

ABSTRACT
Autophagy is a process whereby a double-membrane structure (autophagosome) engulfs unnecessary cytosolic proteins, organelles, and invading pathogens and delivers them to the lysosome for degradation. We examined the fate of cytosolic Salmonella targeted by autophagy and found that autophagy-targeted Salmonella present in the cytosol of HeLa cells correlates with intracellular bacterial replication. Real-time analyses revealed that a subset of cytosolic Salmonella extensively associates with autophagy components p62 and/or LC3 and replicates quickly, whereas intravacuolar Salmonella shows no or very limited association with p62 or LC3 and replicates much more slowly. Replication of cytosolic Salmonella in HeLa cells is significantly decreased when autophagy components are depleted. Eventually, hyperreplication of cytosolic Salmonella potentiates cell detachment, facilitating the dissemination of Salmonella to neighboring cells. We propose that Salmonella benefits from autophagy for its cytosolic replication in HeLa cells.
IMPORTANCE
As a host defense system, autophagy is known to target a population of Salmonella for degradation and hence restricting Salmonella replication. In contrast to this concept, a recent report showed that knockdown of Rab1, a GTPase required for autophagy of Salmonella, decreases Salmonella replication in HeLa cells. Here, we have reexamined the fate of Salmonella targeted by autophagy by various cell biology-based assays. We found that the association of autophagy components with cytosolic Salmonella increases shortly after initiation of intracellular bacterial replication. Furthermore, through a live-cell imaging method, a subset of cytosolic Salmonella was found to be extensively associated with autophagy components p62 and/or LC3, and they replicated quickly. Most importantly, depletion of autophagy components significantly reduced the replication of cytosolic Salmonella in HeLa cells. Hence, in contrast to previous reports, we propose that autophagy facilitates Salmonella replication in the cytosol of HeLa cells.
INTRODUCTION
Salmonella enterica serovar Typhimurium is a facultative intracellular pathogen that causes diseases ranging from self-limited gastroenteritis to typhoid fever in humans and mice (1). It contains two type III secretion systems (T3SSs) encoded by Salmonella pathogenicity island 1 (SPI-1) and SPI-2. These T3SSs are necessary for Salmonella pathogenicity and deliver bacterial proteins (effectors) to the host cell cytosol to manipulate host cell signaling, cytoskeletal, and vesicular pathways (2). In mouse infection models, Salmonella can enter specialized intestinal epithelial microfold (M) cells and gallbladder epithelial cells, and both epithelial cell types play important roles in the establishment of Salmonella infection (3, 4). To better understand the mechanisms of Salmonella interactions with epithelial cells, various tissue culture epithelial cell lines have been widely used, including HeLa, Henle 407, and Caco2 epithelial cells (2, 5). Upon invasion into mammalian cells, most Salmonella bacteria remain in a single membrane-bounded compartment called the Salmonella-containing vacuole (SCV). The SCV rapidly acquires late endosomal markers, such as the lysosomal-associated membrane protein 1 (LAMP-1). Although the SCV appears to be the default niche where Salmonella resides, some bacteria escape from the SCV and replicate rapidly in the host cell cytosol (6). However, the mechanism underlying this phenotype remains unclear. It was recently shown that hyperreplicating cytosolic bacteria (defined as >50 bacteria per cell; the doubling time of these bacteria is around 20 min) present in epithelial cells express SPI-1 genes and possess flagella, both of which prime Salmonella for further invasion (5). These invasion-primed Salmonella bacteria are released into the lumen through extrusion of epithelial cells that contain hyperreplicating bacteria, initiating secondary infections in neighboring cells, indicating that invasive Salmonella can disseminate through bacterium-induced extrusion of mucosal epithelia.
Many unnecessary cellular components or invading pathogens can be engulfed by a double-membrane structure (autophagosome) and delivered to the lysosome for degradation (7). This process is called autophagy. Various host components are involved in the autophagy pathway, such as LC3, Atg5, and p62. LC3 is present in two forms, cytosolic LC3-I and autophagosome-associated LC3-II that is converted from a fraction of LC3-I (8). The levels of LC3-II correlate with autophagosome formation, and green fluorescent protein (GFP)-LC3 is therefore traditionally used as a marker for the autophagosomes (8). Atg5, through association with Atg12 to form a conjugation complex, mediates the conjugation of LC3 to autophagosomes (9). p62 (also called sequestosome 1) interacts with LC3 and ubiquitin that usually serves as a molecular tag for protein aggregates, thus functioning as an adaptor protein to target proteins to autophagosomes (7, 10). Many pathogens can be targeted by autophagy in the cytosol. Some pathogens (group A Streptococcus and Mycobacterium tuberculosis) can be killed by autophagy, while others (Shigella flexneri, Listeria monocytogenes, and Yersinia pseudotuberculosis) are able to antagonize the autophagy pathway or exploit autophagosomes for their benefit.
Salmonella can damage the SCV through SPI-1 components (11). It has been suggested that autophagy targets a population of Salmonella found in damaged SCVs or in the cytosol for degradation and hence restricts Salmonella replication. Paradoxically, a recent report showed that knockdown of Rab1, a GTPase required for autophagy of Salmonella, decreases Salmonella replication in HeLa cells (12). These contradictory results prompted us to reexamine the interaction of autophagy with Salmonella and track the fate of Salmonella targeted by autophagy by a live-cell imaging method. Contrary to previous reports, we showed that autophagy facilitates Salmonella replication in the cytosol of epithelial cells.
RESULTS
The association of p62 and/or LC3 with cytosolic Salmonella increases shortly after initiation of intracellular replication.
Like many other groups who have used HeLa cells to study the interaction of Salmonella with autophagy (13–16), we created HeLa cell lines stably expressing GFP-p62 or GFP-LC3 and infected them with Salmonella. Salmonella replicates in the SCV and the cytosol in epithelial cells, but its cytosolic replication accounts for the majority of its net replication (17). We aimed to determine the percentage of autophagy (p62 or LC3)-targeted Salmonella that is present in the cytosol over time and see how it correlates with intracellular Salmonella replication.
Both wild-type (WT) and ∆sifA Salmonella strains were used to infect HeLa cells, as the ∆sifA strain (compared to the WT) escapes more frequently from the SCV into the cytosol, where it is more likely to be targeted by autophagy (15, 18). The association of p62 and LC3 with both Salmonella strains was measured at 1, 5, and 8 h postinfection (hpi). As shown in Fig. 1A, a subpopulation of Salmonella was associated with GFP-p62 (p62+) but not GFP alone. The percentage of p62+ Salmonella gradually decreased over time (from 1 hpi to 8 hpi) (Fig. 1B), consistent with the previous report where the association of endogenous p62 with Salmonella has been examined (19).
FIG 1 .

The association of p62 with cytosolic Salmonella increases shortly after initiation of intracellular replication. (A) GFP-p62- or GFP-expressing HeLa cells were infected with wild-type (WT) or ∆sifA Salmonella. Cells at 1 hpi and 5 hpi were stained with LAMP-1 (red) and DAPI (cyan). “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. (B to D) Quantification of the percentage of p62+ WT or p62+ ∆sifA bacteria (B), the percentage of p62+ WT or p62+ ∆sifA bacteria that are not associated with LAMP-1 (C), and the percentage of LAMP-1− WT or LAMP-1− ∆sifA bacteria (D) in GFP-p62-expressing cells at 1, 5, and 8 hpi (mean ± SD, n = 3).
