

ABSTRACT
Pervasive transcription is observed in a wide range of organisms, including humans, mice, and viruses, but the functional significance of the resulting transcripts remains uncertain. Current genetic approaches are often limited by their emphasis on protein-coding open reading frames (ORFs). We previously identified extensive pervasive transcription from the murine gammaherpesvirus 68 (MHV68) genome outside known ORFs and antisense to known genes (termed expressed genomic regions [EGRs]). Similar antisense transcripts have been identified in many other herpesviruses, including Kaposi’s sarcoma-associated herpesvirus and human and murine cytomegalovirus. Despite their prevalence, whether these RNAs have any functional importance in the viral life cycle is unknown, and one interpretation is that these are merely “noise” generated by functionally unimportant transcriptional events. To determine whether pervasive transcription of a herpesvirus genome generates RNA molecules that are functionally important, we used a strand-specific functional approach to target transcripts from thirteen EGRs in MHV68. We found that targeting transcripts from six EGRs reduced viral protein expression, proving that pervasive transcription can generate functionally important RNAs. We characterized transcripts emanating from EGRs 26 and 27 in detail using several methods, including RNA sequencing, and identified several novel polyadenylated transcripts that were enriched in the nuclei of infected cells. These data provide the first evidence of the functional importance of regions of pervasive transcription emanating from MHV68 EGRs. Therefore, studies utilizing mutation of a herpesvirus genome must account for possible effects on RNAs generated by pervasive transcription.
IMPORTANCE
The fact that pervasive transcription produces functionally important RNAs has profound implications for design and interpretation of genetic studies in herpesviruses, since such studies often involve mutating both strands of the genome. This is a common potential problem; for example, a conservative estimate is that there are an additional 73,000 nucleotides transcribed antisense to annotated ORFs from the 119,450-bp MHV68 genome. Recognizing the importance of considering the function of each strand of the viral genome independently, we used strand-specific approaches to identify six regions of the genome encoding transcripts that promoted viral protein expression. For two of these regions, we mapped novel transcripts and determined that targeting transcripts from these regions reduced viral replication and the expression of other viral genes. This is the first description of a function for these RNAs and suggests that novel transcripts emanating from regions of pervasive transcription are critical for the viral life cycle.
INTRODUCTION
Gammaherpesviruses are oncogenic herpesviruses that undergo productive replication and can establish latent infection in their hosts. Human gammaherpesviruses, Epstein-Barr Virus (EBV; also called human herpesvirus 4 [HHV-4]) and Kaposi’s sarcoma-associated herpesvirus (KSHV; also called HHV-8), are associated with malignancies, including Burkitt’s and primary effusion lymphomas, nasopharyngeal carcinoma, and Kaposi’s sarcoma. Murine gammaherpesvirus 68 (MHV68; also called γHV68 and MuHV4) is genetically related to EBV and KSHV and causes lymphomas and lymphoproliferative disease in immunocompromised mice (1–5), providing a tractable model system in which to study productive infection in vitro and viral pathogenesis in vivo.
MHV68 and other herpesviruses can serve not only as models for viral infection and pathogenesis but also as systems in which to unravel the complexity of pervasive transcription and to probe the function of its products. Using high-density tiled arrays and RNA sequencing, we and others have shown that widespread transcription occurs outside annotated open reading frames (ORFs) during lytic MHV68 infection, generating regions of transcription termed expressed genomic regions (EGRs) (6, 7). We termed these EGRs rather than genes because the signal in a tiled array analysis may represent multiple different transcripts and because they were not initially investigated for functional importance. These EGRs contain no ORFs with significant homology to known proteins and, as such, may encode noncoding RNAs (ncRNAs), spliced transcripts, novel small polypeptides, and/or long 5′ or 3′ untranslated regions of annotated ORFs (6). While there are several well-studied examples of herpesvirus ncRNAs expressed during lytic or latent infection (reviewed in reference 8), new sequencing and array technologies reveal a substantially more complex transcriptional landscape for both betaherpesviruses and gammaherpesviruses than was previously appreciated (6, 7, 9–14, 64). Intergenic and antisense transcription has also been widely detected in a range of other organisms, including humans and mice (15–17, 63). However, the function of these viral transcripts remains an open question. The implications of these findings for genomic mutagenesis studies targeting ORFs within herpesvirus genomes are incompletely understood. Importantly, while initial findings of pervasive transcription of the MHV68 genome (6) have been independently confirmed (7), whether these RNAs have any function is unknown. One interpretation is that the products of pervasive transcription are irrelevant to the viral life cycle and might merely be the result of failed termination of functionally important, often protein-coding, transcripts. Studies relying on disruption of the viral genome in ways that alter transcripts from both genomic strands cannot resolve this question.
In this study, we used a flow cytometry-based screen combined with strand-specific knockdown of candidate RNAs to assess the importance of RNAs encoded within EGRs. We found that targeting several EGR-encoded transcripts altered the expression of other viral genes, and we selected two adjacent regions (EGRs 26 and 27) for detailed analysis. We characterized the transcript architecture, localization, and effects on other viral genes for RNAs emanating from EGRs 26 and 27. Herein, we report that EGR 26 and 27 transcripts were enriched in the nuclei of infected cells and that targeting transcripts emanating from EGR 27 altered expression of multiple viral genes and proteins and inhibited viral replication. To our knowledge, this is the first proof of functional significance of novel viral transcripts identified using a transcriptome-based approach.
RESULTS
Establishment of a system to target MHV68 transcripts: antisense targeting of a known essential gene alters late viral protein expression.
Since many EGRs are antisense to other EGRs or known ORFs, it was necessary to design a strategy to determine the functionality of transcripts from a single strand of the viral genome. To disrupt viral transcripts in a strand-specific manner, we designed single-stranded antisense oligonucleotide (ASO) gapmer probes to known MHV68 viral genes. Gapmer ASOs, which are reported to disrupt gene expression by RNase-H mediated degradation of their target transcript (18), have been previously used to target herpesvirus ncRNAs (19, 20).
An ASO to the single-stranded DNA binding protein, ORF 6 (21), significantly reduced transcript (Fig. 1A) and protein (Fig. 1B) levels of its target. An ASO to a noncellular, nonviral transcript (green fluorescent protein [GFP]) did not alter ORF 6 transcript or protein abundance (Fig. 1A and B) and served as a negative control. Additionally, the ORF 6 ASO reduced the abundance of the ORF 4-ORF 6 bicistronic transcript (Fig. 1A) (22). We observed a smaller (~1-kb) band in cells transfected with the ASO to ORF 6, which may represent a stable product of the degraded ORF 6 transcript (Fig. 1A). These data demonstrated that targeting a viral gene with an ASO decreased both transcript and protein levels of its target, confirming this as an effective strategy for knocking down specific transcripts in MHV68 infected cells.
FIG 1 .
