

Abstract
Electron spin relaxation times at 295 K were measured at frequencies between 250 MHz and 34 GHz for perdeuterated 2,2,6,6-tetramethyl-4-piperidone-1-oxyl (PDT) in five solvents with viscosities that result in tumbling correlation times, τR, between 4 and 50 ps and for three 14N/15N pairs of nitroxides in water with τR between 9 and 19 ps. To test the impact of structure on relaxation three additional nitroxides with τR between 10 and 26 ps were studied. In this fast tumbling regime T2−1 ∼ T1−1 at frequencies up to about 9 GHz. At 34 GHz T2−1 > T1−1 due to increased contributions to T2−1 from incomplete motional averaging of g-anisotropy, and T2−1 − T1−1 is proportional to τR. The contribution to T1−1 from spin rotation is independent of frequency and decreases as τR increases. Spin rotation dominates T1−1 at 34 GHz for all τR studied, and at all frequencies studied for τR = 4 ps. The contribution to T1−1 from modulation of nitrogen hyperfine anisotropy increases as frequency decreases and as τR increases; it dominates at low frequencies for τR > ∼ 15 ps. The contribution from modulation of g anisotropy is significant only at 34 GHz. Inclusion of a thermally-activated process was required to account for the observation that for most of the radicals, T1−1 was smaller at 250 MHz than at 1 to 2 GHz. The significant 15N/14N isotope effect, the small H/D isotope effect, and the viscosity dependence of the magnitude of the contribution from the thermally-activated process suggest that it arises from intramolecular motions of the nitroxide ring that modulate the isotropic A values.
1. Introduction
Nitroxide T2 has been studied extensively, both experimentally and theoretically, to understand motional dynamics in fluid solutions [1-4]. Early studies of T1 focused primarily on ∼9.5 GHz [5-7]. The utility of T1 to measure oxygen concentrations in membranes [8-10] and tissues [11] and the use of nitroxides for in vivo imaging [12,13] has motivated characterization of the frequency dependence of T1. In 1990 it was reported that T1 was smaller at 2.5 GHz than at ∼9.5 GHz for stearic acid spin labels in lipids at 27°C [14]. Subsequently, it was shown that T1 for spin-labeled lipids and for the small molecules CTPO (3-carbamoyl-2,2,5,5-tetramethyl-3-pyrroline-1-oxyl) and tempone (2,2,6,6-tetramethyl-4-piperidone-1-oxyl) increased from ca. 2.5 GHz to 35 GHz, but then became smaller at ca. 95 GHz [15,16]. The frequency dependence up to 35 GHz was modeled in terms of spin rotation, modulation of nitrogen hyperfine anisotropy and g anisotropy, and generalized spin diffusion [17]. Multifrequency measurements for triarylmethyl (trityl) radicals showed that modulation of intra- and inter-molecular electron-proton interactions had significant impact on T1 at 250 MHz but not at ∼9.5 GHz [18], which highlights the need to study the frequency dependence of relaxation.
Extrapolation of the trend for nitroxide T1 between 2.5 and 35 GHz [15,16] predicts that in the frequency range used for in vivo EPR (250 MHz to 1 GHz) T1 would be less than 0.1 μs. However, it has long been known that lines in the spectra of Fremy's salt ((SO3)2NO2−) in aqueous solution at 60 MHz are Lorentzian with T1= T2 = 0.5 μs [19]. Rapid-scan EPR of perdeuterated 2,2,6,6-tetramethyl-4-piperidone-1-oxyl (PDT, 1a) in aqueous solution at 250 MHz [20] found T2 = 0.5 μs. Since T2 ≤ T1, T2 puts a lower limit on T1. These results motivate examination of nitroxide T1 at frequencies lower than 2.5 GHz to determine the dominant relaxation mechanisms at these lower frequencies.
Recently we reported T1 and T2 for aqueous solutions of a nitronyl nitroxide and of PDT at five frequencies between 250 MHz and 34 GHz [21] in the fast motional regime where the tumbling correlation time, τR, is less than about 25 ps. These studies have now been extended to a wider range of nitroxides and solvents to investigate i) the relative importance of spin rotation and modulation of nitrogen hyperfine anisotropy when τR = 4 to 50 ps at ambient temperature, ii) whether additional mechanisms contribute to relaxation at frequencies lower than ∼9.5 GHz, and iii) the thermally-activated process that is proposed to have maximum impact on T1 at 1 to 2 GHz, and has been observed previously in both the fast [21] and slow tumbling regimes [22]. T1 and T2 were measured by pulsed methods for the nitroxides shown in Figure 1, at frequencies between 250 MHz and 34 GHz.
Figure 1.
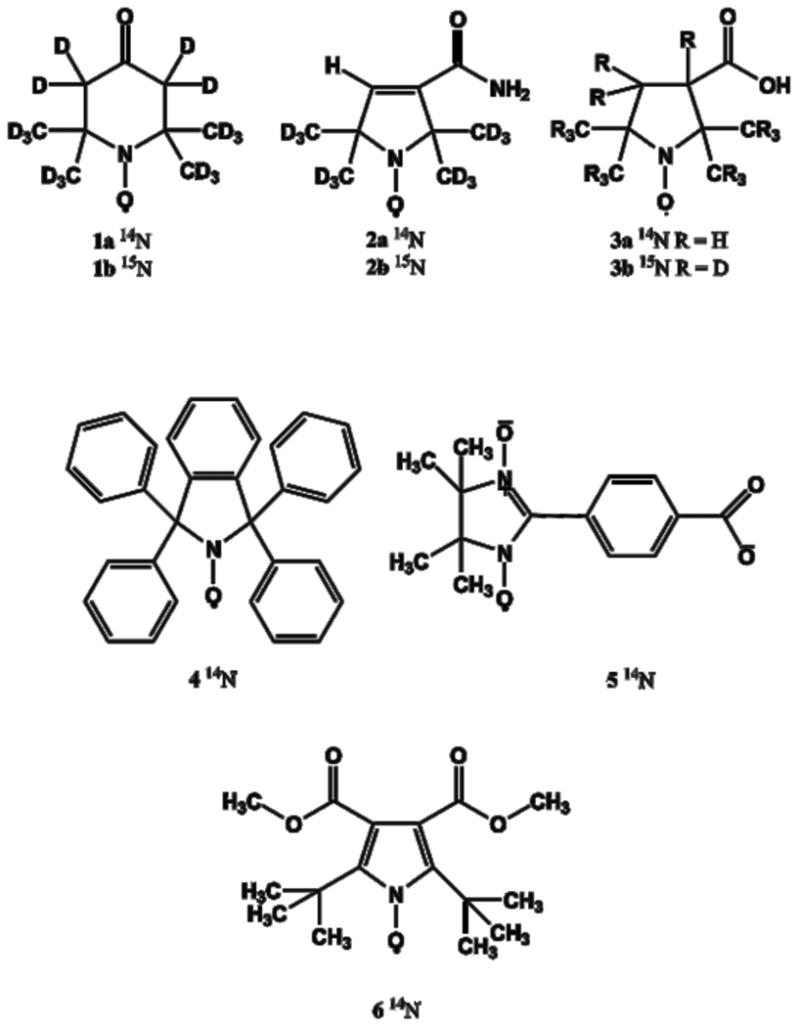
Nitroxide Structures.
Spin-lattice relaxation mechanisms
The importance of spin-rotation (Eq. 1) in the fast tumbling regime was recognized in early discussions of T1−1 for nitroxides and semiquinones [23-25].
| (1) |
where i = x,y,z and ge is 2.0023. (T1SR)−1 is independent of resonance frequency and decreases as τR increases.
Modulation of the large anisotropic (dipolar) nitrogen nuclear hyperfine coupling by molecular tumbling (the END process, Eq. 2) is a major contributor to nitroxide relaxation in certain motional regimes [5,6,17].
| (2) |
where Ai is a component of the nitrogen hyperfine coupling in angular frequency units, Ā is the average nitrogen hyperfine, I is the nitrogen nuclear spin, ω is the resonance frequency, and J(ω) is the Bloembergen, Purcell, Pound (BPP) spectral density function (Eq. 3).
| (3) |
In the rapid-tumbling regime the BPP spectral density function is indistinguishable from the Cole-Davidson spectral density function that has been found to give better agreement than BPP with the temperature dependence of nitroxide T1−1 [26,27].