We then determined the intracellular location of p62+ Salmonella by examining the colocalization of these bacteria with LAMP-1, a marker for the SCV membrane (6). As shown in Fig. 1C, the percentage of LAMP-1− p62+ bacteria (cytosolic) out of total p62+ Salmonella increased from ~15% (WT) and ~22% (∆sifA strain) at 1 hpi to ~42% (WT) and ~52% (∆sifA strain) at 5 hpi. The presence of cytosolic p62+ Salmonella was confirmed by immunostaining for ubiquitin, a marker for Salmonella present in the host cell cytosol (20) (see Fig. S1A in the supplemental material). Since it is known that Salmonella replication in epithelial cells commences at 4.5 to 6 hpi (21), we conclude that the increased presence of cytosolic p62+ Salmonella at 5 hpi correlates with intracellular replication of Salmonella. It appears that such an increased presence of cytosolic p62+ Salmonella is proportional to the increased presence of cytosolic bacteria in the total Salmonella population (from 1 hpi to 5 hpi) (Fig. 1D). In line with this, the ∆sifA strain that showed a slightly higher (although insignificantly) percentage of LAMP-1− bacteria than the WT at 5 hpi had a higher percentage of cytosolic p62+ bacteria (Fig. 1B to D). Interestingly, although the percentage of p62+ Salmonella decreased significantly from 5 hpi to 8 hpi, the percentage of LAMP-1− p62+ Salmonella and the percentage of LAMP-1− Salmonella did not change significantly between these two time points (Fig. 1C and D).
LC3 associated with Salmonella (WT and ∆sifA strains) in a manner that mimicked p62-Salmonella association (see Fig. S1A to D in the supplemental material). Most Salmonella-associated LC3 signals represent autophagosome structures (see Fig. S1E), since the association of LC3 with Salmonella (LC3+ Salmonella) is dependent on Atg5, a protein required for autophagosome formation (9). An electron microscopy study suggested that autophagosome structures were associated with Salmonella that is present in the cytosol (the bacterium indicated by the black arrowhead in Fig. S1F to Fiii) or partially exposed to the cytosol (bacteria indicated by black arrowheads in Fig. S1Fi) through damaged SCVs, consistent with previous reports (15, 16). The association of LC3 with Salmonella also requires p62, since cells treated with p62 small interfering RNA (siRNA) showed decreased colocalization of LC3 with Salmonella (see Fig. S1E). In support of this result, p62 and LC3 colocalized extensively, targeting the same bacterium (see Fig. S1G and H). This is also consistent with the previous report where the association of endogenous p62 with transiently expressed LC3 has been examined (19).
Collectively, these results not only confirm that p62 and LC3 participate in the autophagy pathway and associate with Salmonella but also suggest that the association of p62 and/or LC3 with cytosolic Salmonella correlates with Salmonella replication.
Dynamic association of p62 and/or LC3 correlates with Salmonella replication in the cytosol of HeLa cells.
To further probe this area, we examined the fate of Salmonella associated with p62 and/or LC3 by a live-cell imaging analysis method.
First, GFP-LC3-expressing HeLa cells were infected with monomeric Kusabira orange fluorescent (mKO)-expressing WT Salmonella (WT-mKO). As shown in Fig. 2A and Movie S1 in the supplemental material, LC3 associated with Salmonella (GFP-LC3+) in multiple patterns. In one case, GFP-LC3 puncta (Fig. 2A, arrowhead labeled “2”) gradually expanded and associated with Salmonella from two poles of the same bacterium or one pole of each dividing bacterium. These puncta then transiently fused with each other, completely surrounding the bacterium. In less than 30 min, they disappeared from one pole, accompanied by the completion of cell division of this bacterium. In other cases, GFP-LC3 puncta associated with Salmonella at one side or one pole of the bacterium (Fig. 2A, arrowheads labeled “1” and “3”). Overall, GFP-LC3 puncta moved rapidly in the cell and appeared to transiently associate with Salmonella, highlighting the dynamic association of LC3 with Salmonella. Some bacteria, appearing in light red (e.g., arrowheads labeled “1,” “2,” and “3”), associated with GFP-LC3 extensively and replicated in less than 30 minutes, as shown previously for hyperreplicating cytosolic bacteria (5). In contrast, other bacteria, appearing in bright red (arrowheads labeled “5” and “6”), did not associate with GFP-LC3 and were not replicating; these bacteria are likely in SCVs. LAMP-1 staining of Salmonella-infected cells suggested the light-red GFP-LC3+ bacteria represent Salmonella present in damaged SCVs or in the cytosol of host cells, whereas the bright-red GFP-LC3− bacteria represent Salmonella in intact SCVs (see Fig. S2A). Real-time analysis of GFP-LC3-expressing cells infected with wild-type cyan fluorescent protein (CFP)-expressing Salmonella (WT-CFP) in the presence of LysoTracker (which labels acidic compartments, including the SCV) also showed that GFP-LC3+ Salmonella was not in an intact SCV (see Movie S2).
FIG 2 .

Dynamic association of p62 and/or LC3 correlates with Salmonella replication in the cytosol of HeLa cells. (A) GFP-LC3-expressing cells were infected with mKO-expressing Salmonella. After infection for 4.5 h (0 min), cells were imaged at 30-s intervals with a spinning-disc confocal microscope. A series of live-cell imaging data is shown, and the elapsed time (minutes) is shown in the corner of each picture. Arrowheads 1, 2, 3, and 4 indicate replicating bacteria, whereas arrowheads 5 and 6 indicate nonreplicating bacteria. Scale bar, 10 µm. (B) GFP-p62-expressing cells were infected with mKO-expressing Salmonella. After infection for 4.5 h (0 h), the cells were imaged at 20-s intervals with a spinning-disc confocal microscope. Boxed areas indicate where p62 associates with cytosolic Salmonella. Arrows 1 to 5 indicate bright bacteria that are not replicating; arrowheads 6 and 7 in column 1 (0 h) indicate light-red bacteria that start to replicate. Scale bar, 5 µm.
Next, GFP-p62-expressing cells were infected with WT-mKO. During 4 h of real-time imaging, a subset of cytosolic Salmonella associated with p62 extensively and replicating rapidly (Fig. 2B, bacteria in light red in boxed areas; see also Movie S3). In contrast to p62+ bacteria, most Salmonella bacteria in intact SCVs (bright red) showed very limited association with p62 (Fig. 2B, bacteria labeled by arrows 2 to 5; see also Movie S3). These bacteria were not replicating.
Last, cells expressing GFP-p62 and mRFP-LC3 were infected with WT-CFP. The same bacterium clearly associated with both p62 and LC3 and was replicating in less than 30 min (see Fig. S2B, bacteria indicated by white arrows, and Movie S4). Many mRFP-LC3 puncta frequently and rapidly moved along the surface of Salmonella bacteria, with GFP-p62 signals being redistributed to dividing bacteria. Future work is to determine if/how the redistribution of p62 affects the traffic pattern of LC3 (autophagosomes) to the surface of Salmonella bacteria.
Overall, live-cell imaging analysis demonstrates that a subpopulation of Salmonella is dynamically associated with p62 and LC3 while replicating in the cytosol of epithelial cells.
p62 and autophagy support Salmonella replication in the cytosol of HeLa cells.
To investigate whether Salmonella benefits from p62 and LC3 for its replication in the cytosol, we determined if depletion of autophagy proteins by siRNA would decrease the cytosolic replication of Salmonella, using a gentamicin protection assay and a microscopic analysis that quantifies the percentage of infected cells containing different numbers of bacteria per cell.
