circuit of adult mice. Exp Neurol. 2011;227:237–51. doi: 10.1016/j.expneurol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 41.Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB. Distribution and characterization of tumor necrosis factor-alpha-like immunoreactivity in the murine central nervous system. J Comp Neurol. 1993;337:543–67. doi: 10.1002/cne.903370403. [DOI] [PubMed] [Google Scholar]
- 42.Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–81. [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Tsuchihashi Y, Kawai Y, Naruse S, Fujita S. Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. J Neuroinflammation. 2011;8:70. doi: 10.1186/1742-2094-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149–52. doi: 10.1016/s0304-3940(96)12460-5. [DOI] [PubMed] [Google Scholar]
- 45.Pirzadeh A, Mammen A, Kubin J, Reade E, Liu H, Mendoza A, Greeley WJ, Wilson DF, Pastuszko A. Early regional response of apoptotic activity in newborn piglet brain following hypoxia and ischemia. Neurochem Res. 2011;36:83–92. doi: 10.1007/s11064-010-0267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tchelingerian JL, Quinonero J, Booss J, Jacque C. Localization of TNF alpha and IL-1 alpha immunoreactivities in striatal neurons after surgical injury to the hippocampus. Neuron. 1993;10:213–24. doi: 10.1016/0896-6273(93)90312-f. [DOI] [PubMed] [Google Scholar]
- 47.Colbourne F, Corbett D. Delayed postischemic hypothermia: a six month survival study using behavioral and histological assessments of neuroprotection. J Neurosci. 1995;15:7250–60. doi: 10.1523/JNEUROSCI.15-11-07250.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nozari A, Safar P, Stezoski SW, Wu X, Henchir J, Radovsky A, Hanson K, Klein E, Kochanek PM, Tisherman SA. Mild hypothermia during prolonged cardiopulmonary cerebral resuscitation increases conscious survival in dogs. Crit Care Med. 2004;32:2110–6. doi: 10.1097/01.ccm.0000142700.19377.ae. [DOI] [PubMed] [Google Scholar]
- 49.Hewlett KA, Corbett D. Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience. 2006;141:27–33. doi: 10.1016/j.neuroscience.2006.03.071. [DOI] [PubMed] [Google Scholar]
- 50.Fagan SC, Edwards DJ, Borlongan CV, Xu L, Arora A, Feuerstein G, Hess DC. Optimal delivery of minocycline to the brain: implication for human studies of acute neuroprotection. Exp Neurol. 2004;186:248–51. doi: 10.1016/j.expneurol.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 51.Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2182–90. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McAllister JP, 2nd, Miller JM. Minocycline inhibits glial proliferation in the H-Tx rat model of congenital hydrocephalus. Cerebrospinal Fluid Res. 2010;7:7. doi: 10.1186/1743-8454-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang WC, Qiao Y, Xu L, Kacimi R, Sun X, Giffard RG, Yenari MA. Direct protection of cultured neurons from ischemia-like injury by minocycline. Anat Cell Biol. 2010;43:325–31. doi: 10.5115/acb.2010.43.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–29. doi: 10.1038/jcbfm.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, Ferriero DM, Vexler ZS. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2005;25:1138–49. doi: 10.1038/sj.jcbfm.9600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keilhoff G, Schweizer H, John R, Langnaese K, Ebmeyer U. Minocycline neuroprotection in a rat model of asphyxial cardiac arrest is limited. Resuscitation. 2011;82:341–9. doi: 10.1016/j.resuscitation.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 57.Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke. 2007;38:146–52. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- 58.Sairanen TR, Lindsberg PJ, Brenner M, Carpen O, Siren A. Differential cellular expression of tumor necrosis factor-alpha and Type I tumor necrosis factor receptor after transient global forebrain ischemia. J Neurol Sci. 2001;186:87–99. doi: 10.1016/s0022-510x(01)00508-1. [DOI] [PubMed] [Google Scholar]
- 59.Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R. The effect of hyperoxia following cardiac arrest - A systematic review and meta-analysis of animal trials. Resuscitation. 2012;83:417–22. doi: 10.1016/j.resuscitation.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 60.Yeh ST, Aune SE, Wilgus TA, Parent AE, Angelos MG. Hyperoxemic reperfusion after prolonged cardiac arrest in a rat cardiopulmonary bypass resuscitation model. Resuscitation. 2013;84:114–20. doi: 10.1016/j.resuscitation.2012.08.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–35. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fujii Y, Shirai M, Tsuchimochi H, Pearson JT, Takewa Y, Tatsumi E, Taenaka Y. Hyperoxic Condition Promotes an Inflammatory Response During Cardiopulmonary Bypass in a Rat Model. Artif Organs. 2013 doi: 10.1111/aor.12125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Migration of macrophages labeled with micrometer sized paramagnetic iron oxide particles s into the brain at 24 h after the injury. No difference between shams (panel A) and rats subjected to hypothermic CA (panel B) was appreciated, while rats subjected to TBI showed massive macrophage infiltration (panel C). These results argue against a role for circulatory inflammatory cells in our model.