The frequency dependence of (T1A)−1 depends on τR (Eq. 3). For τR in the range of 4 to 50 ps, ωτR changes from < 1 at 250 MHz to > 1 at 34 GHz. For ωτR > 1, (T1A)−1 increases as ω decreases. For ωτR << 1, (T1A)−1 is independent of ω and increases as τR increases. Because (T1A)−1 is proportional to γ2 I(I+1), where γ is the gyromagnetic ratio, the ratio [(T1A)−1(14N)]/[(T1A)−1(15N)] = 1.36. The 15N/14N isotope effect on T1−1 provides a test of the contribution from the END mechanism [27,28].
Modulation of g anisotropy by molecular tumbling is described by Eq. (4) [5,6].
| (4) |
where Δg = gzz − 0.5(gxx+gyy), δg = 0.5(gxx − gyy), and μB is the electron Bohr magneton. This contribution enhances relaxation for radicals with larger g anisotropy, particularly at higher microwave frequency.
The enhancement of electron spin T1−1 by thermally-activated methyl rotation has been demonstrated in a variety of solid samples [29-32]. Nuclear spin-lattice relaxation enhancement by thermally-activated processes has been studied extensively in NMR [33-36]. For nitroxides in viscous water/glycerol solutions at 1.9, 3.1, and 9.2 GHz at room temperature, T1−1 was larger than predicted by Eqs. (1) – (4) [22]. The additional process dominated relaxation for τR > ∼3×10−9 s, and the magnitude of the contribution increased with decreasing frequency between 9.2 and 1.9 GHz. This contribution was modeled as a thermally-activated process, Eq. (5).
| (5) |
where τtherm = τc0 exp(Ea/RT), Ea is the activation energy, τc0 is the pre-exponential factor, Ctherm is the coefficient for the contribution of the thermally-activated process, and ωref = 9.5 GHz. Data as a function of temperature would be needed to separately evaluate Ea and τc0. Since experiments were performed at a single temperature, these parameters were combined into a single variable τtherm, which was found to be about 1.0×10−10 s. This thermally-activated process was also required to model the frequency dependence of T1−1 for two nitroxides in the rapid-tumbling regime [21].
Nitroxides studied
Several of the radicals selected for this study (Figure 1) have been used for in vivo imaging: PDT (1a) to monitor redox activity [37], mono-hydrogenated deuterated CTPO (mHCTPO, 2a) [38] for in vivo imaging of local oxygen concentration [39,40], and 3-carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl radical (carboxy-proxyl, 3a) for in vivo O2 quantitation in mouse brain [41-43]. T1 and T2 for 1a – 3a, along with their 15N derivatives (denoted as 1b-3b) have been investigated previously at ∼9.5 GHz [28]. Other nitroxides with different ring structures, and g- and A- anisotropies were selected for comparison. The phenyl isoindoline nitroxide (4)[26] contains no methyl groups and the central ring is expected to be more rigid than in 1a – 3a. Nitroxides that are similar to 4, including some that are isotopically labeled, have recently been investigated for in vivo O2 quantitation [44,45]. The nitronyl nitroxide 5 was included because the nitrogen hyperfine splittings are smaller than for 1a – 3a and the g anisotropy is larger [21]. 6 has smaller nitrogen hyperfine splittings than 1a – 3a due to extensive delocalization of the unpaired electron into the aromatic ring, the ring is expected to be more rigid, and the four ring methyls of 1a – 3a are replaced by two t-butyl groups.
Relaxation times T1 and T2 for 1 to 6 were measured at frequencies between 250 MHz and 34 GHz. For 1a τR was varied by changing solvent, including both nonpolar organics and water:glycerol mixtures.
2. Experimental
Samples
1b with > 98% isotope purity were purchased from CDN Isotopes (Quebec, Canada). 1a [28], 3a [46], 3b [46], 4 [26], and 5 [21] were prepared as reported. 2a and 2b were prepared as previously reported [47] and provided by Prof. Howard Halpern (University of Chicago). 6 was prepared as reported [48], and was a gift from Prof. Andre Rassat.
For slower tumbling radicals, 4, 1a in 44% glycerol, and 1a in 69% glycerol, solutions were 0.2 mM. Most solutions were 0.4 to 0.5 mM (Table 1). Between 0.2 mM and 0.5 mM in aqueous solution of 1a – 3a in water there is only a 5 to 15% increase in T1−1 due to Heisenberg exchange [28], so the higher concentrations were used to improve signal-to-noise (S/N), particularly at the lower frequencies. The contribution from Heisenberg exchange is expected to be independent of resonance frequency so it will not alter the frequency dependence of T1−1 that is the focus of this study.
Table 1. Nitroxide Parameters.
| FW | Solvent | Conc. mM |
τC ps |
Aiso G |
Axx G |
Ayy G |
Azz G |
gxx | gyy | gzz | Lit. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 186 | Toluene | 0.4 | 4 | 14.5 | 4.1 | 5.1 | 33.6 | 2.0094 | 2.0063 | 2.0023 | [67] |
| 1a | 186 | Water | 0.5 | 9 | 16.3 | 5.5 | 6.3 | 35.9 | 2.0092 | 2.0061 | 2.0022 | [56] |
| 1a | 186 | Light Mineral Oil | 0.5 | 9 | 14.5 | 4.1 | 5.1 | 33.6 | 2.0094 | 2.0063 | 2.0023 | [67] |
| 1a | 186 | 44% Glycerol | 0.2 | 19 | 16.3 | 5.5 | 6.3 | 35.9 | 2.0092 | 2.0061 | 2.0022 | [56] |
| 1a | 186 | 69% Glycerol | 0.2 | 50 | 16.3 | 5.5 | 6.3 | 35.9 | 2.0092 | 2.0061 | 2.0022 | [56] |
| 1b | 187 | Water | 0.5 | 9 | 22.6 | 7.7 | 8.8 | 50.3 | 2.0092 | 2.0061 | 2.0022 | [56] |
| 2a | 195 | Water | 0.5 | 13 | 16.3 | 5.5 | 6.3 | 35.9 | 2.0092 | 2.0061 | 2.0022 | [68] |
| 2b | 196 | Water | 0.5 | 13 | 22.6 | 7.7 | 8.8 | 50.3 | 2.0092 | 2.0061 | 2.0022 | [68] |
| 3a | 186 | Water | 0.5 | 19 | 16.3 | 5.5 | 6.3 | 35.9 | 2.0092 | 2.0061 | 2.0022 | [68] |
| 3b | 202 | Water | 0.5 | 19 | 22.9 | 7.7 | 8.8 | 50.3 | 2.0092 | 2.0061 | 2.0022 | [68] |
| 4 | 410 | Toluene | 0.2 | 26 | 13.4 | 3.2 | 3.2 | 31.8 | 2.0090 | 2.0058 | 2.0020 | [26,27] |
| 5 | 276 | 2 mM NaOH | 0.5 | 25 | 8.1 | 3.6 | 3.6 | 18.6 | 2.0110 | 2.0065 | 2.0021 | [69] |
| 6 | 310 | Toluene | 0.5 | 10 | 4.7 | 0.4 | 0.4 | 13.2 | 2.0094 | 2.0060 | 2.0024 | [26] |
O2 was removed by one of three methods, depending on sample tube size [21]. Briefly, for large-diameter samples used at lower microwave frequencies, the samples were bubbled with N2 and then flame-sealed. Samples in 4 or 6 mm OD tubes for experiments at 1 to 3 GHz were degassed by several freeze-pump-thaw cycles. N2 was passed over the outside of samples in thin Teflon tubing supported in quartz EPR tubes for experiments at 9 or 34 GHz.
Spectroscopy
Relaxation times were measured at 250 MHz [49], L-band (1.0, 1.5, and 1.9 GHz) [50], S-band (2.5 and 3.0 GHz) [50], X-band (∼9.5 GHz) [51] and Q-band (34.5 GHz) using locally-constructed pulsed EPR spectrometers and resonators and on a Bruker E580 with 9 and 34 GHz bridges and resonators [21]. Measurements were made at 21° ± 2° C. The relaxation times for 1a in 69% glycerol and for 4 at ∼640 MHz were obtained on a spectrometer that will be described separately. Each measurement was performed at least three times. The S/N for T1 was 80-100, except for 1a in 69% glycerol at 630 MHz and for 4 at 640 MHz and 250 MHz where S/N was lower because relaxation times were of the same order as the resonator ringdown time. T2 was measured as the time constant of 2-pulse echo decays. Most T1 measurements were by 3-pulse inversion recovery, and some were also measured by long-pulse saturation recovery. The spin echo decay and saturation recovery curves fit well with a single exponential, using a least squares criterion. A potential complication in the measurement of T1 by inversion recovery is the need to distinguish T1 from nitrogen T1N, which also can contribute to the return of magnetization to equilibrium [16]. At long τR, T1N is shorter than T1, but becomes equal to T1 for short τR [6]. If the S/N is low, it may be difficult to distinguish between one- and two-component decays. At lower resonance frequencies, the inversion recovery curves fit well with a single exponential, as expected when ωτR ≤ ∼1 [6]. For slower tumbling radicals at ∼9.5 GHz and most radicals at 34 GHz, two components were observed in the fits to the inversion recovery curves, as expected for ωτR > 1 [52,53]. The longer relaxation time is assigned as T1. The uniform penalty (UPEN) fitting procedure [54,55] was used to confirm the presence of two contributions to the exponential decays at 34 GHz. Values of T1 at ∼9.5 GHz for 1a in 69% glycerol, 3a, and 4 obtained by saturation recovery with pump times that are long relative to T1 were in good agreement with values obtained by inversion recovery, which confirmed that the inversion recovery experiments were accurately reporting T1.