The replication of WT Salmonella in HeLa cells treated with control siRNA or siRNA targeting p62, LC3 (including isoforms LC3A, LC3B, and LC3C) (22), Atg5, or p62 and LC3 was first determined. Western blot analysis confirmed that LC3, Atg5, and p62 were effectively knocked down in cells treated with corresponding siRNA (Fig. 3A). Knockdown of LC3, Atg5, or p62 and LC3 by siRNA significantly decreased autophagy, as evidenced by the decreased levels of LC3-II (Fig. 3A). In contrast, knockdown of p62 did not affect autophagy, as the level of LC3-II remained similar in control siRNA- and p62-siRNA-treated cells. Knockdown of p62, LC3, Atg5, or p62 plus LC3 did not affect the invasion ability of Salmonella, as assessed by the population of infected cells containing different numbers of bacteria at 2 hpi (Fig. 3B, top). However, the replication of Salmonella in cells treated with p62, LC3, Atg5, or p62 and LC3 siRNA was significantly reduced compared to that in cells treated with control siRNA at 6 and 8 hpi (Fig. 3C). Knockdown of Atg16L1, another subunit of the large protein complex containing the Atg5-Atg12 conjugate (23), decreased Salmonella replication to a similar extent as knockdown of Atg5 (see Fig. S3A and B). The decreased replication of Salmonella in cells treated with p62, LC3, Atg5, or p62 and LC3 siRNA was not due to detachment of infected cells from plates at 6 hpi, as all cells showed similar cell viability (see Fig. S3C). The knockdown of p62 and LC3 did not have an additive effect on the replication of Salmonella compared to the knockdown of p62 or LC3 alone (Fig. 3A), indicating that p62 and LC3 are involved in the same pathway that facilitates Salmonella replication. Additionally, control siRNA-treated cells showed a greater percentage of cells with high numbers of bacteria than autophagy protein siRNA-treated cells at 6 hpi, concomitant with a decreased percentage of cells with low numbers of bacteria (Fig. 3B, bottom). At 8 hpi, a decrease in the bacterial replication was observed, suggesting that some of the infected cells may have detached from plates.
FIG 3 .

p62 and autophagy favors cytosolic replication of Salmonella. (A) Western blot analyses of HeLa cells treated with control siRNA (si-NT) or siRNA targeting p62 (si-p62), LC3 (si-LC3), Atg5 (si-Atg5), and p62 and LC3 (si-p62+LC3). These cells were infected with WT Salmonella. Calnexin was used as a loading control. (B) Microscopic analyses of infected cells containing 1 to 5, 6 to 10, 11 to 20, or greater than 20 bacteria per cell at 2 hpi (top) and 6 hpi (bottom). The number of bacteria in at least 50 infected cells was enumerated, and the percentage of cells containing the indicated number of bacteria was presented (mean ± SD, n = 3). (C) Replication of Salmonella was compared in HeLa cells used in panel A. The fold replication was calculated as fold increases of the number of bacteria at late time points compared to the number of bacteria at 2 hpi (mean ± SD, n = 3). (D) HeLa cells were treated with control siRNA (si-NT) or siRNA targeting TBK1 (TANK-binding kinase-1) (si-TBK1), TBK1 and LC3 (si-TBK1+LC3), and TBK1 and ATG5 (si-TBK1+ATG5) and infected with Salmonella for 2 h (top) or 6 h (bottom). The percentage of cells containing the indicated number of bacteria was quantified as in panel B (mean ± SD; n = 3). (E) GFP-LC3-expressing HeLa cells were treated with si-NT, si-TBK1, si-TBK1+LC3, or si-TBK1+Atg5. The percentage of LC3+ bacteria or LAMP-1− bacteria at 5 hpi was quantified (mean ± SD, n = 3). (F) Western blot analyses of HeLa cells used in panel D.
As discussed above, a larger fraction of ∆sifA bacteria (compared to WT bacteria) was present in the cytosol and associated with LC3. As expected, ∆sifA bacteria exhibited the same replication defect as WT Salmonella in LC3 siRNA-treated cells (see Fig. S3D and E).
TBK1 helps to maintain the integrity of vacuolar membranes during intracellular bacterial infection, and knockdown of TBK1 leads to the disruption of SCVs, resulting in release of Salmonella into the host cell cytosol, where Salmonella hyperreplicates (24). GFP-LC3-expressing and regular HeLa cells were treated with siRNA that targets TBK1, TBK1 and LC3 (TBK1-LC3), or TBK1 and Atg5 (TBK1-Atg5), followed by infection with Salmonella. Consistent with the previous report (24), TBK1 siRNA-treated cells showed a higher percentage of LAMP-1− Salmonella at 5 hpi and had a significantly higher percentage of cells containing more than 20 bacteria than control siRNA-treated cells at 6 hpi (Fig. 3D, bottom; Fig. 3E, right). Furthermore, TBK1 siRNA-treated cells showed a slightly but insignificantly higher percentage of LC3+ Salmonella bacteria than control siRNA-treated cells (Fig. 3E, left). However, the percentage of LC3+ bacteria dropped dramatically when cells depleted of TBK1 were further treated with LC3 siRNA or Atg5 siRNA; this correlated with a significant decrease in the percentage of cytosolic bacteria (LAMP-1−) and cells containing more than 20 bacteria in TBK1-LC3 siRNA- or TBK1-Atg5 siRNA-treated cells (compared to TBK1 siRNA-treated cells) at late time points (Fig. 3D, bottom; Fig. 3E, right). These differences were not caused by a different invasion rate of Salmonella to cells, as the numbers of intracellular bacteria at 2 hpi are similar among different siRNA-treated cells (Fig. 3D, top). The knockdown efficiency of each protein was confirmed by Western blotting (Fig. 3F). Thus, knockdown of autophagy components decreases the hyperreplication of cytosolic Salmonella in TBK1 siRNA-treated cells.
Taken together, these results demonstrate that p62 and autophagy support Salmonella replication in the cytosol of HeLa cells.
The effector SopB is required for Salmonella to associate with autophagosomes.
It has been shown that Salmonella autophagy requires the SPI-1 T3SS (16). As multiple effectors are translocated via the SPI-1 T3SS, we attempted to determine which effector(s) could increase the association of autophagosomes with Salmonella and thus promote Salmonella replication in HeLa cells.
First, we examined the colocalization of LC3 with the WT and several Salmonella mutants (the ∆invA, ∆sopB, ∆sopE ∆sopE2, ∆sptP, ∆sipA, ∆sipB, and ∆ssaR mutants). The ∆invA (invA encodes an inner membrane protein) and ∆sipB mutants do not translocate any SPI-1 effectors (including SipB itself) (25), whereas the ∆ssaR mutant does not translocate any SPI-2 effectors into host cells (26). ∆invA bacteria barely associated with LC3, while ∆ssaR bacteria associated with LC3 to the same extent as the WT (16). These two mutants were used as controls. The remaining mutants lack one or two SPI-1 effectors that can promote Salmonella replication rate or invasion ability (27, 28). Unlike other mutants, ∆invA and ∆sipB bacteria are unable to invade into epithelial cells (data not shown) and therefore are used in a coinfection model as described previously (21). It is believed coinfection of cells with WT and ∆invA (or ∆sipB) bacteria facilitates the SPI-1-dependent cointernalization of ∆invA (or ∆sipB) bacteria with the WT. As shown in Fig. 4A, ∆sipB and ∆invA mutants had little association with LC3, whereas ∆sopE ∆sopE2, ∆sptP, ∆sipA, ∆ssaR, and WT strains were associated with LC3 at similar levels. ∆sopB bacteria were also able to associate with LC3, albeit less frequently than the WT. The decreased association of LC3 with ∆sopB could be rescued by overexpression of SopB in the ∆sopB mutant (∆sopB/pACDE mutant).
FIG 4 .

The effector SopB is required for Salmonella to associate with autophagosomes. (A) GFP-LC3-expressing cells were infected with the indicated strains. The ∆sopB/pACDE mutant expresses the wild-type SopB in a ∆sopB background. Unlike other strains that are individually used to infect cells, ∆invA and ∆sipB bacteria were used in a coinfection model. The percentage of LC3+ bacteria at 5 hpi was quantified for each strain (mean ± SD, n = 3). (B) Western blot analysis of HeLa cells used in panel A. (C) HeLa cells treated with si-NT or si-LC3 were infected with the indicated strains. The fold replication of these strains at 6 hpi was determined (mean ± SD, n = 3).