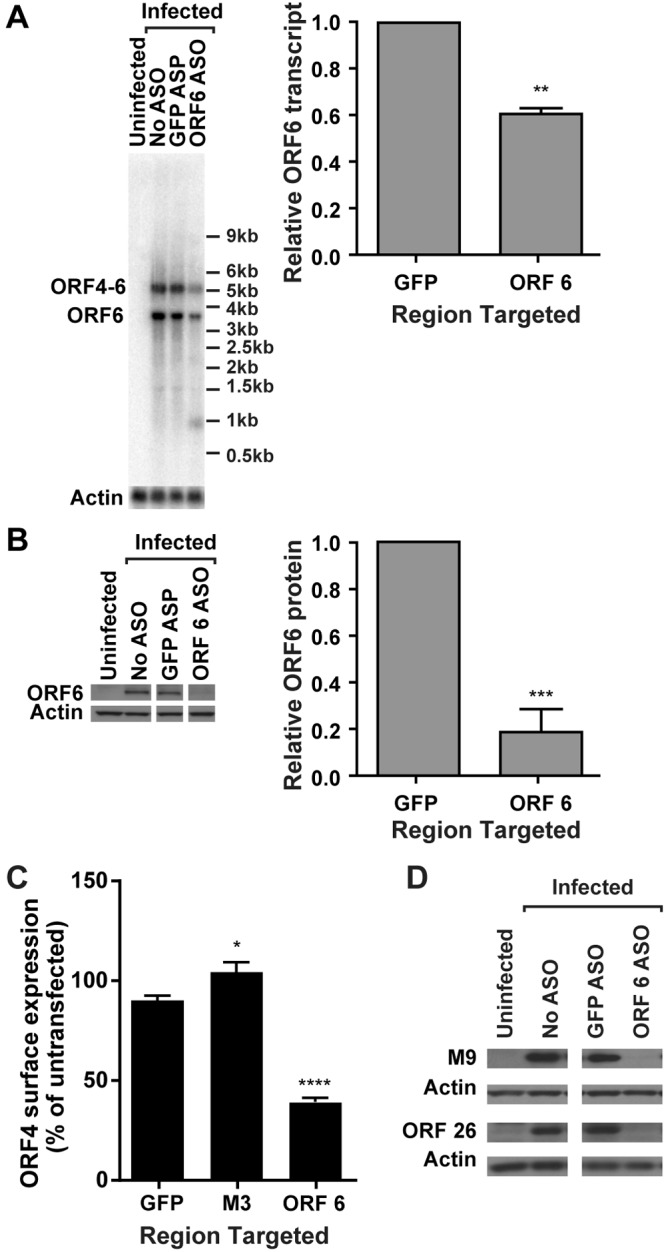
An antisense oligonucleotide to ORF 6 decreases ORF 6 transcript and protein expression and late-gene expression. 3T12 cells transfected with ASOs targeting ORF 6, M3, or GFP (negative control) or left untransfected (No ASO) were infected with MHV68. (A) Representative Northern blot for ORF 6 or actin transcripts at 14 hpi and corresponding quantification of ORF 6 monocistronic transcript levels normalized to those of actin (MOI = 10; values are means and standard errors of the means [SEMs] from 3 experiments; **, P < 0.01 by paired t test). (B) Representative Western blot for ORF 6 and actin protein at 18 hpi and corresponding quantification of ORF 6 normalized to actin (MOI = 10; data are means and SEMs from 8 experiments; ***, P < 0.001 by paired t test). Relevant lanes of representative blots are shown. (C) Flow cytometry analysis of ORF 4 surface expression at 24 hpi (MOI = 5 or 10). Flow cytometry data are graphed as the percentage of ORF 4-positive cells for each condition normalized to the value from untransfected cells (data are means and SEMs for 34 to 35 replicates; statistically significant results relative to GFP are indicated; ****, P < 0.0001 by one-way ANOVA with Dunnett’s posttest). (D) Western blot analysis of M9 or ORF 26 at 18 hpi. Relevant lanes of representative blots are shown. Data are representative of four independent experiments.
We next validated a flow cytometry-based assay to rapidly assess the effect of knockdown of transcripts on viral protein expression. Viral genes are expressed as a cascade of immediate-early, early, and late genes. Disrupting an essential gene of either immediate-early or early classes will broadly disrupt expression of all late genes. To enable us to capture potential effects on multiple aspects of the viral life cycle, we therefore selected expression of a late gene, ORF 4, as our target for assay development. ORF 4 encodes the viral complement regulatory protein (v-RCA) which is expressed on the surfaces of infected cells (22). An ASO to the essential gene ORF 6 (21) significantly inhibited the surface expression of ORF 4 (Fig. 1C). An ASO designed to be complementary to the nonessential gene M3 (23) did not reduce ORF 4 surface expression (Fig. 1C). Since ORF 4 is transcribed as a bicistronic transcript with ORF 6, it is possible that the reduction in ORF 4 surface expression observed was due to direct knockdown of the transcript that encodes ORF 4 by the ASO designed to target ORF 6. To confirm that knocking down ORF 6 altered expression of other proteins, we evaluated protein expression of ORF 26 and M9 (ORF 65) (24, 25) and confirmed that knocking down ORF 6 decreased expression of multiple viral late proteins (Fig. 1D). These data demonstrated that disrupting the expression of an essential gene transcript was detectable by a change in ORF 4 surface expression and indicated that this method could be used as a screening tool for the function of EGR transcripts.
Targeting EGR transcripts alters surface expression of the late viral protein encoded by ORF 4.
To determine whether transcripts encoded within EGRs played a role in productive infection, we designed ASOs to regions within 13 EGRs (EGR nomenclature is as in reference 6). We selected 12 EGRs that might be important for viral replication (EGRs 9, 10, 11, 15, 16, 18, 20, 21, 23, 25, 26, and 27) and one EGR that might be dispensable for viral replication (EGR 30) based on results from a genome-wide transposon-based mutagenesis screen of the MHV68 genome (26). Since transposon mutagenesis disrupts genetic elements on both DNA strands, some of the phenotypes previously attributed to transposon insertions in known ORFs could also be explained by a function of EGR-encoded transcripts from the opposite strand. Cells were transfected with ASOs to regions of the 13 selected EGRs or the known viral genes ORF 6 and M3, infected with MHV68, and then analyzed for ORF 4 surface expression by flow cytometry (see Table S1 in the supplemental material for ASO sequences and targeted genomic coordinates). ASO targeting of EGRs 9, 16, 18, 23, 26, and 27 showed statistically significant reductions in ORF 4 surface expression (Fig. 2). For EGRs 9, 18, and 23, which were targeted with multiple ASOs, not all ASOs altered ORF 4 expression, suggesting that these ASOs may differ in knockdown efficiency or may target distinct transcripts encoded by the EGR. In fact, we have found that there is extensive splicing within the MHV68 transcriptome, including within EGR 23, which may account for the differences observed (L. S. Johnson, S. P. Canny, and H. W. Virgin, unpublished data). Cells transfected with ASOs targeting EGRs 11 and 20 had increased ORF 4 surface expression (Fig. 2), which might suggest a role for transcripts emanating from these EGRs in suppressing viral replication.
FIG 2 .
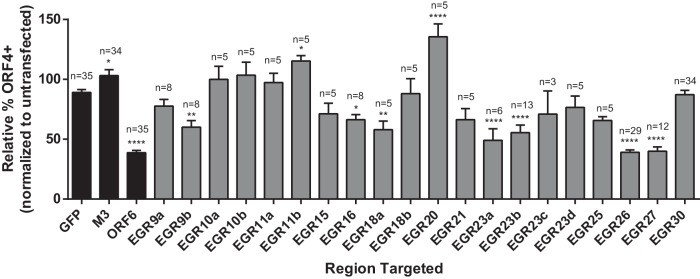
Targeting of EGRs 9, 16, 18, 23, 26, and 27 reduces ORF4 surface expression on infected cells. 3T12 cells transfected with the indicated ASOs or left untransfected were analyzed for cell surface expression of ORF 4 at 24 hpi by flow cytometry. Cells were infected at an MOI of 5 or 10. Controls were included in parallel in each experiment in which EGRs were analyzed. The percentage of ORF 4-positive cells for each condition was normalized to the value from untransfected cells. Data are means of the pooled data and SEMs; n, number of independent experiments. Statistically significant results relative to GFP are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Dunnett’s posttest). Black bars, controls (data reproduced from Fig. 1C); gray bars, EGRs.
Taken together, these results show that several EGR-encoded transcripts were important for ORF 4 protein expression. ASOs to the adjacent EGRs 26 and 27 (EGR 26c and EGR 27b in Table S1 in the supplemental material) on the negative strand of the viral genome had the largest effect on cell surface expression of ORF 4, comparable to the effect of targeting the essential gene ORF 6 (Fig. 2). For this reason, we chose to further characterize EGR 26- and EGR 27-encoded transcripts and to assess the effect of targeting RNAs from this region on viral gene expression.
Mapping 5′ and 3′ ends of EGR 26 and EGR 27 transcripts.
To define the transcript(s) targeted by ASOs to EGRs 26 and 27, we used a combination of RNA sequencing, random amplification of 5′ and 3′ cDNA ends (5′ and 3′ RACE), and Northern blot analysis. First, we confirmed active RNA expression within EGRs 26 and 27 in lytically infected fibroblasts by RNA sequencing and identified a transcriptional signal similar to the signal that we had previously detected using tiled array technology (see Fig. S1 in the supplemental material) (6). Spearman correlations calculated between our previous tiled array data and RNA sequencing datasets from 18 h postinfection (hpi) yielded correlation coefficients ranging from 0.78 to 0.85.
