

Abstract
Expression of MHC class I molecules, which provide immune surveillance against intracellular pathogens, is higher on lymphoid cells than on any other cell types. In T cells, this is a result of activation of class I transcription by the T cell enhanceosome consisting of Runx1, CBFβ and LEF1. We now report that MHC class I transcription in T cells also is enhanced by Foxp3, resulting in higher levels of class I in CD4+CD25+ T regulatory cells than in conventional CD4+CD25− T cells. Interestingly, the effect of Foxp3 regulation of MHC class I transcription is cell-type specific: Foxp3 increases MHC class I expression in T cells but represses it in epithelial tumor cells. In both cell types, Foxp3 targets the upstream IRE and downstream core promoter of the class I gene. Importantly, expression of MHC class I contributes to the function of CD4+CD25+ T regulatory cells by enhancing immune suppression, both in in vitro and in vivo. These findings identify MHC class I genes as direct targets of Foxp3 whose expression augments regulatory T cell function.
Introduction
Major histocompatibility class I molecules function to provide immune surveillance against intracellular pathogens. In the mature immune system, presentation of foreign peptides by class I molecules regulates both innate and adaptive immunity, by inhibiting non-specific NK responses and triggering specific CTL responses, respectively. During development, MHC class I expression on non-hematopoietic thymic epithelial cells is essential for the maturation and survival of CD8+ T cells. Although originally thought to be passive receptors of intracellularly derived peptides, emerging evidence reveals that maintenance of homeostatic levels of MHC class I is critical for a functional immune response (1–4).
MHC class I molecules are ubiquitously expressed in somatic cells, although at different levels in different cell types, and their expression is dynamically regulated by hormones and cytokines. Lymphoid cells express the highest levels of class I, whereas neurons express two-orders of magnitude less. MHC class I expression is controlled transcriptionally by the interaction of tissue-specific transcription factors with cognate DNA sequence elements on the extended class I promoter. The DNA sequence elements that mediate class I regulation have been mapped to distinct domains that mediate tissue specific and hormone specific signals (3, 5–7). Transcription initiates at multiple sites within the core promoter, which integrates upstream signals targeting distinct transcription starts in response to tissue specific or dynamic signals (5, 6).
A variety of DNA-binding transcription factors have been identified that interact either directly or indirectly with tissue specific and hormone specific regulatory elements. For example, a B lymphocyte-specific enhanceosome consisting of the coactivator CIITA and DNA-bound transcription factors RFX, CREB/ATF and NF-Y leads to high cell surface class I and II expression in B cells (8–11). In T cells, the constitutive high level expression of class I is not due to CIITA but is established by a T cell enhanceosome consisting of RUNX1, CBFβ and LEF1 (12). Whether additional factors in the different T cell subsets superimpose on the T cell enhanceosome to differentially regulate MHC class I expression has not been examined previously.
In the present study, we have examined the regulation of MHC class I expression in T cell subsets. We report that class I levels in CD4+CD25+ T regulatory cells are consistently higher than in either conventional CD4+ T effector cells or CD8+ T cells as a result of Foxp3-mediated transcriptional activation. Importantly, these elevated levels of class I contribute to efficient Treg suppressive function. Surprisingly, Foxp3 affects MHC class I transcription differently in different cell types as it represses class I transcription in epithelial tumor cells. Thus, these studies show that Foxp3 is an active, context-dependent regulator of MHC class I expression and function.
Materials and Methods
Animals
β2m−/− and β2m−/−RAG2−/−mice were bred in the NCI CCR animal colony. PD1 transgenic mice and Foxp3 transgenic mice (A10 and T3) have been described previously (13, 14). The PD1 drop-out transgenic mouse was generated in the NCI core facility by microinjection of a genomic clone of PD1 from which −50bp to +3 bp of promoter sequences were deleted. B6 (CD45.2) mice were purchased from The Jackson Laboratory (Bar Harbor, ME), and B6 (CD45.1) mice were obtained from the Frederick Cancer Research Center (Frederick, MD). All mice were cared for in accordance with National Institutes of Health guidelines.