Values of T1 and T2 for each of the nitrogen hyperfine lines at each of the frequencies studied are listed in supplementary Tables S1 and S2. As observed previously, there is a systematic mI dependence of both T1 and T2 [28]. Values of T1 decrease in the order mI = +1 (low-field) ≥ mI = 0 (center field) > mI = −1 (high-field) for 14N and mI = 0.5 > mI= −0.5 for 15N (Table S1). In tumbling regies where the END process (Eq. 2) is the dominant contribution to T1−1 the ratio of T1 for 14N mI = 1 to mI = −1 is about 1.2, which is in good agreement with prior results [28]. The mI dependence is smaller at concentrations above 0.5 mM (Table S3) where Heisenberg exchange may be significant, and in tumbling regimes where the END mechanism (Eq. 2) is a smaller fraction of the combined contributions to T1−1 Since the available models for the END mechanism do not include an mI dependent contribution, the values of T1−1 plotted in Figures 2, 4, and 5, and included in the modeling, are mI = 0 (14N) or the average for mI = ±0.5 (15N).
Figure 2.
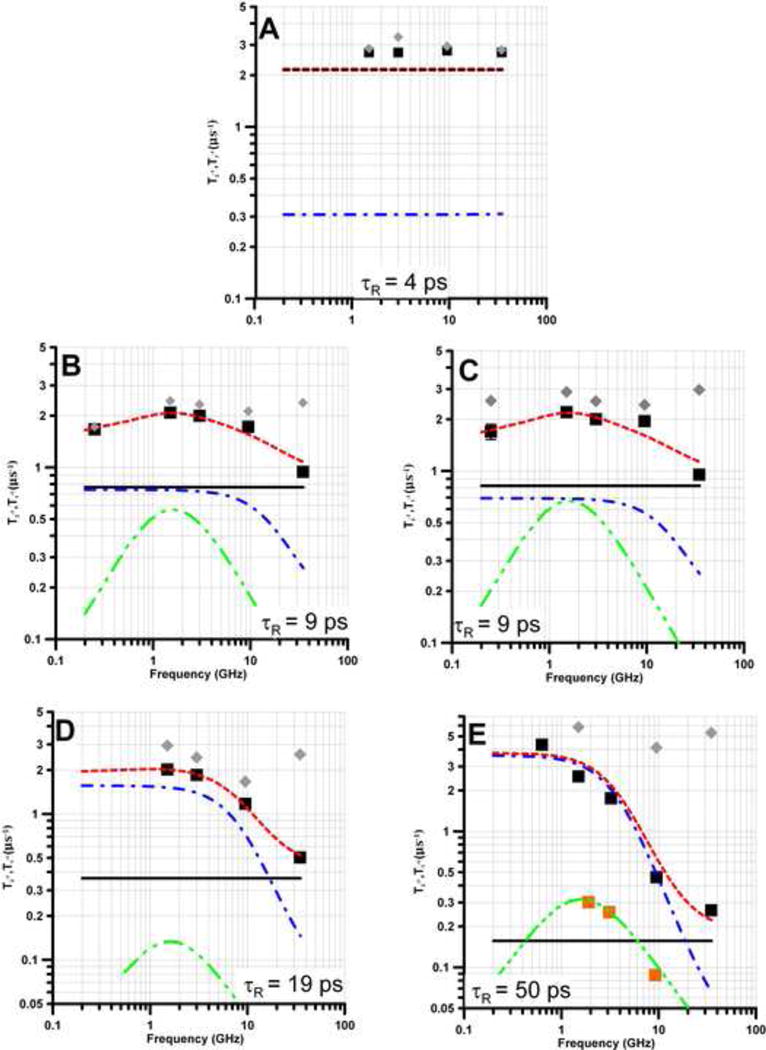
Frequency Dependence of T1−1 (■) and T2−1 (
 ) for PDT (1a) in solvents that result in τR between 4 and 50 ps. (A) toluene, (B) water, (C) light mineral oil, (D) 44% glycerol in water, and (E) 69% glycerol in water. Spin lattice relaxation is modeled as the sum (
) for PDT (1a) in solvents that result in τR between 4 and 50 ps. (A) toluene, (B) water, (C) light mineral oil, (D) 44% glycerol in water, and (E) 69% glycerol in water. Spin lattice relaxation is modeled as the sum (
 ) of contributions from spin rotation (Eq. 1,
) of contributions from spin rotation (Eq. 1,
 ), the modulation of g- and A-anisotropy (Eq. 2 - 4,
), the modulation of g- and A-anisotropy (Eq. 2 - 4,
 ), and a thermally-activated process (Eq. 5,
), and a thermally-activated process (Eq. 5,
 ). In (E) the gold squares and fit line
). In (E) the gold squares and fit line
 are the contribution from the thermally-activated process observed previously for τ > ∼ 3×10−9 s [22]. In (A) the contribution from spin rotation is indistinguishable from the fit line.
are the contribution from the thermally-activated process observed previously for τ > ∼ 3×10−9 s [22]. In (A) the contribution from spin rotation is indistinguishable from the fit line.
Figure 4.
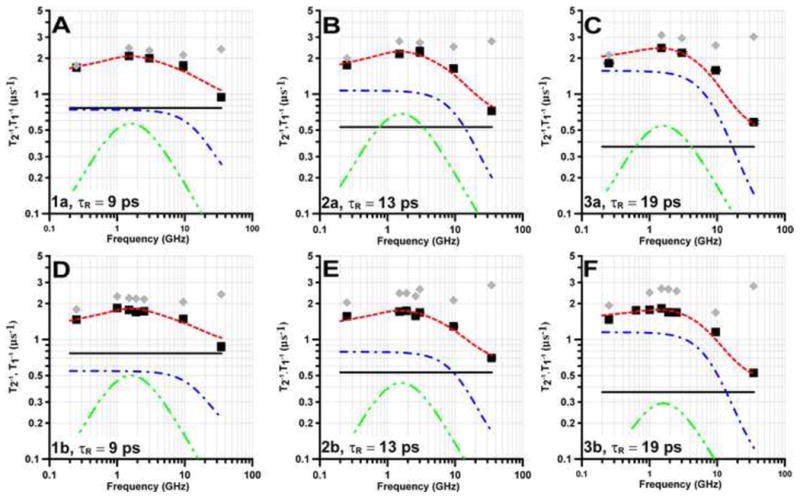
Frequency dependence of T1−1 (■) and T2−1 (
) in water for the mI = 0 lines of (A) 1a, (B) 2a, (C) 3a or the mI = ± 0.5 lines of (D) 1b, (E) 2b, and (F) 3b. Spin lattice relaxation is modeled as the sum (
) of contributions from spin rotation (Eq. 1,
), the modulation of g- and A- anisotropy (Eq. 2 – 4,
) and a thermally-activated process (Eq. 5
).
Figure 5.