To determine the sizes and relative positions of transcripts emanating from EGRs 26 and 27, we used Northern blot analysis. We identified an ~12-kb transcript (referred to here as EGR 26 transcript A) that overlapped EGRs 26 and 27 and ORFs 65 to 67, an ~8-kb transcript (referred to here as EGR 27 transcript A) that overlapped EGR 27 and ORFs 65 to 67, and an ~3-kb transcript (referred to here as EGR 27 transcript B) that overlapped EGR 27 (Fig. 3). We also identified a transcript of >12 kb that overlapped and was detected by probe 1 which we termed EGR 26 transcript B but did not map further. Using a probe to genomic coordinates 101170 to 101208 (probe 12) (Fig. 3A), we detected an ~8-kb transcript and an ~3-kb transcript on the opposite side of the 100-bp internal repeat (Fig. 3B), suggesting that both EGR 27 transcripts A and B overlap the 100-bp internal repeat. We tested two gene-specific primers in the PCR step of 5′ RACE for EGR 27 and obtained comparable results. We identified 9/9 colonies with 5′ ends within one nucleotide of 101225. We were unable to detect transcripts using probe 13, suggesting that ~101225 was the 5′ end for both EGR 27 transcripts A and B (Fig. 3B). Probe 8, but not probe 9 or 10, detected an ~12-kb transcript comparable in size to EGR 26 transcript A (Fig. 3A and B). 5′ RACE analysis identified 6/6 colonies with 5′ ends within four nucleotides of 97790 (1 colony with a 5′ end at 97786, 1 at 97789, 3 at 97790, and 1 at 97791), suggesting that the 5′ end of EGR 26 transcript A was ~97790.
FIG 3 .

EGR 26 and 27 transcriptional architecture. (A) Schematic representation of EGR 26-27/M9 region transcripts. Approximate size and transcript name are listed below the relevant transcript. (B) Northern blot detection of EGR 26-27/M9 region transcripts. RNA harvested at 18 hpi (MOI = 10) was analyzed by Northern blot analysis using the indicated probes. 28S and 18S rRNA bands visualized by ethidium bromide staining to demonstrate equal loading are shown below the corresponding Northern blots. Northern blots analyzed with probe 12 and probe 2 (far right) had 500 ng of poly(A)-selected RNA per lane. *, virus-specific bands that are of comparable size to EGR 27 transcripts A and B. **, smaller EGR 26 transcripts referenced in text.
EGRs 26 and 27 encode polyadenylated transcripts, as shown by Northern blot analysis of polyadenylated RNA (Fig. 3; also data not shown) and confirmed by RNA sequencing of polyadenylated RNA (see Fig. S1 in the supplemental material). To identify 3′ ends of EGR 26 and 27 transcripts, we mined RNA sequencing data from four experiments, two from 18 hpi and two from 6 hpi, using a custom analysis pipeline. By mining RNA sequencing data for polyadenylated reads, we identified 3′ ends within the EGRs 26 and 27 at 85207, 93842, and 98510 and within the intervening ORFs at 95322, 95758, and 95895 (see Fig. S1 and Table S3 in the supplemental material). It is worth noting that 3′ ends at 95322, 95758, and 95895 were not attributable to transcripts detected by Northern blotting, suggesting that there may be additional, less abundant transcripts within this region. This is important because it indicates that Northern blot analysis may not be sensitive enough to detect all transcripts encoded within EGRs that might be targeted by specific ASOs. We performed 3′ RACE analysis to confirm 3′ ends at 98510 (6/6 colonies) and within 3 nucleotides of 93842 (5/5 colonies). In addition, our RNA sequencing approach confirmed six previously known polyadenylation sites as well as 53 novel sites downstream of known protein-coding and EGR-encoded transcripts; the majority of these were detected at both 6 and 18 hpi (see Table S3 in the supplemental material). Together, these data identified three transcripts that overlap the EGR 26-EGR 27 region and suggest that additional low-abundance transcripts from this region that are not readily detectable by RACE or Northern blotting may exist. This highlights the profound transcriptional complexity across the MHV68 genome (Fig. 3) (6, 7, 27).
EGR 26 and EGR 27 transcripts are enriched in the nuclei of infected cells.
To determine if transcripts that overlap EGRs 26 and 27 were retained in the nuclei or exported to the cytoplasm of infected cells, we separated nuclear and cytoplasmic fractions, extracted RNA, and probed for EGR 26 transcript A using probe 2 and EGR 27 transcripts A and B using probe 11. We confirmed adequate separation of nuclei from cytoplasm by Western and Northern blot analyses of known cytoplasmic and nuclear proteins and RNAs with only modest contamination of the cytoplasmic fraction with nuclear protein and RNA (Fig. 4). We found that the EGR 26 transcript A, EGR 27 transcript A, and EGR 27 transcript B were enriched in the nuclear fraction (Fig. 4B). Nuclear enrichment of EGR 26 and EGR 27 transcripts was comparable to that of the well-characterized mouse nuclear ncRNA, nuclear enriched abundant transcript 2 (Neat2, also known as Malat1 [28]) (Fig. 4B). Analysis of representative viral (ORF 6) and host (actin) protein-coding transcripts demonstrated that these translated transcripts accumulated in the cytoplasm (Fig. 4B). The nuclear distribution of EGR 26 transcript A, EGR 27 transcript A, and EGR 27 transcript B was consistent with a nuclear RNA and not with protein-coding transcripts, suggesting that these EGR transcripts may be nuclear noncoding ncRNAs.
FIG 4 .
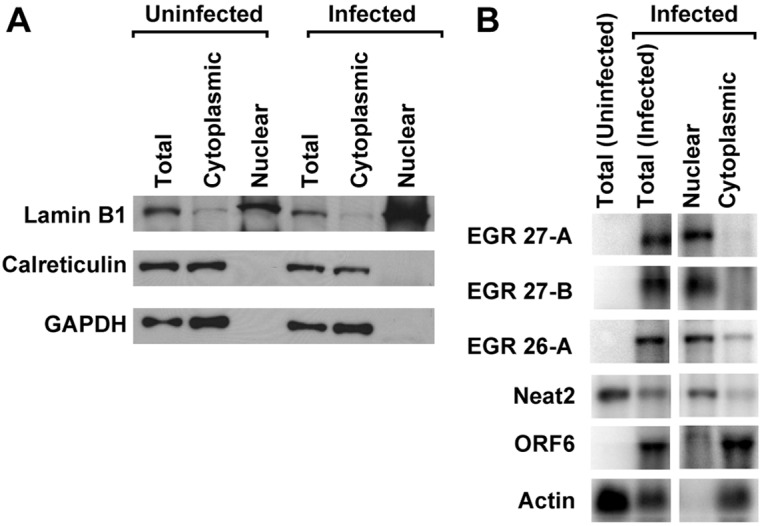
EGR 26 and 27 transcripts are enriched in nucleus of infected cells. Protein or RNA extracted from nuclear or cytoplasmic fractions or unfractionated cells (Total) was analyzed by Western blotting (A) or Northern blotting (B) at 18 hpi (MOI = 10). (A) Four micrograms of protein was loaded per sample. (B) Five micrograms of RNA for each total sample and approximate cellular equivalents for nuclear (0.5 µg) or cytoplasmic (4.5 µg) fractions were analyzed. Data are representative of three independent experiments. EGR 26-A indicates EGR 26 transcript A (as shown in Fig. 3) detected using Northern blot probe 2. EGR 27-A and 27-B indicate EGR 27 transcripts A and B, respectively (as shown in Fig. 3) detected using Northern blot probe 11.
Targeting EGR 27 transcripts alters surface expression of ORF 4 protein and viral replication.
To determine the effect of knockdown of RNAs encoded within EGR 27 on ORF 4 surface expression, we targeted this region with four ASOs, two of which we predicted would target both EGR 27 transcripts detectable by Northern blot and two of which we predicted would specifically target EGR 27 transcript A. Because EGR 27 transcript A overlaps the entire length of EGR 27 transcript B, it was not possible to specifically target the smaller transcript. EGR 27a and EGR 27b ASOs reduced the abundance of both EGR 27 transcripts as predicted (Fig. 5B and C). Surprisingly, EGR 27d ASO also significantly reduced EGR 27 transcript B, although it was not predicted to target this transcript (Fig. 5C), raising the possibility that an additional transcript might be derived from this region. In support of this hypothesis, we identified additional 3′ ends at 95322, 95758, and 95895, which have not been ascribed to specific transcripts (see Table S3 in the supplemental material), and were not attributable to RNAs detected by Northern blot (see above), suggesting the presence of additional as-yet-unmapped transcripts within this region. As predicted, EGR 27c ASO reduced EGR 27 transcript A but not EGR 27 transcript B (Fig. 5B and C). All EGR 27 ASOs reduced ORF 4 surface expression (Fig. 5E). While we cannot attribute the effect on ORF 4 surface expression to a specific transcript because the transcripts overlap, these data confirm, using independent ASOs, the results of our initial screen (Fig. 2) by showing that RNAs encoded in this region of the genome are functionally important for viral gene expression.
FIG 5 .