Reagents for flow cytometry and protein immunoblotting
mAbs with the following specificities were used in this study: CD4 (RM4.5 and GK1.5), CD25 (PC61 and 7D4), H-2Kb (AF6-88.5), H-2Db (28-14-8), H-2Kd (SF1-1.1), H-2Dd (34–2–12), CD45.1 (A20), CD45.2 (104), CD45RB (16A), CD152 (CTLA-4; UC10-4F10-11) all obtained from BD Biosciences (San Diego, CA); PD1 and HLA class I (PT85A with specificity for both porcine and human MHC class I, but not mouse MHC class I) from VMRD (Pullman, WA) and Foxp3 (FJK-16s) obtained from eBioscience (San Diego, CA). Stained cells were analyzed on a BD LSR flow cytometer (Becton Dickinson). Where indicated, fluorescence was quantified and expressed as mean fluorescent intensity (MFI). Antibodies with the following specificities were used for protein immunoblotting: FLAG from Sigma (St. Louis, MO); and HSP60 from Santa Cruz (Santa Cruz, CA). Total cell lysates from transfected HeLa or Jurkat cells were resolved by SDS-PAGE followed by blotting.
Cells lines and cultivation
HeLa epithelial cells were grown in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 20 mM HEPES (pH 7.2), and gentamicin sulfate (10ng/ml). Jurkat (T cell) line was maintained in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 55 µM 2-ME, 100 µM minimal essential amino acids, 1 mM sodium pyruvate, and gentamicin sulfate (10 ng/ml). MCF-7 human breast adenocarcinoma cells were grown in EMEM supplemented with 10% FBS, 0.1mM non-essential amino acids, 10µg/ml Insulin and 1mM Sodium pyruvate. All Cell lines were maintained in a humidified, 7.5% CO2 incubator at 37°C.
Plasmids and cloning strategies
The MHC class I promoter construct used in these studies derived from the swine class I gene PD1 (13, 15). −416LUC was generated by ligating class I promoter sequences, extending from the 5’ XbaI site to the HindIII site at position −14 of −416WT (12), into the NheI/HindIII sites of the pGL2B luciferase expression vector (Promega). The PD1 promoter truncation series, ligated to the luciferase reporter, were previously described (12). Drop-out constructs were previously described (16). The IRE mutant was generated by replacing the wild-type IFN responding element “TTTCAC” with “TCTCGC” (Stratagene).
Luciferase reporter assay
For HeLa or MCF-7 cell transfection, 2×105 Hela cells or MCF-7 cells were seeded 1 day before transfection to reach 80% confluency and were cotransfected with Foxp3 and various PD1 luciferase reporter constructs together with a control plasmid construct, either pRL-TK renilla or GFP construct. For Jurkat cell transfection, 2×106 Jurkat cells were cotransfected with Foxp3 and PD1 luciferase reporter constructs together with a control plasmid construct, either pRL-TK renilla or GFP construct. Cells were harvested 24 h after transfection and analyzed with a Dual Assay Reporter Kit (Promega). Where indicated, RUNX1 cDNA was cotransfected with the Foxp3 and PD1 luciferase reporter constructs into Hela or MCF-7 cells. Data were analyzed by comparing luciferase activity to renilla activity or GFP and adjusted to the fold increase over background.
Quantitative RT-PCR
RNA was purified from the indicated FACS sorted cell types with the RNeasy MiniKit (Qiagen) and reverse transcribed to cDNA, using a SuperScript III Kit (Invitrogen). Relative quantities of mRNA expression were analyzed using real-time PCR (Applied Biosystems ABI Prism 7700 Sequence Detection System, Applied Biosystems), with SYBR (Qiagen) green fluorescence dye. The primer sequences for MHC class I: 5’- CTTGCTCTGGTTGTAGTAGC -3’ and 5’- ATGGAAGTCGGCTACGTGGAC -3’; for CTLA-4: 5’- TCTGAAGCCATACAGGTGACCC -3’ and 5’- CATAAATCTGCGTCCCGTTGC -3’; for TGF-β: 5’- TGGAGCAACATGTGGAACTC -3’ and 5’- GTCAGCAGCCGGTTACCA -3’; and for IL-10: 5’- AGAGCAAGGCAGTGGAGCAG -3’ and 5’- GGGATGACAGTAGGGGAACC -3’. Relative transcript abundance was determined by using the ΔCt method after normalization with 18S (Ambion Inc, TX ). Amplification of beta-2-microglobulin products was achieved using TaqMan probes (Mm00437762_m1), and relative transcript abundance was normalized with RPL13A (Mm01612986_gH).