Next, the replication of the above-described mutants, except the ∆sipB mutant in control siRNA- and LC3 siRNA-treated cells, was measured. We reasoned that if a mutant showed low (compared to that of the WT) or no association with autophagosomes, its replication would be independent of autophagy. Western blot analysis confirmed that LC3 was effectively depleted by siRNA treatment (Fig. 4B). As shown in Fig. 4C, LC3 siRNA treatment compared to control siRNA treatment decreased the replication of all mutants except the ∆invA mutant, although the decrease for the ∆sopB mutant was not as large as that observed for other mutants and the WT. Complementation of the ∆sopB mutant with a plasmid (pACDE) expressing SopB partially restored the replication of the ∆sopB mutant in control siRNA-treated cells but not in LC3 siRNA-treated cells. Thus, SopB is able to enhance Salmonella replication through increasing the interaction of Salmonella with autophagy. Unlike the WT and other mutants, ∆invA bacteria did not show a replication defect (or benefit) in LC3 siRNA-treated cells in a coinfection model (Fig. 4C, right). This suggests that even within the same cell, the replication/survival of the ∆invA mutant, but not the WT, is independent of autophagy.
Collectively, these data suggest that, together with the T3SS apparatus proteins InvA and SipB, the effector SopB is required for Salmonella to associate with autophagosomes for replication in HeLa cells.
Cells containing hyperreplicating bacteria and showing decreased autophagy are positive for caspase-1 and caspase-3/7 activation and undergo cell death.
Cells containing hyperreplicating Salmonella can be extruded out of the epithelial monolayer and undergo cell death, releasing intracellular bacteria into the lumen that facilitate the dissemination of Salmonella to neighboring cells (5). Based on this concept, we tracked the fate of the hyperreplicating bacterium-containing (HRBC) cells. A real-time analysis of a Salmonella-infected GFP-LC3-expressing cell showed that a subset of Salmonella was hyperreplicating in the cytosol where it frequently associated with LC3 at 4.5 hpi (see Movies S1 and S5). The hyperreplication of Salmonella was accompanied by the decrease of LC3 levels in the cell at late time points. Such decreased LC3 signals correlated with lower levels of autophagosome formation (i.e., decreased autophagy), as bafilomycin (a drug used to block the fusion of autophagosomes with lysosomes) treatment did not increase LC3 signals in HRBC cells (data not shown). The decreased autophagy was observed in more than 90% of HRBC cells. As shown in Movie S5, the HRBC cell eventually detached from the plate. We speculated that the cell could be undergoing cell death, which led to its detachment from the plate. Consistent with this hypothesis, most (~86%) HRBC cells displaying decreased autophagy (as judged by the low level of p62 signals colocalized with Salmonella) showed abnormal (such as chromatin condensation and DNA fragmentation) nuclear morphology at 8 hpi (Fig. 5A and B). In contrast, only ~33% of HRBC cells having normal autophagy (as judged by a relatively high level of p62 staining colocalized with Salmonella) contained abnormal nuclei (Fig. 5B). Figure 5C shows an HRBC cell with relatively enhanced autophagy exhibiting normal nuclear morphology.
FIG 5 .

Cells containing hyperreplicating bacteria and showing decreased autophagy are positive for caspase-1 and caspase-3/7 activation and undergo cell death. (A) GFP-p62-expressing HeLa cells were infected with Salmonella for 8 h and stained with DAPI (blue). A cell containing hyperreplicating bacteria (more than 50 bacteria per cell, i.e., HRBC cells) and showing abnormal nuclear morphology is shown. Scale bar, 10 µm. (B) At least 100 HRBC cells were divided into two groups. The first group exhibited a low level of p62 signals colocalized with Salmonella (<1% of intracellular bacteria), and the second group exhibited a relatively high level of p62 signals (>2% of intracellular bacteria). The number of cells showing abnormal nuclear morphology (such as chromatin condensation and DNA fragmentation) was further quantified and expressed as the percentage of cells with abnormal nuclei in each group of cells (mean ± SD, n = 3). (C) A GFP-p62-expressing HRBC cell shows a relatively enhanced autophagy and normal nuclear morphology. Scale bar, 10 µm. (D) Cells stably expressing monomeric red fluorescent protein (mRFP)-LC3 were infected with WT Salmonella for 7 h and stained with DAPI (blue) and active caspase-1 probe FAM-YVAD-FMK (green) or caspase-3/7 probe FAM-DEVD-FMK (similar to caspase-1 staining [data not shown]). (E) Quantification of the percentage of casapase-1 (left)- or caspase-3/7 (right)-positive cells in uninfected cells, HRBC cells showing decreased autophagy, and HRBC cells showing normal autophagy at 8 hpi (mean ± SD, n = 3).
We next sought to determine if HRBC cells with decreased autophagy were positive for active caspase-1 and caspase-3/7, both of which have been shown to be activated and capable of inducing cell death in extruding HRBC cells (5). As shown in Fig. 5D and E, HRBC cells with normal autophagy (as measured by the level of mRFP-LC3 associated with bacteria) and uninfected cells were negative for casapse-1 or caspase-3/7 staining, whereas the majority of HRBC cells with decreased autophagy were positive for active casapase-1 and caspase-3/7.
Taken together, these results suggest that hyperreplication of Salmonella accompanied by decreased autophagy and enhanced caspase-1 and caspase-3/7 activation promotes cell death, potentiating cell detachment at later stages of infection.
DISCUSSION
We propose that the p62-dependent autophagy pathway favors the replication of Salmonella in the cytosol of HeLa cells (Fig. 6). This concept is based on the following observations: (i) the increased association of p62 or LC3 with cytosolic Salmonella correlates with Salmonella replication; (ii) the replication of WT Salmonella and the ∆sifA mutant (a strain that escapes into the cytosol more often than the WT at 5 hpi) was reduced when p62, LC3, Atg5, or Atg16L1 was depleted; (iii) the replication of cytosolic Salmonella in TBK1 siRNA-treated cells (which increases the number of cytosolic bacteria) was reduced when autophagy was inhibited; (iv) p62 and LC3 belong to the same pathway to facilitate Salmonella replication; (v) most convincingly, live-cell imaging analyses demonstrated that a subset of cytosolic Salmonella associated extensively with p62 and/or LC3 and replicated quickly (in contrast, most intravacuolar Salmonella bacteria ineffectively associated with p62 and/or LC3 and replicated at much lower rates than cytosolic Salmonella); and (vi) hyperreplication of Salmonella in the cytosol concomitant with decreased autophagy and enhanced casapase-1 and caspase-3/7 activation potentiates cell death, leading to the detachment of HRBC cells at later times postinfection.
FIG 6 .