Effect of targeting EGR 26 and EGR 27 transcripts on ORF 4 surface expression and viral replication. (A) Schematic of EGR 26s and 27. See Table S1 in the supplemental material for specific coordinates targeted by ASOs. (B to D) 3T12 cells transfected with the indicated ASOs were analyzed for EGR 27 transcript A (B), EGR 27 transcript B (C), or EGR 26 transcript A (D) at 14 hpi by Northern blotting. EGR 27 transcripts A and B were detected using Northern blot probe 11, and EGR 26 transcript A was detected using Northern blot probe 2. Depending on the experiment, 1.5 to 5 µg RNA was used per sample. The graphs are quantitations of signal intensity of the indicated transcript normalized to actin and presented as a fraction of the signal from untransfected cells. Representative Northern blots are shown. The actin for EGR 27 transcripts is reproduced for panels B and C, as the same blot is displayed. The pound sign indicates the location of a tear in the agarose gel. Data are the means of the pooled data (3 to 7 experiments) and SEMs. (E) 3T12 cells transfected with the indicated ASOs were analyzed for cell surface expression of ORF4 at 24 hpi by flow cytometry. As for Fig. 2, the percentage of ORF4-positive cells for each condition was normalized to the value from untransfected cells. Data are means of the pooled data (5 to 35 experiments) and SEMs. (F) 3T12 cells transfected with the indicated ASOs were analyzed for viral titer at 24 hpi by plaque assay. Data are means (3 experiments) and SEMs. Statistically significant results relative to GFP are shown (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with Dunnett’s posttest). Black bars, controls; blue bars, EGR 26 ASOs; red bars, EGR 27 ASOs.
We also targeted EGR 26 with six ASOs, five predicted to target EGR 26 transcript A (EGR 26a to 26e) and one directed 3′ of EGR 26 transcript A (EGR 26f). ASOs to EGRs 26b, 26c, and 26d significantly reduced EGR 26 transcript A levels (Fig. 5D). Although the EGR 26d ASO reduced EGR 26 transcript A, it did not significantly reduce ORF 4 surface expression (Fig. 5E), suggesting that the effect on ORF 4 surface expression by other EGR 26 ASOs may be due to other smaller transcripts in this region (Fig. 3B). The effect of EGR 26f ASO, which did not target EGR 26 transcript A but likely targeted other RNAs derived from EGR 26, such as EGR 26 transcript B, further supports the idea that RNAs derived from EGR 26 are important for viral protein expression. These data suggest that similar to the transcripts emanating from EGR 27, RNAs from EGR 26 are also functionally relevant and that the functional significance of EGR transcription is not restricted to a single region of the genome.
To determine whether the observed changes in ORF 4 protein expression were associated with a detectable change in viral replication, we analyzed viral titers at 24 hpi in ASO-transfected cells. We detected a significant reduction in viral titer in cells transfected with EGR 27a, EGR 27b, EGR 27d, and EGR 26c ASOs, comparable to the reduction in viral titer in cells transfected with an ASO to the essential gene ORF 6 (Fig. 5F). Given that target messages were only partially reduced by ASOs (Fig. 1A and B and 5B to D), it is notable that both ORF 6 and EGR ASOs significantly reduced viral titers.
Targeting of EGR 27 transcripts alters expression of multiple viral genes.
Having shown that RNAs derived from EGRs can be functionally important, as measured by the effects on a viral late protein and viral replication, we next assessed the extent of ASO effects on different aspects of viral transcription and protein expression. To confirm that EGR 27 ASOs reduced late-gene expression, we assessed the effect of EGR 27 ASOs on ORF 26 and M9 (ORF 65) protein expression by Western blot analysis and spliced ORF 29 transcripts by quantitative reverse transcription-PCR (qRT-PCR) (29). All EGR 27 ASOs reduced ORF 26 and M9 protein expression and ORF 29 transcript expression (Fig. 6A and B). These data show that targeting EGR 27 transcripts broadly altered the expression of multiple late genes and did not exert effects restricted to surface expression of the late protein encoded by ORF 4.
FIG 6 .

Effect on immediate-early, early, and late genes by EGR 27. 3T12 cells transfected with GFP or ASOs targeting EGR 27 or left untransfected (No ASO) were infected with MHV68 (MOI = 10) and analyzed for protein (A and D) or transcript levels (B, C, and E). See Fig. 5 and also Table S1 in the supplemental material for ASO locations. (A) Representative Western blots for M9 and ORF 26 proteins at 18 hpi (2 or 3 experiments). (B) ORF 29 transcript levels at 14 hpi. RNA (1 µg) was reverse transcribed (RT), and cDNA was analyzed by qPCR using primers designed to detect spliced ORF 29 transcripts or GAPDH. Data are relative ORF 29 abundance normalized to GAPDH transcript abundance and compared to untransfected cells by the ∆∆CT method (means and SEMs from 3 to 8 experiments). (C) Representative Northern blot for ORF 6 and actin at 14 hpi and corresponding quantification of ORF 6 transcript levels normalized to actin and compared to the value for untransfected cells (means and SEMs from 5 to 7 experiments). (D) Representative Western blot for ORF 6 and actin at 18 hpi (3 experiments). The representative experiment is the one whose results are shown in panel A. (E) ORF 50 transcript levels at 14 hpi measured by qRT-PCR, as for panel B. Statistical analyses were performed by one-way ANOVA with Dunnett’s posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Next, we tested the effect of EGR 27 ASOs on the expression of the early gene ORF 6 (30) or the immediate-early gene ORF 50, also known as the replication and transcription activator (RTA) (31). We found that EGR 27a, 27b, and 27d ASOs, but not the EGR 27c ASO, significantly reduced ORF 6 transcript levels (Fig. 6C). However, all EGR 27 ASOs reduced ORF 6 protein levels (Fig. 6D). Furthermore, we found that EGR 27a and 27d ASOs, but not the EGR 27b or 27c ASO, significantly reduced the abundance of spliced ORF 50 transcripts (Fig. 6E). In summary, these data suggest that a transcript or transcripts targeted by EGR 27a and/or 27d ASOs act early in the viral life cycle, altering gene expression of specific viral genes from each kinetic class. Furthermore, a transcript(s) targeted by EGR 27c ASO may act later in the viral life cycle.
The EGR 26c ASO decreases specific genes of all kinetic classes.
We also evaluated the EGR 26c ASO, the ASO used in our initial screen (Fig. 2), for its effect on the viral life cycle. To confirm that EGR 26c ASO altered the expression of multiple late genes, we analyzed its effect on the expression of early-late (M3) and late (M9) genes (30, 32, 33) using Northern and Western blot analyses. We found that EGR 26c ASO reduced M9 and M3 protein levels (Fig. 7A and C) as well as M9 and M3 transcript levels (Fig. 7B and data not shown). Taken together with the effect of EGR 26c ASO on ORF 4 cell surface expression, these data demonstrate that transcript(s) targeted by EGR 26c ASO were important for expression of several early-late and late genes and suggested that it acted upstream of their expression. Interestingly, EGR 26c ASO did not alter the level of EGR 27 transcript A, detected by the Northern blot probe 7 (data not shown), indicating that EGR 26c ASO altered the levels of specific viral RNAs rather than overall transcription of the viral genome.
FIG 7 .