Cell preparations
CD4+ lymph node T (LNT) cells with a purity of >97% were obtained by antibody mediated magnetic bead depletion and further fractionated into CD4+CD25+, CD4+CD25− and CD4+CD45RBhi T cell populations with a purity >99% by electronic cell sorting.
In vitro functional assays
For anti-TCR induced T cell proliferation, responder T cells (3–5 × 104/well) were placed in 96-well round bottom plates (0.2 ml) together with irradiated T cell-depleted B6 spleen cells (2000R) as accessory cells (APC) and stimulated with anti-CD3 mAb (1µg/ml) and/or rIL-2 (200U/ml) for 72h. For in vitro suppression assays, CD4+CD25− responder T cells (3–5 × 104/well) were cultured with an equal number of CD4+CD25+ T cells, APC, and anti-CD3 mAb (1µg/ml) for 72h. Where indicated, cultures were pulsed with [3H]-thymidine 8h prior to harvest. Alternatively, CFSE-labeled CD4+CD25− responder T cells were cocultured with CD4+CD25+ and APCs (which expressed a different CD45 allele from the Tconv cells) and stimulated with anti-CD3 (1 µg/mL) for 72 hours. At the end of culture, CFSE fluorescence of the responder T cells was determined.
T cell reconstitution and in vivo induction of inflammatory bowl disease (IBD)
β2m−/−RAG2−/− mice were injected i.v. with 4×105 purified CD4+CD45RBhigh T cells either alone or in combination with 4×105 CD4+CD25+ T cells from the indicated sources. Following T cell transfer, mice were weighed weekly and monitored for clinical signs of diarrhea.
Intracellular staining
To detect intracellular CTLA-4 and Foxp3, thymocytes and purified LN cells were surface stained with anti-CD4, anti-CD8 and anti-CD25, fixed and then stained for CTLA-4 and Foxp3.
Analysis of Chip-Seq data
SRA files were obtained from GEO Series Accession GSE40684 (http://www.ncbi.nlm.nih.gov/geo/) (17). Sequence reads were aligned to the UCSC mm9 genome using Bowtie (v1.1.2) (18) using default parameters allowing for 2 mismatches (−m 1 −v2). Peak calling was performed using MACSv1.4 (19) with default parameters (1.86E8 effective genome size, tag size 26bp, band width 300). A p value of 1e-05 was used for peak detection compared to input. Histogram distribution of sequence tag reads were visualized using Integrative Genomics Viewer (Version 2.0).
Results
Cell surface expression of MHC class I is higher on CD4+CD25+ T regulatory cells than on conventional CD4+ T cells
To examine the possibility that MHC class I expression is differentially regulated in different T cell subsets, we first compared cell-surface class I levels on CD4+ and CD8+ cells in the thymus and lymph nodes (Figure 1A, lower panel). There was no difference in class I in either the thymus or lymph nodes between CD4+ and CD8+ cells. To further characterize class I expression among CD4+ cells, we asked whether CD4+CD25+ T regulatory cells (Tregs) differed in their MHC class I expression from conventional CD4+CD25− T cells (Tconv). Surprisingly, we found that cell surface H-2Kb protein levels on both thymic and peripheral Tregs were approximately 2-fold higher than on Tconv cells (Figure 1A, upper panels). The elevated class I cell surface expression on Tregs, relative to CD4+ Tconv cells, was reflected in the amount of total class I protein (Figure 1B, right). To determine if regulation of MHC class I in Tregs is pre-translational, we compared MHC class I RNA levels in CD4+ Tregs and Tconv cells (Figure 1B, left). Consistent with the cell surface protein expression, MHC class I RNA levels were significantly higher in CD4+ Tregs than CD4+ Tconv.