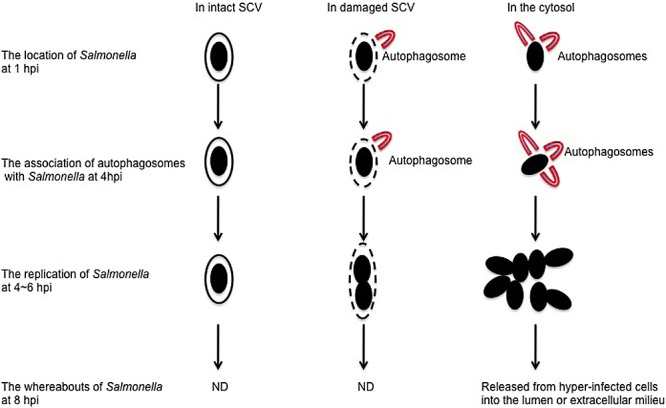
A proposed model for p62-dependent autophagy favoring Salmonella replication in the cytosol of HeLa cells. In this proposed model, Salmonella likely acquires nutrients supplied by autophagy for its replication. At least three populations of bacteria are present in infected HeLa cells. The first population of bacteria remains in intact Salmonella-containing vacuoles (SCVs). These bacteria replicate slowly, as they do not associate with autophagosomes and are difficult to acquire nutrients from the cytosol. The second population of bacteria is partially exposed to the cytosol through the gaps between damaged SCVs (16). These bacteria are ineffectively coated with ubiquitin and associated with autophagosomes to a low level. They acquire a limited amount of nutrients for replication and replicate faster than the first population of bacteria. The third population of bacteria is completely exposed to the cytosol. These bacteria are effectively coated with ubiquitin and associated with autophagosomes to a high level. They acquire sufficient nutrients from the cytosol and replicate the fastest. Notably, some cells containing the third population of bacteria will undergo cell death and detach from the epithelial layer, resulting in the release of hyperreplicating bacteria that are capable of initiating secondary infections in neighboring cells. The whereabouts of the first and second populations of bacteria at 8 hpi were not determined (ND).
This model challenges the current dogma that p62 and autophagy protect host cells against Salmonella infection (16, 19, 29). The differing conclusions could be due to off-target effects of siRNA when used at high concentrations (~20 to 50 nM) by other groups as opposed to 1 nM in our study or incomplete dissociation of membrane-associated Salmonella when quantifying intracellular bacterial numbers. For instance, it is possible that the 0.1% Triton X-100 lysis buffer used in several seminal studies (14, 22, 30) dissociates autophagosome-associated Salmonella less efficiently than the 1% Triton X-100 and 0.1% SDS buffer used in this study. The time point at which bacteria were quantified may also be important. Zheng et al. (19) demonstrated that p62 limits Salmonella replication at 10 hpi, whereas we quantified bacteria at 6 hpi to avoid analysis when the host cells begin to detach or die at late time points (5). Additionally, the fate of autophagy-targeted Salmonella could be cell type dependent. In the first study examining the role of autophagy in Salmonella replication (16), Birmingham et al. examined the colocalization between autophagy markers and bacteria in HeLa cells and mouse embryonic fibroblasts (MEFs) but quantified only the effect of autophagy inhibition in MEFs. Thus, it is possible that inhibiting autophagy increases Salmonella replication in MEFs as they reported but has the opposite effect in HeLa cells, as we observed in this study.
Interestingly, Huang et al. recently showed that knockdown of Rab1, a GTPase required for autophagy of Salmonella, decreases Salmonella replication in HeLa cells (12). Although they interpreted this result as a distinct role for Rab1 in supporting Salmonella replication in vacuoles, a recent live-cell imaging study (17) convincingly shows that cytosolic replication of Salmonella accounts for the majority of the net replication of Salmonella. Thus, their observation with Rab1 knockdown could be explained by our model in which inhibiting autophagy reduces cytosolic Salmonella replication.
Autophagy also benefits several other bacterial pathogens, including Legionella pneumophila, Coxiella burnetii, and Yersinia pseudotuberculosis (29). Two mechanisms are proposed to contribute to the beneficial effects of autophagy on bacterial pathogens. First, through autophagolysosomal fusion, autophagosomes may serve as a source of nutrients for intracellular pathogens (7). This mechanism could partly explain why a previous study found that inhibition of autophagy by chloroquine (a drug that may block autophagolysosomal fusion, in addition to blocking acidification of the SCV) decreases Salmonella replication (31, 32). In support of this hypothesis, we were able to identify that oleic acid (a type of free fatty acid), but not amino acids, is likely to support Salmonella replication through autophagy (see Fig. S4A to C). However, the free fatty acid is not the limiting factor for Salmonella replication, since supplementation of the free fatty acid into autophagy-deficient cells does not rescue the replication defect of Salmonella (see Fig. S4C). There are likely other nutrients supplied by autophagy for Salmonella replication. Autophagosomes may also serve as a protected niche for intracellular pathogens (7). We speculate that before a complete fusion of the autophagosome and the lysosome, the autophagosome ensures Salmonella replication in a less acidic environment and/or accommodates replicating bacteria through constant membrane elongation. This hypothesis awaits further study.
In addition to confirming that Salmonella autophagy requires the SPI-1 T3SS, this study identifies that SipB is the key player mediating Salmonella-autophagosome association. Possibly through its pore-forming activity, SipB damages the SCV, allowing Salmonella to access the host cell cytosol where it can access the autophagy machinery. Alternatively, SipB translocated into the host cell cytosol induces the formation of autophagosomes (33), which in turn associate with ubiquitinated Salmonella. However, it is also possible that SipB functions as a translocon component to translocate other effectors that mediate Salmonella-autophagosome association. Unlike ∆sipB bacteria that were barely associated with autophagosomes, ∆sopB bacteria showed a moderately decreased association with autophagosomes, suggesting that SopB plays a lesser role than SipB in mediating Salmonella-autophagosome association. Since SopB can be ubiquitinated in host cells over a prolonged period (34), it will be interesting to determine if the ubiquitination of SopB enhances Salmonella-autophagosome association.
All together, our various cell biology-based assays, especially the live-cell imaging analyses, provide compelling evidence that Salmonella benefits from p62-dependent autophagy for its replication in the cytosol of HeLa cells.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Wild-type Salmonella (Salmonella enterica serovar Typhimurium strain SL1344) and its isogenic mutant strains were cultured as described previously (21). Details of various Salmonella strains and plasmids are summarized in Table S1 in the supplemental material.
Cell culture.
HeLa human epithelial cells (ATCC CCL-2) and 293T/17 human kidney epithelial cells (ATCC CRL-11268) were maintained in Dulbecco’s modified Eagle medium (DMEM) high glucose (Thermo Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% nonessential amino acids (Invitrogen), and 1% GlutaMax (Gibco). Cells were used from passages 5 to 15.
Generation of stable cell lines with lentivirus infection.
Lentivirus constructs pLVX-GFP-LC3WT, pLVX-GFP, pLVX-GFP-p62, and pLVX-mRFP-LC3 were individually packaged into lentivirus particles in 293T/17 cells using the lentiviral packaging mix (Sigma). HeLa cells were infected with these lentivirus particles in the presence of 8 µg/ml Polybrene (Millipore) and selected in DMEM containing 3 µg/ml puromycin (Sigma) for 2 weeks. A single-cell-derived stable cell line was further obtained by culturing cells in a 96-well plate with a limiting dilution procedure.
Bacterial infection and gentamicin protection assay.
Salmonella was cultured in Luria broth (LB) overnight at 37°C with shaking (200 rpm), followed by dilution into 10 ml of fresh LB (1:33), and continued to grow under the same conditions for 3 h. One milliliter of bacteria was then centrifuged at 8,000 × g for 2 min and resuspended in 1 ml of phosphate-buffered saline (PBS) containing Ca2+ and Mg2+ (PBS+/+, pH 7.4). This suspension was diluted in DMEM (no antibiotics) at 1:500, and 335 µl of diluents was added directly to HeLa cells to give a multiplicity of infection (MOI) of 10. After 10 min of infection, the monolayers were washed three times in PBS+/+ and then incubated in fresh culture medium. After 20 min, fresh culture medium containing 100 µg/ml gentamicin was added. At 2 hpi, the gentamicin concentration was reduced to 10 µg/ml. For coinfection experiments, wild-type Salmonella and mutant strains were mixed at a ratio of 1:40 before infection. At designated time points, cells were washed three times with PBS and lysed in 250 µl of lysis buffer (PBS with 1% Triton X-100 and 0.1% SDS). Bacteria released from cells were plated onto an LB agar plate (with 100 µg/ml streptomycin and 50 µg/ml kanamycin to select mutant strains) to obtain the bacterial CFU. The number of bacteria recovered at 2 hpi defines the invasion ability of Salmonella, whereas the fold increase of CFU at later time points versus the CFU at 2 hpi is referred to as fold replication and defines the replication ability of Salmonella.