EGR 26c ASO decreases specific genes of all kinetic classes. 3T12 cells transfected with GFP or EGR 26c ASO or untransfected (No ASO) were infected with MHV68 (MOI = 10) and analyzed for protein (A, C, and E) or transcript levels (B, D, and F). (A) Representative Western blots for M9 and actin at 18 hpi (2 experiments). (B) Representative Northern blot using a probe to M3 or actin at 14 hpi and corresponding quantification of M3 transcript levels normalized to those of actin (means and SEMs from 3 experiments). A 0.5-µg portion of RNA was used per lane for M3 Northern blots. (C) Representative Western blot for M3 protein at 18 hpi and corresponding quantification of M3 protein levels normalized to those of actin (means and SEMs from 4 experiments). (D) Representative Northern blot for ORF 6 and actin at 14 hpi and corresponding quantification of ORF 6 transcript levels normalized to actin for cells transfected with GFP or EGR 26 ASOs (means and SEMs from 3 to 5 experiments). (E) Representative Western blot for ORF 6 and actin at 18 hpi and corresponding quantification of ORF 6 protein normalized to actin (means and SEMs from 5 experiments). Representative Western blots for ORF 6 are the same as in Fig. 1. (F) ORF 50 transcript levels at 14 hpi. RNA (1 µg) was reverse transcribed, and cDNA was analyzed by qPCR using primers designed to detect spliced ORF 50 transcripts or GAPDH. Data are relative ORF 50 abundances normalized to GAPDH transcript abundance and compared to untransfected cells by the ∆∆CT method (means and SEMs from 4 experiments). Statistical analyses were performed by paired t test (A, B, C, and E) or one-way ANOVA with Dunnett’s posttest (D and F). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next, we tested the effect of EGR 26 ASOs on the expression of ORF 6 and found that EGR 26a, 26b, and 26c ASOs reduced ORF 6 transcript levels, while EGR 26d, 26e, and 26f ASOs did not (Fig. 7D), suggesting that a transcript targeted by EGR 26a, 26b, and 26c ASOs acts to reduce viral gene expression. EGR 26c ASO also reduced ORF 6 protein levels (Fig. 7E). Finally, we tested whether EGR 26c ASO altered the expression of ORF 50 by qRT-PCR. We found that EGR 26c ASO significantly reduced the abundance of spliced ORF 50 transcripts (Fig. 7F). Northern blot analysis using probe 2 on RNA selected for polyadenylated transcripts revealed the presence of smaller transcripts that might be targeted by EGR 26c ASO (see Fig. 3B). The fact that EGR 26a, 26b, and 26c ASOs all reduced ORF 6 transcript abundance (Fig. 7D) suggests that a transcript that overlaps genomic coordinates 85576 to 85891 acts early in the viral life cycle, affecting gene expression of multiple specific viral genes.
DISCUSSION
In this paper, we provide the first data demonstrating that pervasive transcription of a herpesvirus genome, identified by pangenomic analysis of RNA expression using both tiled arrays and RNA sequencing, can generate functionally important RNAs for viral gene and protein expression. This is significant because it was unknown whether extensive antisense transcription observed by many groups from various herpesvirus genomes is functionally important or is due to “read-through” transcription that results in large amounts of functionally irrelevant RNA. We identified six EGRs that generated transcripts that altered ORF 4 surface expression. Upon examining transcripts encoded by two EGRs in more detail, we confirmed that targeting transcripts from the strand of the genome antisense to known protein coding genes altered multiple aspects of the viral transcriptional and translational program.
These data have fundamental implications for the approach to and interpretation of genetic studies and highlight the importance of mapping transcripts derived from the strand opposite ORFs. A limitation of traditional genetic approaches is the emphasis on ORFs and the utilization of genetic approaches that alter the sequences of both strands of the viral genome. This approach has been very fruitful but misses an important layer of complexity of viral gene regulation revealed here. One of our most notable findings is the extreme complexity of transcripts emanating from EGRs and the fact that many of these transcripts were polyadenylated but remained concentrated in the nucleus. Furthermore, our data suggest that in some cases there may be important contributions of less abundant transcripts whose presence may be difficult to detect using classical methods (EGR 26 is an example). Taken together, our results support the use of sensitive technologies, such as next-generation RNA sequencing, in transcript analysis and the necessity of considering antisense transcripts in genetic analyses of herpesviruses and designing functional experiments to evaluate the function of each strand of the viral genome independently.
Implications of transcriptional complexity of EGR-encoded RNAs.
It is interesting to reconsider previous findings in view of the transcriptional complexity underlying the MHV68 genome. Here, we report a region of the MHV68 genome in which detailed analysis reveals at least five transcripts overlapping the late capsid protein M9 (ORF 65), each of which may play distinct roles for the virus (34, 35). Previous studies using RNase protection assays and qRT-PCR have identified the M9 region as (i) a candidate region of latent gene expression, (ii) a component of the virion, and (iii) producing an RNA with immediate-early kinetics (30, 32, 36). We note that previous analyses are complicated by the presence of multiple overlapping transcripts because probes designed to M9 detect five independent transcripts (including EGR 26 transcript A and EGR 27 transcript A).
Additionally, our results suggest that caution should be used for interpretation of transposon screens (26, 37). Because ~90% of EGR nucleotides overlap regions where transcription occurs on the opposite strand (6) (see also Fig. S1 in the supplemental material), any mutagenesis strategy that disrupts both strands may alter functionally relevant transcripts derived from either strand. For example, transcripts encoded by EGRs 26 and 27 are antisense to known protein-coding ORFs 63, 64, 68, and 69. ORFs 63 and 64 are predicted to encode essential tegument proteins, ORF 68 an essential glycoprotein, and ORF 69 an essential gene of unknown function (1, 26). Thus, while these transposon screens provide invaluable functional data, defining the genetic elements responsible for observed phenotypes will need to incorporate the transcriptional complexity of each region and subsequently utilize strand-specific approaches for functional analysis.
Potential mechanism for EGRs 26 and 27 transcript function.
Our studies show that transcripts encoded by EGRs are functionally important and affect multiple parts of the viral life cycle but do not define specific mechanisms by which these transcripts act. EGR transcripts may act as ncRNAs, may encode small peptides or novel proteins, or in some cases may act as long UTRs for already-identified ORFs. Interestingly, both EGR 26 transcript A and EGR 27 transcript A overlap ORFs for several small proteins, namely M9, ORFs 66 and 67. While we cannot exclude a contribution of peptides or proteins encoded within EGRs, the reduction in ORF 50 transcript expression that we observed following targeting with EGR 27a and 27d ASOs and the nuclear localization of EGR 27 transcripts is consistent with the hypothesis that transcripts targeted by these ASOs function to epigenetically regulate the ORF 50 promoter(s). There is a growing consensus in the mammalian ncRNA literature that epigenetic modification of chromatin is a key function of long ncRNAs (38–42). Interestingly, the KSHV ncRNA, polyadenylated nuclear (PAN) RNA, associates with the ORF 50 promoter as well as the demethylases JMJD3 and UTX and the methyltransferase MLL2 (43). Many studies highlight the importance of epigenetic modifications in controlling herpesvirus gene expression, and herpesvirus proteins can interact with histone deacetylases (HDACs) to prevent gene silencing (44–48). We and others have identified several important modifications that silence ORF 50 in macrophages, B cells, and cells from latently infected mice, including methylation of the distal ORF 50 promoter (49, 50) and recruitment of HDACs and the nuclear receptor corepressor (NCoR) to the core ORF 50 promoter (51, 52). It is intriguing to speculate whether transcripts targeted by EGR 27a, 27d, and/or 26c ASO may act as a molecular scaffold for chromatin-modifying proteins or prevent the association of known repressive complexes with ORF 50 promoters in lytically infected fibroblasts.
In contrast, the EGR 27c ASO, which specifically targeted EGR 27 transcript A, did not reduce ORF 50 transcript expression, suggesting that this RNA may act later in the viral life cycle, playing a role in viral DNA replication or virion assembly. Interestingly, a role for an EBV RNA, the BHLF1 transcript, in DNA replication was recently described; the BHLF1 transcript forms an RNA-DNA hybrid molecule at the origin of lytic replication (OriLyt) and is important for recruitment of the viral single-stranded binding protein BALF2 to OriLyt (53). These results suggest that RNAs derived from EGRs may play a range of different roles in viral replication and merit further investigation and consideration in functional studies.
Many new transcripts have been identified not only in MHV68 but also in important human pathogens, including KSHV and HCMV (9–12, 64). Our results suggest that transcripts in each of these viruses may play crucial roles in the viral life cycle and/or in viral pathogenesis and that strand-specific approaches combined with detailed transcriptional analysis similar to ours will allow identification of novel transcripts critical for viral gene expression and infection.
MATERIALS AND METHODS
Cells, viruses, and virus assays.
NIH 3T12 fibroblasts (ATCC CCL-164) were grown in Dulbecco’s modified Eagle medium (DMEM) containing 5% fetal calf serum (FCS), 2 mM l-glutamine, and 10 mM HEPES. Cells were infected with MHV68 clone WUMS (ATCC VR-1465) for 1 h at 37°C at a multiplicity of infection (MOI) of 5 or 10, as indicated in the text. Viral passaging and titer determinations were performed on NIH 3T12 fibroblasts as described previously (54) except that cells were overlaid with 2% methylcellulose in MEM supplemented with 5% FCS and 2 mM l-glutamine.
Antisense oligonucleotides.