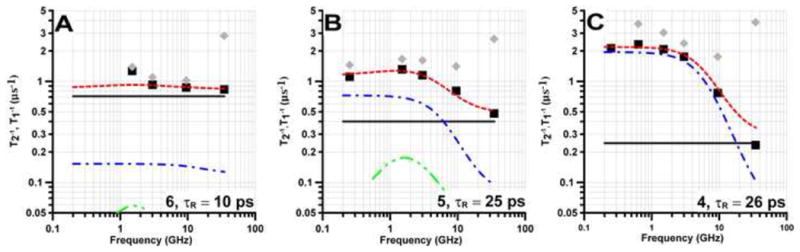
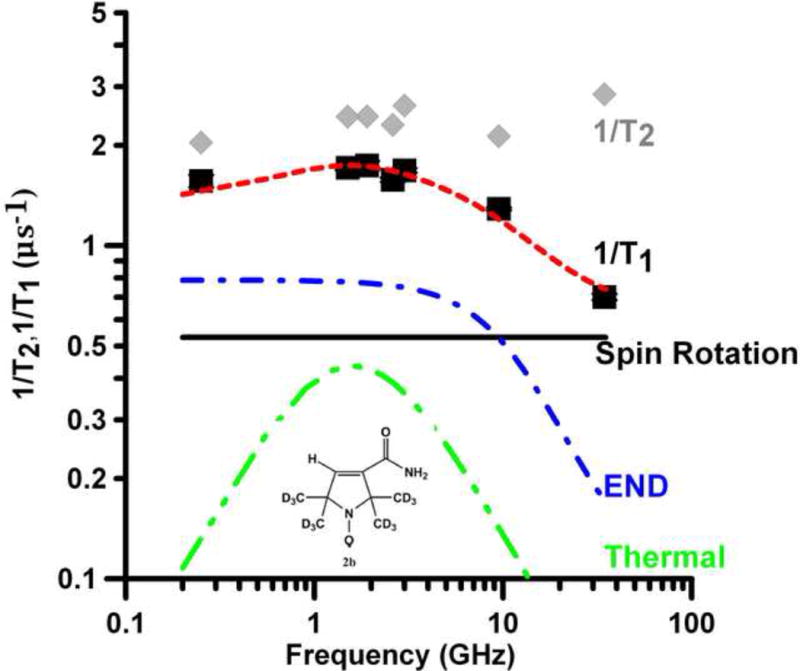
Frequency dependence of T1−1 (■) and T2−1(
) for the mI =0 lines of (A) 6 in toluene, (B) 5 in 2 mM aqueous NaOH and (C) 4 in toluene. Spin lattice relaxation is modeled as the sum (
) of contributions from spin rotation (
), the modulation of g- and A- anisotropy (
) and a thermally activated process (
 ).
).
Modeling the frequency dependence of 1/T1
The g- and nitrogen A-values are summarized in Table 1. A-values for 1b to 3b were calculated from the values for the 14N analogs multiplied by the ratio of the gyromagnetic ratios for 15N/14N = 1.40. For 6 immobilized in sucrose octaacetate, simulation of the ∼9.5 GHz spectrum (unpublished data) gave a well-defined Azz = 13.2 G. Values of Axx = Ayy = 0.4 G were estimated by assuming that Axx = Ayy and using Aiso = 4.7 G in toluene. Values of τR for all radicals, except 5, were calculated from the mI dependence of T2 at X-band [28]. The uncertainties in τR are about ±20%. The value of τR = 4 ps for PDT (1a) in toluene is in good agreement with the literature value of 5 ps [56]. The value of τR for 5 is taken from [21]. The T1−1 data in Figures 2, 4, and 5 are modeled as the sum of contributions from spin rotation ((Eq. 1, solid black line), modulation of A- and g-anisotropy by molecular tumbling (Eq. 2 – 4, blue dashed line) and a thermally-activated process (Eq. 5, green dashed line). The sum of the contributions is the red fit line. The contribution from modulation of g-anisotropy is negligible except at 34 GHz. For 1 - 4 and 6, there are no adjustable parameters in the calculations of the contributions to relaxation from Eq. (1) – (4). The only adjustable contribution to T1−1 was from the thermally-activated process, Eq. (5). Independent variation of Ctherm and τtherm did not give significantly improved fit to the data, so τtherm was fixed at 1.0×10−10 s [21]. The contribution of the END process for nitronyl nitroxide 5 was discussed in [21].
3. Results and Discussion
Much of the prior work on electron spin lattice relaxation of nitroxides has focused on lower temperatures and/or relatively long tumbling correlation times [5,6,15,22,26,27,56,57]. Understanding relaxation in the faster tumbling regime provides exciting new insights for designing small molecule spin probes for applications to in vivo imaging and dynamic nuclear polarization (DNP).
Relaxation times were measured at 0.25, 1.5, 2.5 to 3.0, 9.4 to 9.5 and 34 GHz, with the exception of 1a in 69% glycerol for which the lowest frequency data were measured at ca. 0.63 GHz (Figures 2). For 1b – 3b relaxation times also were measured at 1.0 and 1.9 GHz. Relaxation times for 1a in water and for 5 are reproduced from [21]. At ∼9.5 and 34 GHz the T1 for 1a in water is in good agreement with values for 0.5 mM natural abundance tempone reported in [15]. The values of T1 reported for tempone in water at 2.5 – 3.2 GHz [15] are about 40% shorter than observed for 1a. The greater difference at lower frequencies may be due in part to the factor of ∼1.5 H/D isotope effects previously reported for nitroxide T1 in solution [22,27]. For the natural isotope abundance analogue of 2a, the values of T1 reported in [15] are about 10% longer than observed for 2a in this study, which is smaller than the H/D isotope effect expected [22].
Interpretation of frequency dependence of relaxation
The frequency dependence of T1−1 (■) and T2−1 (
) for PDT (1a) in five solvents (toluene, water, light mineral oil, 44% glycerol in water, and 69% glycerol in water) is shown in Figure 2. For τR < ∼20 ps and resonance frequencies ≤ 9 GHz, T1−1 ∼ T2−1 as expected in the fast tumbling regime. At 34 GHz, T2−1 − T1−1 is substantial (Figure 2) due to increased contributions from incomplete motional averaging of g-anisotropy. The slope of the plot of T2−1 − T1−1 vs τR is 2.3×1017 s−2 (Figure 3). Calculation of the predicted slope of the plot based on the A, B, and C terms in the Kivelson model of linewidths [58,59] and typical g and A hyperfine anisotropy predicts a slope of 1.6×1017 s−2. Given the approximations in the model and the range of solvents and nitroxides included in the plot, the agreement is good, which lends credence to the values of τR.
Figure 3.
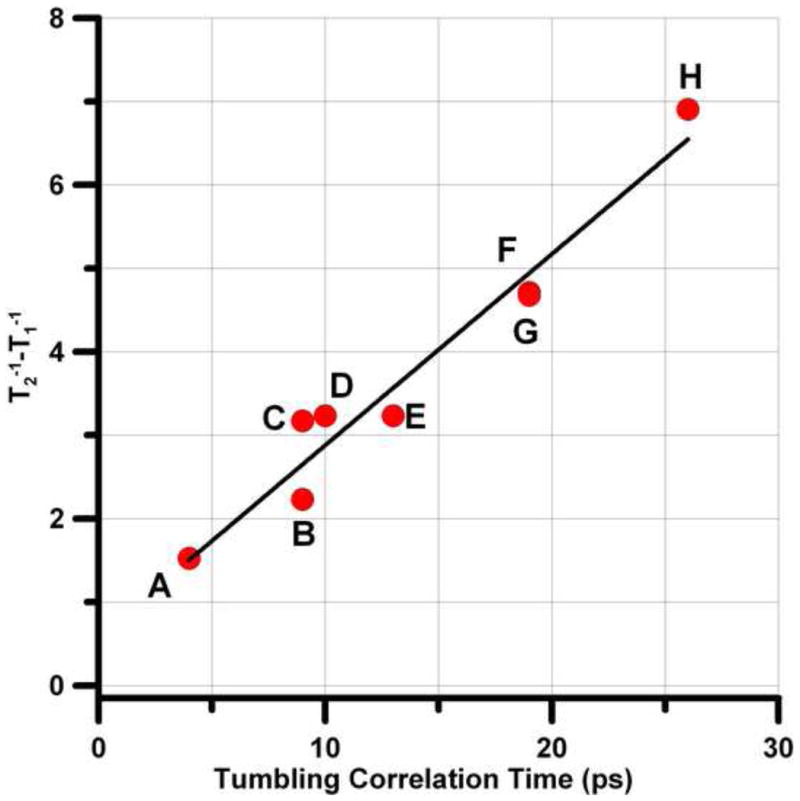
Dependence of T2−1 − T1−1 on τR for the high-field 14N hyperfine line at Q-band for: A, 1a in toluene; B, 1a in water; C, 1a in light mineral oil; D, 6 in toluene; E, 2a in water; F, 3a in water; G, 1a in 44% glycerol; H, 4 in toluene. The slope of the least-squares fit line is 2.3×1017 s−2.