Transient transfection with siRNA.
All small interfering RNAs (siRNAs) are from a pool of four single siRNAs that targets the same gene (SMARTpool siRNA; Dharmacon RNA Technologies). The following siRNA SMARTpools (catalog no.) are used: p62 siRNA (M-010230-00), Atg5 siRNA (M-004374-04), Atg16L1 siRNA (M-021033-02), TBK1 siRNA (M-003788-02), LC3A siRNA (M-013579-00), LC3B siRNA (M-012846-01), LC3C siRNA (M-032399-01), and nontargeting scrambled control siRNA (D-001206-13). For LC3 knockdown, 1 nM of each LC3A, LC3B, and LC3C siRNA was used for transfection. One day before transfection with siRNA, HeLa cells were seeded at densities of 7.5 × 105 per 10-cm dish in 8 ml DMEM without antibiotics. Cells were transfected with 1 nM siRNA using INTERFERin (Polyplus Transfection) according to the manufacturer’s instructions. One day after transfection with siRNAs, cells were trypsinized and seeded into a 24-well plate at a density of 4 × 104 cells per well. Bacterial infections were performed 48 h posttransfection.
Western blot analysis.
Cells were washed three times with PBS and lysed in NP-40 buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Nonidet P-40, 10 mM Na 4P2O7, 50 mM NaF) supplemented with a complete protease inhibitor tablet (Roche). The protein of interest was then detected by the standard immunoblotting method.
Antibodies and reagents.
Antibodies used for Western blotting and immunofluorescence were anti-p62 (BD Transduction Laboratories), anti-LC3, anti-Atg16L1, and anti-ubiquitin (MBL International Incorporation), anti-Atg5 (Cell Signaling), anti-TBK1 (Imgenex), anti-FLAG and anti-calnexin (Sigma), anti-LAMP-1 (clone H4A3), and Alexa Fluor 488 and Alexa Fluor 568 (Invitrogen). Dimethyl sulfoxide (DMSO) and oleic acid (conjugated to albumin) were purchased from Sigma. Nonessential amino acids at the final concentration of 10× and essential amino acids at the final concentration of 5× (Invitrogen) were supplemented into the medium for 3 h before infection; oleic acid (200 µM) was added into the medium for 15 h before infection. All the above-named reagents remained in the medium throughout the subsequent infection.
Cell viability assay.
Equal volumes of cells (including floating cells in the medium) and 0.4% trypan blue (Invitrogen) were mixed and incubated at room temperature for 3 min. The stained cells were visualized by a bright-field microscope. The viable cell will exclude trypan blue and have a clear cytoplasm, whereas a nonviable cell will take up trypan blue and have a blue cytoplasm. At least 100 cells were counted and used to calculate the percentage of viable cells.
Immunofluorescence staining and fluorescence microscopy.
Cells were fixed by 4% paraformaldehyde (PFA) for 30 min at room temperature, followed by washing with PBS+/+ twice. Excess PFA was quenched by incubation of the cells with 50 mM NH4Cl for 10 min. Fixed cells were blocked and permeabilized by incubation buffer (IB; PBS+/+ containing 0.2% saponin, 10% normal goat serum) for 30 min. Permeabilized cells were sequentially incubated in IB containing primary (1 h) and secondary (1 h) antibody at room temperature. Coverslips that contain the cells were mounted on glass slides using ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) and sealed with nail polish.
All images are shown as representative confocal z-slices unless otherwise indicated. Images were taken with a multiphoton laser scanning microscope (Olympus FV1000MPE) operated by FV10-ASW software. Adobe Photoshop and Adobe Illustrator were used to process pictures exported from the FV10-ASW software. For quantification of bacteria associated with fluorescence signals, a Zeiss 510 metafluorescence microscope was used. For each condition, at least 100 bacteria or 50 infected cells were analyzed.
Live-cell imaging.
Cells were cultured on glass-bottom dishes (MatTek). In most cases, unless otherwise indicated, cells were washed with PBS containing Ca2+ and Mg2+ (pH 7.4; PBS+/+) twice at 2 hpi and imaged in Leibovitz’s L-15 medium (Invitrogen) containing 10% FBS, Oxyrase (1:100) (Oxyrase), and 10 µg/ml gentamicin. Cells were imaged with a high-speed spinning-disc confocal microscope (PerkinElmer) at designated intervals. Images were acquired and analyzed with Volocity three-dimensional (3D) image analysis software (Improvision). Movies were exported from the Volocity software in QuickTime movie format and edited with the iMovie software if necessary.
Live-cell staining.
One day before infection (MOI of 10), 1.5 × 105 cells were seeded into a 35-mm glass-bottom dish (MatTek). One hour before imaging, LysoTracker red (100 nM, Invitrogen) was added into the medium. Right before imaging with a spinning-disc confocal microscope, cells were washed with PBS+/+ twice and replaced with Leibovitz’s L-15 medium containing 10% FBS, Oxyrase (1:100), and 10 µg/ml gentamicin. To stain active caspase-1 or caspase-3/7, cells (at 7 hpi) grown on coverslips were incubated with FAM-YVAD-FMK or FAM-DEVD-FMK (Immunochemistry Technologies), respectively. One hour after the incubation, cells were further processed according to the instructions from the manufacturer and mounted on glass slides using ProLong Gold with DAPI.
Correlative light microscopy-electron microscopy (CLEM).
HeLa cells stably expressing EGFP-LC3 were cultured on glass-bottom dishes and infected with Salmonella for 5 h. The samples were processed similarly to those performed by Kageyama et al. (35). A spinning-disc confocal microscope (PerkinElmer Ultraview VoX) was used to identify cells containing GFP-LC3+ bacteria. A transmission electron microscope (Hitachi 7600) was used to take the electron micrographs of cells containing GFP-LC3+ bacteria.
Statistical analysis.