Custom antisense gapmer oligonucleotides (ASOs) containing a phosphorothioate backbone and locked nucleic acid (LNA) residues to increase probe stability and knockdown efficiency (55–57) were designed and synthesized by Exiqon (Woburn, MA) to target viral transcripts (see Table S1 in the supplemental material). Gapmers contain LNA bases at their ends surrounding a central stretch of DNA enabling RNase H-mediated cleavage of their targets (18). Transfections were performed with 40 pmol of ASO per 105 cells using Lipofectamine 2000 (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. An ASO designed to target GFP was used as a negative control. Cells were infected with MHV68 6 h after transfection and harvested at the indicated times postinfection. Toxicity was assessed by alamarBlue (Life Technologies) at 30 h posttransfection according to the manufacturer’s instructions. There was no association between toxicity relative to untransfected cells as assessed by alamarBlue and phenotype as assessed by ORF 4 surface staining (Spearman’s correlation; P = 0.8250).
Western blot analysis, antisera, and antibodies.
For MHV68 Western blot analysis, samples were lysed at 18 hpi in 2× Laemmli buffer, subjected to protein electrophoresis on 4 to 15% or 4 to 20% Tris gradient gels (Bio-Rad), and then transferred to polyvinylidene difluoride membranes. For Western blot analysis of subcellular fractions, 2× Laemmli buffer was added to samples lysed in the relevant buffer as described below (“Subcellular fractionation”). Then, 4 µg of protein per sample was subjected to protein electrophoresis on a 10% Tris gel and transferred to a polyvinylidene difluoride membrane. Antibodies used were anti-lamin B1 (Abcam), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (clone GAPDH-71.1; Sigma, St. Louis, MO), anti-calreticulin (clone 16/calreticulin) (BD Biosciences), polyclonal rabbit antisera generated to ORF 6 (58), M3 (59), ORF 26 (24), or M9 (24), and a goat anti-rabbit or goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody as appropriate (Jackson Immunoresearch, West Grove, PA). MHV68 Western blots were stripped and reprobed with anti-beta-actin (clone AC-74) (Sigma) and then with a goat anti-mouse HRP-conjugated secondary antibody (Jackson Immunoresearch) to control for loading. Blots were developed with ECL Plus chemiluminescent reagent (GE Healthcare Life Sciences) or Pierce enhanced chemiluminescence (ECL) chemiluminescent reagent (Thermo Scientific) and imaged using film or a Storm 840 phosphorimager. Bands were quantitated using ImageJ (NIH). For each sample, the indicated protein was normalized to the actin loading control.
Flow cytometric analysis for detection of surface expression of ORF 4 protein.
At 24 hpi, cells were removed from tissue culture dishes by gentle scraping after incubation in a 0.02% EDTA solution for 10 min at 4°C. Cells were fixed in 2% formaldehyde and stained using anti-ORF 4 antiserum (22) at 1:10,000 or preimmune rabbit serum (Cocalico Biologicals, Reamstown, PA), followed by donkey anti-rabbit DyLight 649 secondary antibody (Biolegend, San Diego, CA). Flow cytometry was performed with a FACSCalibur (BD Biosciences, San Jose, CA). Data were analyzed by FlowJo (Tree Star, Ashland, OR).
RNA sequencing (RNA-Seq) library construction and expression analysis.
Total RNA was isolated from TRIzol (Life Technologies) as described previously (6) at 6 or 18 hpi. Poly(A)-selected RNA was chemically fragmented using RNA fragmentation reagents (Life Technologies) and purified using RNeasy MinElute cleanup columns (Qiagen). Directional RNA-Seq libraries were generated using 600 ng of fragmented 18-hpi RNA according to Illumina’s directional mRNA-Seq protocol or 200 ng of 6 hpi RNA according to Illumina’s directional Tru-Seq protocol. Nucleotides with a Phred quality score less than 20 were trimmed from the 3′ ends of raw Illumina reads. Trimmed reads with a mean quality score less than 10 or a length less than 20 were discarded. Reads were then mapped to mouse rRNA, MHV68 genome (GenBank accession number U97553), and mouse genome sequences (Build 37) with the short-read aligning program Bowtie-0.12.5 (60). Bowtie alignments were performed using the parameter setting “-best” and the settings “-e 420” and “-e 600” for 76-nucleotide and 100-nucleotide reads, respectively. Bowtie output from the MHV68 genome mapping was converted to WIG (wiggle track format) files of read depth coverage, on a log2 scale and visualized using Gbrowse (http://www.gbrowse.org). Correlation coefficients between read depth coverage, and tiled array signals were calculated using Spearman’s ranked correlation coefficient in the R statistical environment.
Identification of polyadenylated reads from RNA-Seq data sets.
3′-terminal adenosine (A) residues were trimmed from filtered reads with five or more 3′ terminal A’s. Both trimmed and untrimmed reads were mapped to MHV68 with Bowtie with default “-e” settings. Mapped reads that contained at least five nongenomic A’s were considered putative polyadenylated reads. If a potential polyadenylation cleavage site was located within or downstream of a stretch of genomic A’s, the coordinates of this potential site were moved upstream of the genomic poly(A) stretch to maintain consistency. Sites that were located adjacent to a stretch of seven or more genomic A’s were excluded due to the possibility that during RACE validation, such sites could provide false internal priming of the oligo(dT) primer. Sites with an average of at least five supporting reads between biological replicates were clustered. The site reported in Table S3 in the supplemental material is that with the most supporting reads in 30-nucleotide sliding windows. Raw read quality filtering and the identification of putative polyadenylated reads were done using a combination of Linux utilities and custom Perl scripts (available upon request).
Rapid amplification of cDNA ends (RACE).
5′ and 3′ transcript ends were identified by RACE using Invitrogen’s 5′ and 3′ RACE systems according to the manufacturer’s instructions. cDNA was generated from total RNA extracted 18 hpi using a gene-specific primer (for 5′ RACE) or an oligo(dT)-containing adapter primer (for 3′ RACE). PCR amplification was performed using gene-specific primers with Invitrogen’s amplification primers. The following gene-specific primers were used for reverse transcription in 5′ RACE: EGR 26 transcript A, 5′ CGATCAGGTGGCTCAACTGG 3′; EGR 27 5′ GCGAGGAGCAGCACAGCAGA 3′. For 5′ RACE reactions, the PCR primers used were as follows: EGR 26 transcript A, 5′ CTGCTCACATACAAGGTATCTGG 3′; EGR 27, 5′ GCAGAGGTCCGTCCAGTAGCGA 3′ and 5′ GGTCCGTCCAGTAGCGA 3′. For 3′ RACE reactions, the PCR primers used were as follows: EGR 27 transcript A, 5′ GCCAGACATTCGCACAACAC 3′; EGR 27 transcript B (B1), 5′ CGAGATACAATGTTGAAGCATTCA 3′. The resulting PCR products were gel purified and ligated into a pCR4-TOPO TA sequencing vector (Life Technologies). Universal M13 forward and reverse primers were used for sequencing.
Northern blot analysis.
Total RNA was isolated as described (6) at 14 or 18 h postinfection (hpi) as indicated. Templates for Northern probes were amplified by PCR from viral or mouse genomic DNA (using PCR primers listed in Table S2 in the supplemental material), prepared as described in reference 6 for probes 2 and 7 (formerly EGR 26 probes 1 and 4 [6], respectively), or obtained from a commercial vendor for actin (Life Technologies). Northern blotting using Ambion’s NorthernMax kit (Life Technologies, Grand Island, NY) and generation of single-stranded P32-labeled RNA probes using the Maxiscript Sp6/T7 kit (Life Technologies) were performed as described previously (6). Probe 12 was generated using the mirVANA miRNA probe construction kit (Life Technologies) according to the manufacturer’s instructions. Five micrograms of total RNA was used for all Northern blot analyses unless otherwise stated. Membranes were scanned using a Storm 840 Phosphorimager and quantitated using ImageQuant TL (GE Healthcare Biosciences, Pittsburgh, PA). For quantitation of each sample, the indicated transcript signal was normalized to the signal from the actin loading control.
Subcellular fractionation.
Nuclei were separated from cytoplasm using a protocol adapted from published methods (61). Briefly, 3T12 cells were removed from plates by trypsinization at 18 hpi and centrifuged to pellet cells. After a washing with phosphate-buffered saline (PBS), the pellet was resuspended in 150 mM NaCl, 50 mM HEPES, 1% NP-40, and 1 U/µl SUPERase-In RNase inhibitor (Ambion) to disrupt nonnuclear membranes and incubated on ice for 30 min. The lysate was centrifuged at a relative centrifugal force (RCF) of 7,000 to pellet the nuclei, and the supernatant removed for RNA and protein analysis (cytoplasmic fraction). After washing with PBS, the nuclear pellet was resuspended in cold 150 mM NaCl, 50 mM HEPES, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 1 U/µliter SUPERase-In RNase Inhibitor (Life Technologies) to disrupt the nuclear membrane and incubated for 2 h at 4°C with rotation. RNA was extracted from lysates using TRIzol L.S. (Life Technologies) and analyzed by Northern blotting as described above. Approximate cellular equivalents were calculated based on the relative amounts of RNA recovered from nuclear and cytoplasmic fractions. For comparison, unfractionated cells were collected in TRIzol (Life Technologies) and then processed to isolate RNA according to the manufacturer’s instructions. Samples were also analyzed by Western blot to ensure adequate separation of fractions as described above.