As τR increases the frequency-independent contribution from spin rotation (Eq. (1) decreases (Figure 2). At τR = 4 ps spin rotation dominates T1−1 and relaxation is independent of frequency (Figure 2A). The agreement between experiment and calculation at τR = 4 ps is not as good as at longer τR. For the small PDT molecule (1a) in low-viscosity toluene, Heisenberg exchange due to nitroxide-nitroxide collision, which is not included in the simulation, may contribute more to relaxation than for other samples studied. In contrast to the dependence for spin rotation, as τR increases the contribution from modulation of A and g-anisotropy increases. As τR increases from 4 ps to 50 ps, the relaxation at frequencies less than 34 GHz changes from domination by spin rotation to domination by modulation of A anisotropy. The frequency at which the contribution from modulation of A changes from frequency independent to frequency dependent is a strong function of τR (Eq. 2-4). The longer the value of τR, the lower the frequency at which the change in slope occurs. Thus the agreement between the observed and calculated frequency dependence of T1−1 is a validation of the model and of the values of τR. For τR = 9 ps, in either water (Fig. 2B) or light mineral oil (Fig. 2C), T1-1 is smaller at 250 MHz than at 1-2 GHz. This frequency dependence is not predicted by the spin rotation (Eq. 1) or modulation of A or g-anisotropy (Eq. 2-4). Modeling of the relaxation rates therefore included a contribution from a thermally-activated process, Eq. (5). The effect of the thermally-activated process is maximum when ω = 1/τtherm, which was observed to occur at about 1.6 GHz (Fig. 2 and 4), and corresponds to τtherm = 1.0×10−10 s. This is the same value of τtherm that was used to model the frequency dependence of T1−1 for tempol with τR > ∼3×10−9 s (300 ps) in viscous water:glycerol mixtures [22]. The value of τtherm was fixed at 1.0×10−10 s and the coefficient Ctherm (Table 3) was adjusted to match the experimental data for T1−1 The value of Ctherm was similar in water (Fig. 2B) or light mineral oil (Fig. 2C), which indicates that the process does not require a protic solvent. Ctherm is smaller in 44% glycerol (Fig. 2D), which demonstrates a dependence on viscosity. For comparison, the contributions to T1−1 from the thermally-activated process for tempol with τR > ∼3×10−9 s in viscous water:glycerol mixtures [22] is shown as orange squares in Fig. 2E, and is negligibly small relative to the contributions from modulation of A and g anisotropy at τR = 50 ps.
Table 3. Values of Ctherma.
| Ctherm (×1016) s−2 | |||||
|---|---|---|---|---|---|
| Solvent |
14N MI = 0 |
14N average MIb |
15N average MIc |
14N/15Nd | |
| 1a | Water | 7 | 7.7 | 1.4 | |
| Light mineral oil | 8.6 | 8.5 | |||
| 44% Glycerol | 3.8 | 4.5 | |||
| 1b | Water | 5.7 | |||
| 2a | Water | 8.2 | 9.0 | 1.7 | |
| 2b | Water | 5.2 | |||
| 3a | Water | 6.5 | 9.0 | ||
| 3b | Water | 3.5 | 2.6 | ||
| 4 | Toluene | <0.7 | |||
| 5 | 2 mM NaOH | 2.8 | |||
| 6 | Toluene | 0.7 | |||
Uncertainties are about 10% for τR ∼ 10 ps and increase to ∼25% for τR > 25 ps.
Based on modeling of average T1−1 for mI = +1,0,−1
Based on modeling of average T1−1 for mI = ±1/2
Calculated from values obtained for average T1−1 for all mI lines
The frequency dependence of relaxation in water for 14N/15N pairs of radicals is shown in Fig. 4. These radicals have τR in the range of 9 to 19 ps. At 34 GHz spin rotation (Eq. 1) dominates T1−1 which is about the same for 14N and 15N (Fig. 4). As seen in Fig. 2 the contribution from (T1A)−1 (Eq. 2) increases as frequency decreases from 34 to about 3 GHz. If the END process were the only relaxation mechanism, the dependence on nuclear spin I and the hyperfine coupling constants, A, would cause the ratio of [T1−1(14N)]/[T1−1(15N)] to be 1.36 [27]. For the pairs 1a/1b, 2a/2b, and 3a/3b the ratios of relaxation rates increase, as expected, as the contribution from the END process increases (Table 2) and is in the range of 1.3 to 1.4 where the END process dominates. The limiting value of (T1A)−1 at low frequency increases as τR increases (Eq. 2). Because of the increasing role of the END process at lower frequencies, T1−1 becomes more frequency dependent as τR increases.
Table 2.
Frequency dependence of (T1−1 for 14N mI = 0)/(T1−1 for 15N mI = ±0.5)a and of the relative contributions from relaxation mechanisms.b,c
| Freq. (GHz) | 1a/1b | 2a/2b | 3a/3b | |||
|---|---|---|---|---|---|---|
| 33.9 | 1.05 | SR > END > THM | 1.00 | SR > END > THM | 1.12 | SR > END > THM |
| 9.4-9.5 | 1.16 | SR > END > THM | 1.28 | END > SR > THM | 1.38 | END > SR > THM |
| 2.5-3.0 | 116 | SR ∼ END > THM | 1.35 | END > SR ∼ THM | 1.32 | END > SR ∼ THM |
| 1.5 | 1.18 | SR∼ END ∼ THM | 1.27 | END > SR ∼ THM | 1.34 | END > SR ∼ THM |
| 0.25 | 1.13 | SR ∼ END > THM | 1.12 | END > SR > THM | 1.25 | END > SR > THM |
Uncertainties in ratios of T1 are 5 to 7%.
Abbreviations for relaxation mechanisms are spin rotation (SR), modulation of A anisotropy (END), and thermally-activated process (THM).
The contributions from relaxation mechanisms are displayed quantitatively in Figure 4.
For the two nitroxides with smaller nitrogen hyperfine couplings, 6 and nitronyl nitroxide 5, the contribution to T1−1 from (T1A)−1 is substantially reduced (Fig. 5A,B). Even though τR for 5 in water (25 ps) is about the same as for 4 in toluene (26 ps), T1−1 for 5 at low frequencies is about a factor of two smaller than for 4, due to the decreased contribution from (T1A)−1. Nitroxide 6 has nitrogen hyperfine couplings that are less than 1/3 of those for 1 – 4. The τR for 6 in toluene (10 ps) (Figure 5A) is intermediate between those for 1a (τR = 9 ps) (Fig. 4A) and 2a (τR = 13 ps) (Fig. 4B) in water, so the contributions from spin rotation are similar. However, the contribution from (T1A)−1 for 6 in toluene is so much smaller than for 1 or 2 in water that T1−1 for 6 is approximately equal to the value predicted for spin rotation alone (Fig. 5A).
Analogous to what was observed for 1a in water and mineral oil (Fig. 2B,C) T1−1 for 1b, 2a, 2b, 3a, 3b, and 5 at 250 MHz is smaller than at 1-2 GHz (Fig. 4 and 5), which cannot be accounted for by Eq. (1) – (4). The enhanced relaxation at 1-2 GHz is attributed to a thermally-activated process. Ctherm is larger for 1a - 3a (containing 14N) than for 1b - 3b (containing 15N) (Table 3), which indicates a nitrogen isotope effect on Ctherm. The nitrogen isotope effect is observed independent of whether the T1−1 for 14N is analyzed for mI = 0 or for the average of all three nitrogen hyperfine lines. For the pairs 1a/1b and 2a/2b the only substitution is 14N to 15N. For the pair 3a/3b H is substituted by D as well as 14N by 15N so the larger ratio of values of Ctherm for 3a/3b (2.6) than for 1a/1b or 2a/2b (1.4 or 1.7) suggests that there is an additional H/D isotope effect.
Potential assignments of the thermally-activated process
The characteristic time for the thermally-activated process is about 1×10−10 s, which is longer than the τR of (10 to 50)×10−12 s. Thus the thermally-activated process is modulating an interaction that is not averaged by τR, which therefore excludes modulation of anisotropic dipolar couplings. The values of Ctherm decrease with increasing solvent viscosity, in the order 1a in water > 1a in 44% glycerol > tempol in highly viscous solvents [22]. Values of Ctherm for 1a – 3a are substantially larger than for 4, 5, or 6. The nitroxide rings for 4 - 6 are expected to be less flexible than for 1 – 3 because of bulkier substituents for 4 and 5 and the additional double bond in 6. Because of the higher molecular weight of 4, τR = 26 ps even in low-viscosity toluene. The contribution from (T1A)−1 is so large for 4 that it is difficult to determine whether the contribution from the thermally-activated process is small or absent, but it cannot be as large as for 1a – 3a. These observations suggest that the thermally-activated process is modulation of the nitrogen isotropic A.