All analyses were performed with a 95% confidence interval using GraphPad Prism version 4.0, and the data were expressed as mean values ± standard deviations (SD). To calculate the P values, a two-tailed Student t test was used for Fig. 1B to D, 4C, 5B, and Fig. S1C, D, and G and S3D in the supplemental material; a one-way analysis of variance (ANOVA) with Dunnett’s post hoc test was used for Fig. 3C and E, 4A, 5E, and Fig. S1E and S3A and C in the supplemental material; a two-way ANOVA with Bonferroni posttest was used for Fig. 3B and D and Fig. S4A to C in the supplemental material (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
SUPPLEMENTAL MATERIAL
The association of LC3 with cytosolic Salmonella increases shortly after initiation of intracellular replication. (A) GFP-LC3-expressing HeLa cells were infected with WT Salmonella. Cells at 5 hpi were stained with DAPI (blue) and ubiquitin (Ub; red). (B) GFP-LC3-expressing HeLa cells were infected with WT or ∆sifA bacteria. Cells at 1 hpi and 5 hpi were stained with LAMP-1 (red) and DAPI (blue). In panels A and B, “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. (C and D) Quantification of the percentage of LC3+ bacteria (WT and ∆sifA strains) at 1 hpi, 5 hpi, and 8 hpi (C) and the percentage of LC3+ bacteria that are not associated with LAMP-1 at 1 hpi and 5 hpi (D) in GFP-LC3-expressing cells (mean ± SD, n = 3). (E) Quantification of the percentage of LC3+ bacteria in GFP-LC3-expressing cells treated with the indicated siRNAs at 5 hpi (mean ± SD, n = 3). (F) HeLa cells stably expressing GFP-LC3 were infected with the WT for 5 h. GFP-LC3+ bacteria were located with a spinning-disc confocal microscope and further processed for the electron microscopy. Black arrowheads indicate autophagosome structures. The boxed regions are magnified and shown in panels ii and iv. Panel ii shows an autophagosome-enclosed bacterium in a damaged SCV, whereas panel iv shows a dividing cytosolic bacterium close to autophagosomes. Scale bars, 500 nm. (G and H) HeLa cells expressing GFP-p62 and mRFP-LC3 were infected with WT Salmonella for 5 h. Cells were stained with DAPI (blue). (G) The percentage of LC3+ or LC3− bacteria associated with p62 (left) and the percentage of p62+ or p62− bacteria associated with LC3 (right) were quantified (mean ± SD, n = 3). (H) “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. Download
Dynamic association of p62 and/or LC3 correlates with Salmonella replication in the cytosol of HeLa cells. (A) HeLa cells infected with WT-mKO were stained with LAMP-1 (green) at 5 hpi. Bacteria completely surrounded by LAMP-1 (intact SCV) were bright red, whereas bacteria showing no (cytosolic) or discontinuous (likely within damaged SCV) LAMP-1 staining were light red. “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. (B) HeLa cells expressing GFP-p62 and mRFP-LC3 were infected with cyan fluorescent protein (CFP)-expressing Salmonella (gray). A snapshot of live-cell imaging data was shown. Arrows indicate dividing bacteria. Scale bar, 5 µm. Download
p62 and autophagy facilitate Salmonella replication in the cytosol of HeLa cells. (A) HeLa cells were treated with the indicated siRNAs and infected with Salmonella. The fold replication at 6 hpi was presented (mean ± SD, n = 3). (B) Western blot analysis of HeLa cells used in panel A. (C) Cells were treated with different siRNAs and infected with Salmonella. Viable cells are quantified by a trypan blue assay (mean ± SD, n = 3). (D) HeLa cells were treated with indicated siRNAs and infected with WT or ∆sifA bacteria. The fold replication at 6 hpi was presented (mean ± SD, n = 3). (E) Western blot analysis of HeLa cells used in panel D. Calnexin was used as a loading control. Download
Oleic acid, but not amino acids, is likely to promote Salmonella replication through autophagy. (A to C) HeLa cells were treated with control siRNA or LC3 siRNA and infected with WT Salmonella for 6 h in the absence of (UT) or the presence of different types of nutrients (nonessential amino acids [NEAA] [A]; essential amino acids [EAA] [B]; oleic acid, free fatty acid [FFA] [C]). The fold replication at 6 hpi was presented (mean ± SD, n = 3). The data showed that oleic acid (but not NEAA) enhanced Salmonella replication in control siRNA-treated cells but not in LC3 siRNA-treated cells. Download
Bacterial strains and plasmids used in this study.
Time-lapse micrographs of GFP-LC3-expressing cells infected with mKO-expressing Salmonella. Images were taken at 4.5 hpi. Arrows indicate dividing bacteria. Download
Time-lapse micrographs of GFP-LC3-expressing cells infected with CFP-expressing Salmonella (gray). One hour before imaging (at 3.5 hpi), cells were incubated with the LysoTracker (red). The GFP-LC3+ bacterium (indicated by the arrow) did not colocalize with the LysoTracker, indicating that it is in the cytosol. Download
Time-lapse micrographs of GFP-p62-expressing cells infected with mKO-expressing Salmonella. Cells were imaged at 4.5 hpi (00:00:00). Arrows indicate dividing bacteria. Download
Time-lapse micrographs of GFP-p62- and mRFP-LC3-expressing cells infected with CFP-expressing Salmonella (gray). Images were taken at 4.5 hpi. Arrows indicate dividing bacteria associated with GFP-p62 and mRFP-LC3. Download
Time-lapse micrographs of GFP-LC3-expressing cells infected with mKO-expressing Salmonella. Images taken at different time points were collected and edited with iMovie software. Download
ACKNOWLEDGMENTS
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) (B.B.F. and L.J.F.), a postdoctoral fellowship from CIHR and Inimex Pharmaceuticals, Inc. (H.B.Y.), a postdoctoral fellowship from the CIHR (R.B.R.F.), a Canadian Association of gastroenterology/CIHR/Ferring Pharmaceuticals postdoctoral fellowship (M.A.C.).
B.B.F. is the University of British Columbia Peter Wall Distinguished Professor. L.J.F. is the Canada Research Chair in quantitative proteomics. No competing interests exist for this work.
We thank members of the Finlay lab for critical reading of the manuscript, T. Ratajczak for providing the plasmid pCDNA3-GFP-p62, and G. Martens, B. Ross, and K. Hodgson for technical help on immunofluorescence microscopy and electron microscopy.
Footnotes
Citation Yu HB, Croxen MA, Marchiando AM, Ferreira RBR, Cadwell K, Foster LJ, Finlay BB. 2014. Autophagy facilitates Salmonella replication in HeLa cells. mBio 5(2):e00865-14. doi:10.1128/mBio.00865-14.
REFERENCES
- 1. Ohl ME, Miller SI. 2001. Salmonella: a model for bacterial pathogenesis. Annu. Rev. Med. 52:259–274. 10.1146/annurev.med.52.1.259 [DOI] [PubMed] [Google Scholar]
- 2. Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6:53–66. 10.1038/nrmicro1788 [DOI] [PubMed] [Google Scholar]
- 3. Jones BD, Ghori N, Falkow S. 1994. Salmonella Typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J. Exp. Med. 180:15–23. 10.1084/jem.180.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menendez A, Arena ET, Guttman JA, Thorson L, Vallance BA, Vogl W, Finlay BB. 2009. Salmonella infection of gallbladder epithelial cells drives local inflammation and injury in a model of acute typhoid fever. J. Infect. Dis. 200:1703–1713. 10.1086/646608 [DOI] [PubMed] [Google Scholar]
- 5. Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. 2010. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. U. S. A. 107:17733–17738. 10.1073/pnas.1006098107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brumell JH, Tang P, Zaharik ML, Finlay BB. 2002. Disruption of the Salmonella-containing vacuole leads to increased replication of Salmonella enterica serovar Typhimurium in the cytosol of epithelial cells. Infect. Immun. 70:3264–3270. 10.1128/IAI.70.6.3264-3270.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668. 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deretic V. 2010. Autophagy in infection. Curr. Opin. Cell Biol. 22:252–262. 10.1016/j.ceb.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roy D, Liston DR, Idone VJ, Di A, Nelson DJ, Pujol C, Bliska JB, Chakrabarti S, Andrews NW. 2004. A process for controlling intracellular bacterial infections induced by membrane injury. Science 304:1515–1518. 10.1126/science.1098371 [DOI] [PubMed] [Google Scholar]
- 12. Huang J, Birmingham CL, Shahnazari S, Shiu J, Zheng YT, Smith AC, Campellone KG, Heo WD, Gruenheid S, Meyer T, Welch MD, Ktistakis NT, Kim PK, Klionsky DJ, Brumell JH. 2011. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy 7:17–26. 10.4161/auto.7.1.13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birmingham CL, Brumell JH. 2006. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2:156–158 [DOI] [PubMed] [Google Scholar]