Quantitative reverse transcriptase PCR (qRT-PCR).
For analysis of ORF 50 and ORF 29 transcripts, cDNA was synthesized from 1 µg of RNA using SuperScript III (Life Technologies) and random hexamers (Life Technologies) as described previously (29). qPCR was performed using Power SYBR green master mix (Applied Biosystems) and the primer sequences 5′ TGCCCCCATGTTTGTGATG 3′ and 5′ TGTGGTCATGAGCCCTTCC 3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (51), 5′ GATTCCCCTTCAGCCGATAAG 3′ and 5′ CAGACATTGTAGAAGTTCAGGTC 3′ for spliced ORF 50 transcript, and 5′ TTCTCATTGGCATCTTTGAGG 3′ and 5′ GGAAAATGGGGTGATCCTGT 3′ for spliced ORF 29 transcript (29) on the StepOnePlus System (Life Technologies). Transcript levels were normalized to GAPDH within each sample and compared to untransfected cells using the ∆∆CT method, where CT is the threshold cycle (62).
Statistical analysis.
Data were analyzed statistically with Prism 6 software (GraphPad Software, La Jolla, CA). All experimental conditions were compared to the corresponding untransfected or GFP ASO-transfected controls, as noted in the text. Data were analyzed by two-tailed paired t test or one-way analysis of variance (ANOVA), as indicated in the text. All significant differences are noted.
Nucleotide sequence accession numbers.
Sequencing data are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession numbers SRX403400 to SRX403403 for samples at 6 hpi and SRX403404 to SRX403407 for samples at 18 hpi.
SUPPLEMENTAL MATERIAL
Genome-wide RNA expression in MHV68 as detected by RNA-Seq. RNA-Seq RNA expression measurements were highly reproducible, strongly correlated with our earlier tiled array results, and confirmed the expression of all 30 expressed genomic regions (EGRs). Transcriptional signal from infected 3T12 cells at 18 hpi was plotted on a log2 scale underneath the annotated gene positions. Read depth from two independent RNA-Seq experiments was plotted, on a log2 scale, as blue/light blue and red/light red lines for the positive and negative genomic strands, respectively. Blue and red arrows represent annotated genes on the positive and negative strands of the genome, respectively. Gray boxes represent the two internal repeat regions of the MHV68 genome that were excluded from our analysis. EGRs 1 to 30 are shown as hatched arrows. (A) Genomic positions 1 to 40 kb. (B) Genomic positions 40 to 80 kb. (C) Genomic positions 80 to 119.45 kb. Download
Antisense oligonucleotide probes.
PCR primers for Northern blot probes.
3′ ends of MHV68 transcripts identified by RNA sequencing. The average numbers of reads per position at 6 hpi and 18 hpi are in bold in the second and third columns, respectively. The numbers in parentheses are the numbers of reads from specific experiments at the indicated time points. Gene region refers to a possible gene associated with the indicated polyadenylation site selected based on the gene with the closest 3′ end to a given site.
ACKNOWLEDGMENTS
This work was supported by R01 CA096511 and U54 AI057160 ARRA Administrative Supplement (to H.W.V.). S.P.C. was supported by an NIH F30 fellowship grant HL099019, and T.A.R. was supported by the Damon Runyon Cancer Research Foundation.
We thank Washington University Genome Technology Access Center (GTAC) for Illumina sequencing and technical advice, Jean E. Schaffer and Ben S. Scruggs for helpful experimental advice and discussions, Lori Xu for assistance with experiments, the Sun lab for kind gifts of M9 and ORF 26 antiserum, Christine Yokoyama for assistance with figure design, Scott Handley for help with data submission, and members of the Virgin lab for helpful review of the manuscript.
Footnotes
Citation Canny SP, Reese TA, Johnson LS, Zhang X, Kambal A, Duan E, Liu CY, Virgin HW. 2014. Pervasive transcription of a herpesvirus genome generates functionally important RNAs. mBio 5(2):e01033-13. doi:10.1128/mBio.01033-13.
REFERENCES
- 1. Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus Saimiri. J. Gen. Virol. 71:1365–1372. 10.1099/0022-1317-71-6-1365 [DOI] [PubMed] [Google Scholar]
- 3. Lee KS, Groshong SD, Cool CD, Kleinschmidt-DeMasters BK, van Dyk LF. 2009. Murine gammaherpesvirus 68 infection of IFNgamma unresponsive mice: a small animal model for gammaherpesvirus-associated B-cell lymphoproliferative disease. Cancer Res. 69:5481–5489. 10.1158/0008-5472.CAN-09-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sunil-Chandra NP, Arno J, Fazakerley J, Nash AA. 1994. Lymphoproliferative disease in mice infected with murine gammaherpesvirus 68. Am. J. Pathol. 145:818–826 [PMC free article] [PubMed] [Google Scholar]
- 5. Tarakanova VL, Suarez F, Tibbetts SA, Jacoby MA, Weck KE, Hess JL, Speck SH, Virgin HW. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79:14668–14679. 10.1128/JVI.79.23.14668-14679.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson LS, Willert EK, Virgin HW. 2010. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7:516–526. 10.1016/j.chom.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng BY, Zhi J, Santana A, Khan S, Salinas E, Forrest JC, Zheng Y, Jaggi S, Leatherwood J, Krug LT. 2012. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J. Virol. 86:4340–4357. 10.1128/JVI.05892-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sullivan CS. 2008. New roles for large and small viral RNAs in evading host defences. Nat. Rev. Genet. 9:503–507. 10.1038/nrg2349 [DOI] [PubMed] [Google Scholar]
- 9. Zhang G, Raghavan B, Kotur M, Cheatham J, Sedmak D, Cook C, Waldman J, Trgovcich J. 2007. Antisense transcription in the human cytomegalovirus transcriptome. J. Virol. 81:11267–11281. 10.1128/JVI.00007-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Y, Ganem D. 2010. Making sense of antisense: seemingly noncoding RNAs antisense to the master regulator of Kaposi’s sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J. Virol. 84:5465–5475. 10.1128/JVI.02705-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandriani S, Xu Y, Ganem D. 2010. The lytic transcriptome of Kaposi’s sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. J. Virol. 84:7934–7942. 10.1128/JVI.00645-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760. 10.1073/pnas.1115861108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Concha M, Wang X, Cao S, Baddoo M, Fewell C, Lin Z, Hulme W, Hedges D, McBride J, Flemington EK. 2012. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J. Virol. 86:1458–1467. 10.1128/JVI.06537-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Juranic Lisnic V, Babic Cac M, Lisnic B, Trsan T, Mefferd A, et al. 2013. Dual analysis of the murine cytomegalovirus and host cell transcriptomes Reveal new aspects of the virus-host cell Interface. PLoS Pathog. 9:e1003611. 10.1371/journal.ppat.1003611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. 2008. The antisense transcriptomes of human cells. Science 322:1855–1857. 10.1126/science.1163853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816. 10.1038/nature05874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engström PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C, RIKEN Genome Exploration Research Group. Genome Science Group. (Genome Network Project Core Group) FANTOM Consortium 2005. Antisense transcription in the mammalian transcriptome. Science 309:1564–1566. 10.1126/science.1112009 [DOI] [PubMed] [Google Scholar]
- 18. Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hökfelt T, Broberger C, Porreca F, Lai J, Ren K, Ossipov M, Koshkin A, Jakobsen N, Skouv J, Oerum H, Jacobsen MH, Wengel J. 2000. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. U. S. A. 97:5633–5638. 10.1073/pnas.97.10.5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cazalla D, Yario T, Steitz JA, Steitz J. 2010. Down-regulation of a host microRNA by a herpesvirus Saimiri noncoding RNA. Science 328:1563–1566. 10.1126/science.1187197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Borah S, Darricarrere N, Darnell A, Myoung J, Steitz JA. 2011. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog. 7:e1002300. 10.1371/journal.ppat.1002300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tibbetts SA, Suarez F, Steed AL, Simmons JA, Virgin HW. 2006. A gamma-herpesvirus deficient in replication establishes chronic infection in vivo and is impervious to restriction by adaptive immune cells. Virology 353:210–219. 10.1016/j.virol.2006.05.020 [DOI] [PubMed] [Google Scholar]