One possible mechanism for modulation of g or A is conformational changes of the geometry at the nitroxide nitrogen. Substantial temperature dependence of nitroxide g and A-values in fluid solution has been observed and attributed to fast exchange between conformations [60]. For 1a, 1b the interconversion of twist-crossover conformations averages axial and equatorial methyl group on the ENDOR timescale above about 230 K in a doped crystal [61] and results in equal hyperfine couplings to all twelve methyl protons in fluid solution [28]. The lifetime of about 1×10−7 s at 230 K predicts a lifetime at 298 K that is substantially longer than 1×10−10 s. For 5-member ring nitroxides interconversion of ring conformations is slow on the EPR time scale at 298K and inequivalent couplings are observed for axial and equatorial methyls [62]. The large differences in rates of interconversion for 5- and 6- member ring nitroxides and estimates of lifetimes substantially longer than 10−10 s at 298 K indicate that interconversion of ring conformations is too slow to be assigned as the thermally-activated process for nitroxide relaxation. Recent molecular dynamics calculations have shown that the intramolecular out-of-plane motion of the N-O moiety is a ‘soft’ mode [63,64]. For both 5- and 6-member ring nitroxides the angle between the N-O group and the plane of the molecule can vary by ±10° within a low-energy range of about 0.5 kcal/mole [64]. This angular variation is sufficient to cause substantial changes in the isotropic g and A values. Modulation of either or both of these parameters is an effective spin-lattice relaxation process. Modulation of isotropic A and g would be consistent with a nitrogen isotope effect on Ctherm. The dependence of Ctherm on viscosity could indicate that a smaller range of orientations is encompassed by these motions when solvent viscosity is higher. The value of τtherm is approximately what is predicted for methyl rotation at room temperature based on ENDOR studies of tempone [61]. In these sterically hindered molecules methyl rotation may couple to the motion of the NO bond and be involved in the bond angle variation that dominates the thermally-activated process.
Other potential contributions to relaxation
The discussion of the role of the thermally-activated process (Eq 5) in the relaxation for these rapidly tumbling nitroxides assumes that this process is the same as the one that contributes to the frequency-dependent relaxation of tempol for τR > ∼3×10−9 s in viscous water:glycerol mixtures [22]. At those long values of τR the contributions to T1−1 from spin rotation and modulation of g and A anisotropy become very small. Robinson and co-workers proposed generalized spin diffusion (GSD) as the process that dominated spin-lattice relaxation for slowly tumbling nitroxides [17], which raises the question whether GSD might also contribute for rapidly tumbling samples. The GSD mechanism contributes to relaxation by modulation of electron-nuclear interspin distances via diffusion of solvent nuclei or intramolecular motions, which would have a large H/D isotope effect. In Ref. [19] it was shown that for Fremy's salt in water at 60 MHz the relaxation rate was the same in H2O and D2O. It was also shown previously that at ∼9.5 GHz for τR < 5×10−9 s there was no difference in relaxation rate for a nitroxide in water:glycerol or in D2O:glycerol-d8 mixtures [22]. Thus, solvent diffusion does not appear to be an effective relaxation mechanism for the nitroxides studied here. Furthermore, the proposed frequency dependence of GSD does not predict smaller relaxation rates at 250 MHz than at higher frequencies, so it is not an alternative to the thermally-activated process.
In an early discussion of potential relaxation processes for Fremy's salt, Lloyd and Pake [19] proposed that modulation of spin-orbit coupling by solid-state-like processes would be significant. The temperature dependence at ∼9.5 GHz of T1−1 for tempone in glycerol, 1:1 water:glycerol, decalin, and 3-methylpentane did not exhibit a discontinuity in the region of the glass transition temperature, which was interpreted as indicating that solid-state mechanisms persisted at higher temperatures [27]. In the slow tumbling regime the temperature dependence was modeled with contributions from Raman and local-mode processes. In glycerol, the most viscous of the solvents studied, these processes dominated relaxation at room temperature. However, in the fast tumbling regime these contributions are much smaller than the contributions from spin rotation and modulation of g- and A-anisotropy.
4. Conclusions
Understanding electron spin relaxation has been a goal of magnetic resonance spectroscopists from the earliest days of the field. Most prior studies of electron spin lattice relaxation in fluid solution were performed at ∼9.5 GHz and τR was varied by changing temperature [5,6,27,56,65,66]. The frequency dependence of relaxation is a powerful way to distinguish contributions from proposed mechanisms. The contribution from spin rotation is independent of ω, and decreases as τR increases. The contribution from modulation of g- and A-anisotropy (END process) depends on both ω and τR. The contribution from the thermally-activated process depends on ω, but not τR. For rapidly tumbling nitroxides, T1 decreases with decreasing ω between about 34 and 3 GHz [15] as previously reported, because of the frequency dependence of the END process. However, this trend does not continue at lower frequencies because the END process becomes frequency independent. The frequency dependence of T1−1 between 34 and 3 GHz is smaller when τR = 10 ps than when τR = 50 ps. The thermally-activated process has maximal impact on T1 in the range of 1 to 2 GHz. These three processes are sufficient to model T1−1 over the full frequency range studied. The net effect of the contributions from spin rotation, modulation of anisotropy by tumbling, and the thermally-activated process is that for small rapidly tumbling nitroxides there is a maximum in relaxation rate at about 1 to 2 GHz and relaxation rates become smaller again at lower frequency. Consequently, relaxation rates at very low frequency (e.g., 60 MHz, 250 MHz) are similar to those at ∼9.5 GHz, which is fully consistent with the earliest estimates of nitroxide relaxation rates.
In addition to understanding relaxation mechanisms, a goal of this work is to guide synthesis of application-specific nitroxide radicals. To maximize nitroxide relaxation times at low microwave frequencies, such as for in vivo studies, the nitroxide should be designed to minimize nitrogen hyperfine, thus reducing the effect of the END process (Eq. 2). If the contributions from the END process are decreased, increasing molecular size to increase τR is advantageous. The radical should be as rigid as possible to minimize the impact of the thermally-activated process.
Supplementary Material
Research highlights.
T1 and T2 were measured for 6 nitroxides between 250 MHz and 34 GHz.
Tumbling correlation times were between 4 and 50 ps.
Spin rotation dominates at 34 GHz.
Modulation of anisotropy and thermally-activated process are important.
Thermally-activated process for 14N > 15N and decreases with increasing viscosity.