- 14. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. 2009. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10:1215–1221. 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- 15. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I. 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333:228–233. 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. 2006. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281:11374–11383. 10.1074/jbc.M509157200 [DOI] [PubMed] [Google Scholar]
- 17. Malik-Kale P, Winfree S, Steele-Mortimer O. 2012. The bimodal lifestyle of intracellular Salmonella in epithelial cells: replication in the cytosol obscures defects in vacuolar replication. PLoS One 7:e38732. 10.1371/journal.pone.0038732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249. 10.1093/emboj/19.13.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. 2009. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183:5909–5916. 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- 20. Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. 2004. Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr. Biol. 14:806–811. 10.1016/j.cub.2004.09.022 [DOI] [PubMed] [Google Scholar]
- 21. Steele-Mortimer O, Brumell JH, Knodler LA, Méresse S, Lopez A, Finlay BB. 2002. The invasion-associated type III secretion system of Salmonella enterica serovar Typhimurium is necessary for intracellular proliferation and vacuole biogenesis in epithelial cells. Cell. Microbiol. 4:43–54. 10.1046/j.1462-5822.2002.00170.x [DOI] [PubMed] [Google Scholar]
- 22. von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F. 2012. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 48:329–342. 10.1016/j.molcel.2012.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. 2003. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116:1679–1688. 10.1242/jcs.00381 [DOI] [PubMed] [Google Scholar]
- 24. Radtke AL, Delbridge LM, Balachandran S, Barber GN, O’Riordan MX. 2007. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 3:e29. 10.1371/journal.ppat.0030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collazo CM, Galán JE. 1997. The invasion-associated type III system of Salmonella Typhimurium directs the translocation of Sip proteins into the host cell. Mol. Microbiol. 24:747–756. 10.1046/j.1365-2958.1997.3781740.x [DOI] [PubMed] [Google Scholar]
- 26. Brumell JH, Rosenberger CM, Gotto GT, Marcus SL, Finlay BB. 2001. SifA permits survival and replication of Salmonella Typhimurium in murine macrophages. Cell. Microbiol. 3:75–84. 10.1046/j.1462-5822.2001.00087.x [DOI] [PubMed] [Google Scholar]
- 27. Ibarra JA, Steele-Mortimer O. 2009. Salmonella—the ultimate insider. Salmonella virulence factors that modulate intracellular survival. Cell. Microbiol. 11:1579–1586. 10.1111/j.1462-5822.2009.01368.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou D, Chen LM, Hernandez L, Shears SB, Galán JE. 2001. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 39:248–259. 10.1046/j.1365-2958.2001.02230.x [DOI] [PubMed] [Google Scholar]
- 29. Mostowy S, Cossart P. 2012. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 22:283–291. 10.1016/j.tcb.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 30. Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482:414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. 1993. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 90:10544–10548. 10.1073/pnas.90.22.10544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. 2007. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6:304–312. 10.1038/nrd2272 [DOI] [PubMed] [Google Scholar]
- 33. Hernandez LD, Pypaert M, Flavell RA, Galán JE. 2003. A Salmonella protein causes macrophage cell death by inducing autophagy. J. Cell Biol. 163:1123–1131. 10.1083/jcb.200309161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patel JC, Hueffer K, Lam TT, Galán JE. 2009. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell 137:283–294. 10.1016/j.cell.2009.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, Imamoto F, Noda T, Yoshimori T. 2011. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol. Biol. Cell 22:2290–2300. 10.1091/mbc.E10-11-0893 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The association of LC3 with cytosolic Salmonella increases shortly after initiation of intracellular replication. (A) GFP-LC3-expressing HeLa cells were infected with WT Salmonella. Cells at 5 hpi were stained with DAPI (blue) and ubiquitin (Ub; red). (B) GFP-LC3-expressing HeLa cells were infected with WT or ∆sifA bacteria. Cells at 1 hpi and 5 hpi were stained with LAMP-1 (red) and DAPI (blue). In panels A and B, “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. (C and D) Quantification of the percentage of LC3+ bacteria (WT and ∆sifA strains) at 1 hpi, 5 hpi, and 8 hpi (C) and the percentage of LC3+ bacteria that are not associated with LAMP-1 at 1 hpi and 5 hpi (D) in GFP-LC3-expressing cells (mean ± SD, n = 3). (E) Quantification of the percentage of LC3+ bacteria in GFP-LC3-expressing cells treated with the indicated siRNAs at 5 hpi (mean ± SD, n = 3). (F) HeLa cells stably expressing GFP-LC3 were infected with the WT for 5 h. GFP-LC3+ bacteria were located with a spinning-disc confocal microscope and further processed for the electron microscopy. Black arrowheads indicate autophagosome structures. The boxed regions are magnified and shown in panels ii and iv. Panel ii shows an autophagosome-enclosed bacterium in a damaged SCV, whereas panel iv shows a dividing cytosolic bacterium close to autophagosomes. Scale bars, 500 nm. (G and H) HeLa cells expressing GFP-p62 and mRFP-LC3 were infected with WT Salmonella for 5 h. Cells were stained with DAPI (blue). (G) The percentage of LC3+ or LC3− bacteria associated with p62 (left) and the percentage of p62+ or p62− bacteria associated with LC3 (right) were quantified (mean ± SD, n = 3). (H) “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. Download
Dynamic association of p62 and/or LC3 correlates with Salmonella replication in the cytosol of HeLa cells. (A) HeLa cells infected with WT-mKO were stained with LAMP-1 (green) at 5 hpi. Bacteria completely surrounded by LAMP-1 (intact SCV) were bright red, whereas bacteria showing no (cytosolic) or discontinuous (likely within damaged SCV) LAMP-1 staining were light red. “Zoom” represents a magnified picture of the boxed area. Scale bar, 10 µm. (B) HeLa cells expressing GFP-p62 and mRFP-LC3 were infected with cyan fluorescent protein (CFP)-expressing Salmonella (gray). A snapshot of live-cell imaging data was shown. Arrows indicate dividing bacteria. Scale bar, 5 µm. Download
p62 and autophagy facilitate Salmonella replication in the cytosol of HeLa cells. (A) HeLa cells were treated with the indicated siRNAs and infected with Salmonella. The fold replication at 6 hpi was presented (mean ± SD, n = 3). (B) Western blot analysis of HeLa cells used in panel A. (C) Cells were treated with different siRNAs and infected with Salmonella. Viable cells are quantified by a trypan blue assay (mean ± SD, n = 3). (D) HeLa cells were treated with indicated siRNAs and infected with WT or ∆sifA bacteria. The fold replication at 6 hpi was presented (mean ± SD, n = 3). (E) Western blot analysis of HeLa cells used in panel D. Calnexin was used as a loading control. Download
Oleic acid, but not amino acids, is likely to promote Salmonella replication through autophagy. (A to C) HeLa cells were treated with control siRNA or LC3 siRNA and infected with WT Salmonella for 6 h in the absence of (UT) or the presence of different types of nutrients (nonessential amino acids [NEAA] [A]; essential amino acids [EAA] [B]; oleic acid, free fatty acid [FFA] [C]). The fold replication at 6 hpi was presented (mean ± SD, n = 3). The data showed that oleic acid (but not NEAA) enhanced Salmonella replication in control siRNA-treated cells but not in LC3 siRNA-treated cells. Download
Bacterial strains and plasmids used in this study.
Time-lapse micrographs of GFP-LC3-expressing cells infected with mKO-expressing Salmonella. Images were taken at 4.5 hpi. Arrows indicate dividing bacteria. Download
Time-lapse micrographs of GFP-LC3-expressing cells infected with CFP-expressing Salmonella (gray). One hour before imaging (at 3.5 hpi), cells were incubated with the LysoTracker (red). The GFP-LC3+ bacterium (indicated by the arrow) did not colocalize with the LysoTracker, indicating that it is in the cytosol. Download
Time-lapse micrographs of GFP-p62-expressing cells infected with mKO-expressing Salmonella. Cells were imaged at 4.5 hpi (00:00:00). Arrows indicate dividing bacteria. Download
Time-lapse micrographs of GFP-p62- and mRFP-LC3-expressing cells infected with CFP-expressing Salmonella (gray). Images were taken at 4.5 hpi. Arrows indicate dividing bacteria associated with GFP-p62 and mRFP-LC3. Download
Time-lapse micrographs of GFP-LC3-expressing cells infected with mKO-expressing Salmonella. Images taken at different time points were collected and edited with iMovie software. Download