- 22. Kapadia SB, Molina H, van Berkel V, Speck SH, Virgin HW. 1999. Murine gammaherpesvirus 68 encodes a functional regulator of complement activation. J. Virol. 73:7658–7670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Berkel V, Levine B, Kapadia SB, Goldman JE, Speck SH, Virgin HW. 2002. Critical role for a high Affinity chemokine binding protein in lethal meningitis caused by a γ-herpesvirus. J. Clin. Invest. 109:905–914. 10.1172/JCI200214358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bortz E, Wang L, Jia Q, Wu TT, Whitelegge JP, Deng H, Zhou ZH, Sun R. 2007. Murine gammaherpesvirus 68 ORF52 encodes a tegument protein required for virion morphogenesis in the cytoplasm. J. Virol. 81:10137–10150. 10.1128/JVI.01233-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ebrahimi B, Dutia BM, Roberts KL, Garcia-Ramirez JJ, Dickinson P, Stewart JP, Ghazal P, Roy DJ, Nash AA. 2003. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J. Gen. Virol. 84:99–109. 10.1099/vir.0.18639-0 [DOI] [PubMed] [Google Scholar]
- 26. Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, Sun R. 2005. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 102:3805–3810. 10.1073/pnas.0404521102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krug LT. 2013. Complexities of gammaherpesvirus transcription revealed by microarrays and RNAseq. Curr. Opin. Virol. 3:276–284. 10.1016/j.coviro.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. 2007. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8:39. 10.1186/1471-2164-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S, Cho HJ, Park JJ, Kim YS, Hwang S, Sun R, Jung M. 2007. The ORF49 protein of murine gammaherpesvirus 68, cooperates with RTA in the regulation of virus replication. J. Virol. 81:9870–9877. 10.1128/JVI.00001-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Virgin HW, Presti RM, Li XY, Liu C, Speck SH. 1999. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 73:2321–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu S, Pavlova IV, Virgin HWt, Speck SH. 2000. Characterization of gammaherpesvirus. 68 gene 50 transcription. J. Virol. 74: 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rochford R, Lutzke ML, Alfinito RS, Clavo A, Cardin RD. 2001. Kinetics of murine gammaherpesvirus 68 gene expression following infection of murine cells in culture and in mice. J. Virol 75:4955–4963. 10.1128/JVI.75.11.4955-4963.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Berkel V, Preiter K, Virgin HWt, Speck SH. 1999. Identification and initial characterization of the murine gammaherpesvirus 68 gene M3, encoding an abundantly secreted protein. J. Virol. 73:4524–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bortz E, Whitelegge JP, Jia Q, Zhou ZH, Stewart JP, Wu TT, Sun R. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 77:13425–13432. 10.1128/JVI.77.24.13425-13432.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nealon K, Newcomb WW, Pray TR, Craik CS, Brown JC, Kedes DH. 2001. Lytic replication of Kaposi’s sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J. Virol. 75:2866–2878. 10.1128/JVI.75.6.2866-2878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Husain SM, Usherwood EJ, Dyson H, Coleclough C, Coppola MA, Woodland DL, Blackman MA, Stewart JP, Sample JT. 1999. Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 96:7508–7513. 10.1073/pnas.96.13.7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moorman NJ, Lin CY, Speck SH. 2004. Identification of candidate gammaherpesvirus 68 genes required for virus replication by signature-tagged transposon mutagenesis. J. Virol. 78:10282–10290. 10.1128/JVI.78.19.10282-10290.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andersen AA, Panning B. 2003. Epigenetic gene regulation by noncoding RNAs. Curr. Opin. Cell Biol. 15:281–289. 10.1016/S0955-0674(03)00041-3 [DOI] [PubMed] [Google Scholar]
- 39. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693. 10.1126/science.1192002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rinn JL, Chang HY. 2012. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81:145–166. 10.1146/annurev-biochem-051410-092902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323. 10.1016/j.cell.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:11667–11672. 10.1073/pnas.0904715106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 8:e1002680. 10.1371/journal.ppat.1002680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. 10.1016/j.chom.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6:211–221. 10.1038/nrmicro1794 [DOI] [PubMed] [Google Scholar]
- 46. Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 83:8182–8190. 10.1128/JVI.00712-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139. 10.1073/pnas.0707266104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U. S. A. 101:17234–17239. 10.1073/pnas.0407933101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gray KS, Allen RD, III, Farrell ML, Forrest JC, Speck SH. 2009. Alternatively initiated gene 50/RTA transcripts expressed during murine and human gammaherpesvirus reactivation from latency. J. Virol. 83:314–328. 10.1128/JVI.01444-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gray KS, Forrest JC, Speck SH. 2010. The de novo methyltransferases DNMT3a and DNMT3b target the murine gammaherpesvirus immediate-early gene 50 promoter during establishment of latency. J. Virol. 84:4946–4959. 10.1128/JVI.00060-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goodwin MM, Molleston JM, Canny S, Abou El Hassan M, Willert EK, Bremner R, Virgin HW. 2010. Histone deacetylases and the nuclear receptor corepressor regulate lytic-latent switch gene 50 in murine gammaherpesvirus 68-infected macrophages. J. Virol. 84:12039–12047. 10.1128/JVI.00396-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang Z, Tang H, Huang H, Deng H. 2009. RTA promoter demethylation and histone acetylation regulation of murine gammaherpesvirus 68 reactivation. PLoS One 4:e4556. 10.1371/journal.pone.0004556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rennekamp AJ, Lieberman PM. 2011. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. J. Virol. 85:2837–2850. 10.1128/JVI.02175-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jepsen JS, Wengel J. 2004. LNA-antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Dev. 7:188–194 [PubMed] [Google Scholar]
- 56. Koshkin AA, Nielsen P, Meldgaard M, Rajwanshi VK, Singh SK, Wegnal J. 1998. LNA (locked nucleic acid): an RNA mimic forming exceedingly stable LNA:LNA duplexes. J. Am. Chem. Soc. 120:13252–13253. 10.1021/ja9822862 [DOI] [Google Scholar]
- 57. Grünweller A, Wyszko E, Bieber B, Jahnel R, Erdmann VA, Kurreck J. 2003. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res. 31:3185–3193. 10.1093/nar/gkg409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mounce BC, Tsan FC, Droit L, Kohler S, Reitsma JM, Cirillo LA, Tarakanova VL. 2011. Gammaherpesvirus gene expression and DNA synthesis are facilitated by viral protein kinase and histone variant H2AX. Virology 420:73–81. 10.1016/j.virol.2011.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Berkel V, Barrett J, Tiffany L, Murphy PM, McFadden G, Speck SH, Virgin HW., IV 2000. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 74:6741–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holden P, Horton WA. 2009. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2:243. 10.1186/1756-0500-2-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 63. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, et al. 2012. Landscape of transcription in human cells. Nature 489:101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lin YT, Kincaid RP, Arasappan D, Dowd SE, Hunicke-Smith SP, Sullivan CS. 2010. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. 16:1540–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome-wide RNA expression in MHV68 as detected by RNA-Seq. RNA-Seq RNA expression measurements were highly reproducible, strongly correlated with our earlier tiled array results, and confirmed the expression of all 30 expressed genomic regions (EGRs). Transcriptional signal from infected 3T12 cells at 18 hpi was plotted on a log2 scale underneath the annotated gene positions. Read depth from two independent RNA-Seq experiments was plotted, on a log2 scale, as blue/light blue and red/light red lines for the positive and negative genomic strands, respectively. Blue and red arrows represent annotated genes on the positive and negative strands of the genome, respectively. Gray boxes represent the two internal repeat regions of the MHV68 genome that were excluded from our analysis. EGRs 1 to 30 are shown as hatched arrows. (A) Genomic positions 1 to 40 kb. (B) Genomic positions 40 to 80 kb. (C) Genomic positions 80 to 119.45 kb. Download
Antisense oligonucleotide probes.
PCR primers for Northern blot probes.
3′ ends of MHV68 transcripts identified by RNA sequencing. The average numbers of reads per position at 6 hpi and 18 hpi are in bold in the second and third columns, respectively. The numbers in parentheses are the numbers of reads from specific experiments at the indicated time points. Gene region refers to a possible gene associated with the indicated polyadenylation site selected based on the gene with the closest 3′ end to a given site.