Acknowledgments
Partial support of this work by NIH grant P41 EB002034 to GMR and GRE, H. J. Halpern, PI, is gratefully acknowledged. Richard Quine provided invaluable assistance with instrumentation and George Rinard designed and built resonators for this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider DJ, Freed JH. Calculating slow motional magnetic resonance spectra: a user's guide. In: Berliner LJ, Reuben J, editors. Biol Magn Reson. 1989. pp. 1–76. [Google Scholar]
- 2.Freed JH. ESR and molecular dynamics. Biol Magn Reson. 2005;24:239–268. [Google Scholar]
- 3.Freed JH. Theory of slow tumbling ESR spectra of nitroxides. In: Berliner LJ, editor. Spin Labeling: Theory and Applications. Academic Press; New York: 1976. pp. 53–132. [Google Scholar]
- 4.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson A. 1996;120:155–189. [Google Scholar]
- 5.Robinson BH, Haas DA, Mailer C. Molecular dynamics in liquids: spin-lattice relaxation of nitroxide spin labels. Science. 1994;263:490–493. doi: 10.1126/science.8290958. [DOI] [PubMed] [Google Scholar]
- 6.Robinson BH, Reese AW, Gibbons E, Mailer C. A unified description of the spin-spin and spin-lattice relaxation rates applied to nitroxide spin labels in viscous liquids. J Phys Chem B. 1999;103:5881–5894. [Google Scholar]
- 7.Huisjen J, Hyde JS. A pulsed EPR spectrometer. Rev Sci Instrum. 1974;45:669–675. [Google Scholar]
- 8.Subczynski WK, Mainali L, Camenisch TG, Froncisz W, Hyde JS. Spin-label oximetry at Q- and W-band. J Magn Reson. 2011;209:142–148. doi: 10.1016/j.jmr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin JJ, Hyde JS. Spin-label saturation-recovery electron spin resonance measurements of oxygen transport in membranes. Zeitschrift fuer Physikalische Chemie. 1987;153:57–65. [Google Scholar]
- 10.Yin JJ, Pasenkiewicz-Gierula M, Hyde JS. Lateral diffusion of lipids in membranes by pulse saturation recovery electron spin resonance. Proc Natl Acad Sci U S. 1987;84:964–8. doi: 10.1073/pnas.84.4.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epel B, Sundramoorthy SV, Barth ED, Mailer C, Halpern HJ. Comparison of 250 MHz electron spin echo and continuous wave oxygen EPR imaging methods for in vivo applications. Medical Physics. 2011;38:2045–2052. doi: 10.1118/1.3555297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RM, Matsumoto S, Bernardo M, Sowers A, Matsumoto KI, Krishna M, Mitchell JB. Magnetic resonance imaging of organic contrast agents in mice: capturing the whole-body redox landscape. Free Rad Biol Med. 2011;50:459–468. doi: 10.1016/j.freeradbiomed.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumoto KI, Hyodo F, Mutsumoto A, Koretsky AP, Sowers AL, Mitchell JB, Krishna M. High-Resolution Mapping of Tumor Redox Status by Magnetic Resonance Imaging Using Nitroxides as Redox-Sensitive Contrast Agents. Clin Cancer Res. 2006;12:2455–2462. doi: 10.1158/1078-0432.CCR-05-2747. [DOI] [PubMed] [Google Scholar]
- 14.Hyde JS, Yin JJ, Feix JB, Hubbell WL. Advances in spin label oximetry. Pure Appl Chem. 1990;62:255–60. [Google Scholar]
- 15.Hyde JS, Yin JJ, Subczynski WK, Camenisch TG, Ratke JJ, Froncisz W. Spin-labeled EPR T1 Values Using Saturation Recovery from 2 to 35 GHz. J Phys Chem B. 2004;108:9524–9529. [Google Scholar]
- 16.Froncisz W, Camenisch TG, Ratke JJ, Anderson JR, Subczynski WK, Strangeway RA, Sidabras JW, Hyde JS. Saturation recovery EPR and ELDOR at W-band for spin labels. J Magn Reson. 2008;193:297–304. doi: 10.1016/j.jmr.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mailer C, Nielsen RD, Robinson BH. Explanation of Spin-Lattice Relaxation Rates of Spin Labels Obtained with Multifrequency Saturation Recovery EPR. J Phys Chem A. 2005;109:4049–4061. doi: 10.1021/jp044671l. [DOI] [PubMed] [Google Scholar]
- 18.Owenius R, Eaton GR, Eaton SS. Frequency (250 MHz to 9.2 GHz) and Viscosity Dependence of Electron Spin Relaxation of Triarylmethyl Radicals at Room Temperature. J Magn Res. 2005;172:168–175. doi: 10.1016/j.jmr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Lloyd JP, Pake GE. Spin relaxation in free radical solutions exhibiting hyperfine structure. Phys Rev. 1954;94:579–591. [Google Scholar]
- 20.Tseitlin M, Dhami A, Quine RW, Rinard GA, Eaton SS, Eaton GR. Electron Spin T2 of a Nitroxyl Radical at 250 MHz Measured by Rapid Scan EPR. Appl Magn Reson. 2006;30:651–656. [Google Scholar]
- 21.Biller JR, Meyer VM, Elajaili H, Rosen GM, Eaton SS, Eaton GR. Frequency Dependence of Electron Spin Relaxation Times in Aqueous Solution for a Nitronyl Nitroxide Radical and Per-deuterated-Tempone between 250 MHz and 34 GHz. J Magn Reson. 2012;225:52–57. doi: 10.1016/j.jmr.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owenius R, Terry GE, Williams MJ, Eaton SS, Eaton GR. Frequency Dependence of Electron Spin Relaxation of Nitroxyl Radicals in Fluid Solution. J Phys Chem B. 2004;108:9475–9481. [Google Scholar]
- 23.Atkins PW. Spin-Rotation Interaction. In: Muus LT, Atkins PW, editors. Electron Spin Relaxation in Liquids. Plenum Press; New York: 1972. pp. 279–312. [Google Scholar]
- 24.Atkins PW, Kivelson D. ESR Linewidths in Solution. II. Analysis of Spin-Rotational Relaxation Data. J Chem Phys. 1966;44:169–174. [Google Scholar]
- 25.Rengan SK, Khakhar MP, Prabhananda BS, Venkataraman B. Electron Spin-Lattice Relaxation in Organic Free Radicals in Solutions. Pure Appl Chem. 1972;32:287–305. [Google Scholar]
- 26.Sato H, Kathirvelu V, Fielding AJ, Bottle SE, Blinco JP, Micallef AS, Eaton SS, Eaton GR. Impact of molecular size on electron spin relaxation rates of nitroxyl radicals in glassy solvents between 100 and 300 K. Mol Phys. 2007;105:2137–2151. [Google Scholar]
- 27.Sato H, Bottle SE, Blinco JP, Micallef AS, Eaton GR, Eaton SS. Electron spin-lattice relaxation of nitroxyl radicals in temperature ranges that span glassy solutions to low-viscosity liquids. J Magn Reson. 2008;191:66–77. doi: 10.1016/j.jmr.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biller JR, Meyer V, Elajaili H, Rosen GM, Kao JPY, Eaton SS, Eaton GR. Relaxation Times and Line Widths of Isotopically-Substituted Nitroxides in Aqueous Solution at X-band. J Magn Reson. 2011;212:370–377. doi: 10.1016/j.jmr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann SK, Hilczer W, Goslar J, Kiczka S, Polus I. Resonance-type effects in free radical electron spin-lattice relaxation and electron spin echo dephasing due to a dynamcs of a homogeneous-chain oligomeric system. Chem Phys Phys Chem. 2002;4:4944–4951. [Google Scholar]
- 30.Harbridge JR, Eaton SS, Eaton GR. Electron spin-lattice relaxation in radicals containing two methyl groups, generated by γ-irradiation of polycrystalline solids. J Magn Reson. 2002;159:195–206. doi: 10.1016/s1090-7807(02)00013-7. [DOI] [PubMed] [Google Scholar]
- 31.Harbridge JR, Eaton SS, Eaton GR. Electron Spin-Lattice Relaxation Processes of Radicals in Irradiated Crystalline Organic Compounds. J Phys Chem A. 2003;107:598–610. [Google Scholar]
- 32.Harbridge JR, Eaton SS, Eaton GR. Electron Spin Relaxation of Radicals in γ-irradiated Malonic Acid and Methyl Malonic Acid, J. R. Harbridge. Appl Magn Reson. 2003;24:261–276. [Google Scholar]
- 33.Yamauchi J, McDowell CA. NMR Study of molecular motion in some molecules containing t-butyl groups. J Chem Phys. 1981;75:1051–1058. [Google Scholar]
- 34.Beckmann JD, McKean MC, Frerman FE. Inhibition of general acyl-CoA dehydrogenase by electron transfer flavoprotein semiquinone. Biochem Biophys Res Commun. 1981;102:1290–1294. doi: 10.1016/s0006-291x(81)80151-9. [DOI] [PubMed] [Google Scholar]
- 35.Beckmann PA, Al-Hallaq HA, Fry AM, Plofker AL, Roe BA, Weiss JA. Solid state proton spin relaxation and methyl and t-butyl reorientation. J Chem Phys. 1994;100:752. [Google Scholar]
- 36.Beckmann PA, Schneider E. Methyl group rotation, 1H spin-lattice relaxation in an organic solid and the analysis of nonexponential relaxation. J Chem Phys. 2012;136:054508. doi: 10.1063/1.3677183. [DOI] [PubMed] [Google Scholar]
- 37.Hyodo F, Matsumoto S, Devasahayam N, Dharmaraj C, Subramanian S, Mitchell JB, Krishna MC. Pulsed EPR imaging of nitroxides in mice. J Magn Reson. 2009;197:181–185. doi: 10.1016/j.jmr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin YJ, Teicher BA, Halpern HJ. Synthesis of 4-protio-3-carbamoyl-2,2,5,5-tetraperdeuteromethyl-3-pyrrolin-1-yloxy (mHCTPO): a selectively isotopically labeled compound for use in T2 spin label oxymetry. J Labelled Compds Radiopharm. 1990;28:621–31. [Google Scholar]
- 39.Elas M, Bell R, Hleihel D, Barth ED, McFaul C, Haney CR, Bielanska J, Pustelny K, Ahn KH, Pelizzari CA, Kocherginsky M, Halpern HJ. Electron Paramagnetic Resonance Oxygen Image Hypoxic Fraction Plus Radiation Dose Strongly Correlates With Tumor Cure in FSa Fibrosarcomas. Int J Radiation Oncology Biol Phys. 2008;71:542–549. doi: 10.1016/j.ijrobp.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elas M, Ichikawa K, Halpern HJ. Oxidative stress in live animals with techniques based on electron paramagnetic resonance Radiat. Res. 2012;177:514–523. doi: 10.1667/rr2668.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen J, Liu S, Miyake M, Liu W, Pritchard A, Kao JPY, Rosen GM, Tong Y, Liu KJ. Use of 3-acetoxymethoxycarbonyl-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl as an EPR oximetry probe: Potential for in vivo measurement of tissue oxygenation in mouse brain. Magn Reson Med. 2006;55:1433–1440. doi: 10.1002/mrm.20894. [DOI] [PubMed] [Google Scholar]
- 42.Shen J, Sood R, Weaver J, Timmins GS, Schnell A, Miyake M, Kao JPY, Rosen GM, Liu KJ. Direct visualization of mouse brain oxygen distribution by electron paramagnetic resonance imaging: Application to focal cerebral ischemia. J Cereb Blood Flow Metabol. 2009;29:1695–1703. doi: 10.1038/jcbfm.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kao JPY, Barth ED, Burks SR, Smithback P, Mailer C, Ahn KH, Halpern HJ, Rosen GM. Very-Low-Frequency Electron Paramagnetic Resonance (EPR) Imaging of Nitroxide-Loaded Cells. Magn Reson Med. 2007;58:850–855. doi: 10.1002/mrm.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen J, Bottle S, Khan N, Grinberg O, Reid D, Micallef A, Swartz HM. Development of Isoindoline Nitroxides for EPR Oximetry in Viable Systems. Appl Magn Reson. 2002;22:357–368. [Google Scholar]
- 45.Khan N, Blinco JP, Bottle SE, Hosokawa K, Swartz HM, Micallef AS. The evaluation of new and isotopically labeled isoindoline nitroxides and azaphenalene nitroxide for EPR oximetry. J Magn Reson. 2011;211:170–177. doi: 10.1016/j.jmr.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burks SR, Bakhshai J, Makowsky MA, Muralidharan S, Tsai P, Rosen GM, Kao JPY. 2H,15N-Substituted Nitroxides as Sensitive Probes for Electron Paramagnetic Resonance Imaging. J Org Chem. 2010;75:6463–6467. doi: 10.1021/jo1011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halpern HJ, Peric M, Nguyen TD, Spencer DP, Teicher BA, Lin YJ, Bowman MK. Selective isotopic labeling of a nitroxide spin label to enhance sensitivity for T2 oxymetry. J Magn Reson. 1990;90:40–51. [Google Scholar]
- 48.Ramasseul R, Rassat A, Ramasseul R, Rassat A. Nitroxydes XXXIII: radicaux nitroxydes pyrroliques encombrés. Un pyrryloxyle stable. Bulletin de la Société Chimique de France. 1970:4330–4341. [Google Scholar]
- 49.Quine RW, Rinard GA, Eaton SS, Eaton GR. A pulsed and continuous wave 250 MHz electron paramagnetic resonance spectrometer. Magn Reson Engineer. 2002;15:59–91. [Google Scholar]
- 50.Quine RW, Rinard GA, Ghim BT, Eaton SS, Eaton GR. A 1-2 GHz pulsed and continuous wave electron paramagnetic resonance spectrometer. Rev Sci Instrum. 1996;67:2514–2527. [Google Scholar]
- 51.Quine RW, Eaton GR, Eaton SS. Pulsed EPR spectrometer. Rev Sci Instrum. 1987;58:1709–23. [Google Scholar]
- 52.Popp CA, Hyde JS. Electron-electron double resonance and saturation-recovery studies of nitroxide electron and nuclear spin-lattice relaxation times and Heisenberg exchanges rates: lateral diffusion in dimyristoyl phosphatidylcholine. Proc Nat Acad Sci US. 1982;79:2559–2563. doi: 10.1073/pnas.79.8.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin YJ, Hyde JS. Use of high observing power in electron spin resonance saturation-recovery experiments in spin-labeled membranes. J Chem Phys. 1989;91:6029–35. [Google Scholar]
- 54.Borgia GC, Brown RJS, Fantazzini P. Uniform-penalty Inversion of Multiexponential Decay Data II. Data Spacing, T2 Data, Systematic Errors, and Diagnostics. J Magn Res. 2000;147:273–285. doi: 10.1006/jmre.2000.2197. [DOI] [PubMed] [Google Scholar]
- 55.Borgia GC, Brown RJS, Fantazzini P. Uniform-penalty Inversion of Multiexponential Decay Data. J Magn Res. 1998;132:65–77. doi: 10.1006/jmre.1998.1387. [DOI] [PubMed] [Google Scholar]
- 56.Hwang JS, Mason RP, Hwang LP, Freed JH. Electron spin resonance studies of anisotropic rotational reorientation and slow tumbling in liquid and frozen media. III. Perdeuterated 2,2,6,6-tetramethyl-4-piperidone-N-oxide and an analysis of fluctuating torques. J Phys Chem. 1975;79:489–511. [Google Scholar]
- 57.Eaton SS, Harbridge J, Rinard GA, Eaton GR, Weber RT. Frequency dependence of electron spin relaxation for three S = 1/2 species doped into diamagnetic solid hosts. Appl Magn Reson. 2001;20:151–157. [Google Scholar]
- 58.Kivelson D. Theory of EPR [electron paramagnetic resonance] line widths of free radicals. J Chem Phys. 1960;33:1094–1106. [Google Scholar]
- 59.Chasteen ND, Hanna MW. Electron Paramagnetic Resonance Line Widths of Vanadyl (IV) α-hydroxycarboxylates. J Phys Chem. 1972;76:3951–3958. [Google Scholar]
- 60.Siri D, Guadel-Siri A, Tordo P. Conformational Analysis of five-membered rings by molecular mechanics: applications to nitroxides. J Mol Struct (Theochem) 2002;582:171–185. [Google Scholar]
- 61.Barbon A, Brustolon M, Maniero AL, Romanelli M, Brunel LC. Dynamics and spin relaxation of tempone in a host crystal. An ENDOR, high field EPR and electron spin echo study. Phys Chem Chem Phys. 1999;1:4015–4023. [Google Scholar]
- 62.Rockenbauer A, Korecz L, Hideg K. Ring pseudorotation of pyrroline N-oxyl radicals: an analysis of carbon-13 hyperfine structure of EPR spectra. JCS Perkin Trans 2. 1993;2:2149–2156. [Google Scholar]
- 63.Pavone M, Biczysko M, Rega N, Barone V. Magnetic Properties of Nitroxide Spin Probes: Reliable Account of Molecular Motions and Nonspecific Solvent Effects by Time-Dependent and Time-Independent Approaches. J Phys Chem B. 2010;114:11509–11514. doi: 10.1021/jp102232c. [DOI] [PubMed] [Google Scholar]
- 64.Savitsky A, Plato M, Mobius K. Temperature Dependence of Nitroxide Spin-Label Interaction Parameters: a High-Field EPR Study of Intramolecular Motional Contributions. Appl Magn Reson. 2010;37:415–434. [Google Scholar]
- 65.Goldman SA, Bruno GV, Freed JH. ESR Studies of anisotropic rotational reorientation and slow tumbling in liquid and frozen media. II. Saturation and nonsecular effects. J Chem Phys. 1973;59:3071–3091. [Google Scholar]
- 66.van der Drift E, Rousseeuw BAC, Smidt J. EPR and ELDOR studies on spin relaxation in perdeuterated 2,2,6,6-tetramethyl-4-piperidone-N-oxyl in liquid solutions: The slowly relaxing local structure mechanism. J Phys Chem. 1984;88:2275–2284. [Google Scholar]
- 67.Earle KA, Budil DE, Freed JH. 250-GHz EPR of Nitroxides in the Slow-Motional Regime: Models of Rotational Diffusion. J Phys Chem. 1993;97:13289–13297. [Google Scholar]
- 68.Labsky J, Pilar J, Lovy J. Magnetic resonance study of 4-amino-2,2,6,6-tetramethylpiperidine-N-oxyl and its deuterated derivatives. J Magn Reson. 1980;37:515–522. [Google Scholar]
- 69.Volodarsky LB. Synthesis and Properties. I. CRC Press; Boca Raton: 1988. Imidazoline Nitroxides. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.