Figure 1. MHC class I expression is elevated on Tregs.
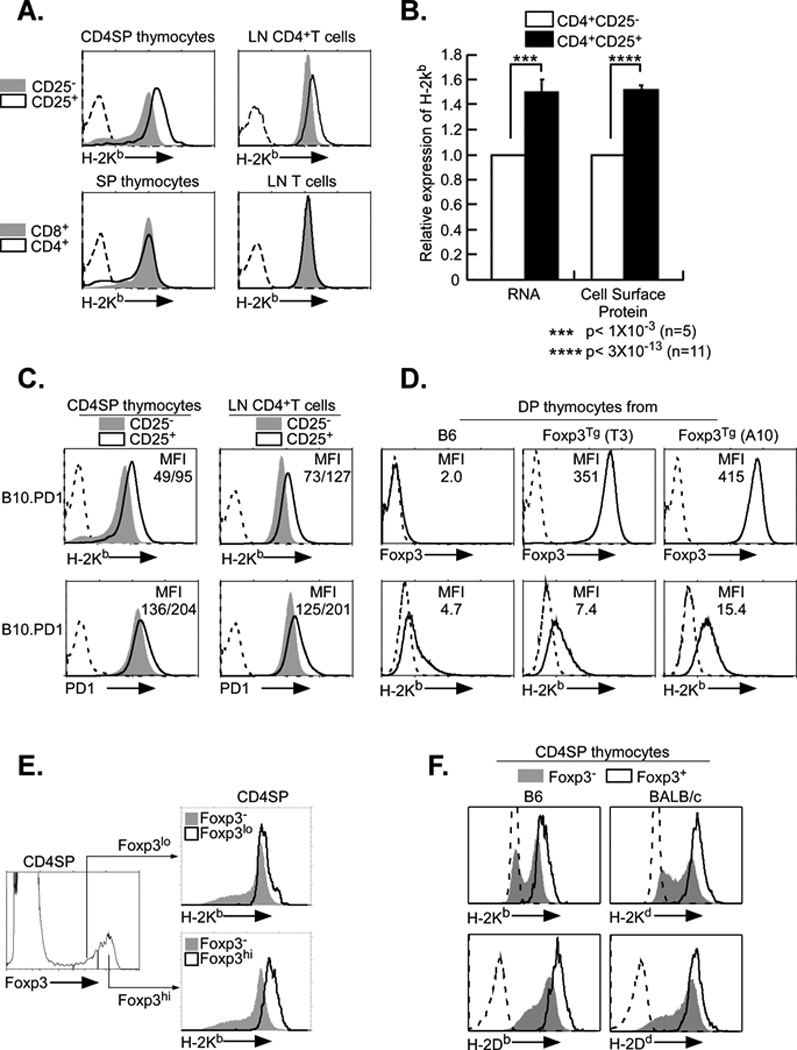
A. Surface expression of MHC class I is elevated on CD4+CD25+ T cells from thymus and lymph node relative to CD4+CD25− T cells. Upper panels: Thymocytes (left) and lymph node cells (LN, right) were stained for H2-Kb, CD4 and CD25, as described in Materials and Methods. FACS profiles of class I expression on the CD4+CD25+ (black line) and CD4+CD25− (shaded) populations are shown. Lower panels. Class I levels on CD8+ (shaded) and CD4+ (black line) thymocytes (left) and lymph nodes (right) were determined by staining for CD4, CD8 and H-2Kb.
B. Class I RNA and cell surface protein levels are elevated in CD4+CD25+ T cells relative to CD4+CD25− T cells. Total RNA was isolated from purified Tregs and Tconv and subjected to RT-PCR (left). Cell surface class I protein levels were calculated from total mean fluorescence intensity (MFI) following FACS profiling with anti-H-2Kb antibody (right). Both sets of results are presented relative to CD4+CD25− Tconv levels. ***p<0.001 (n=5); ****p<0.0001 (n=11).
C. Expression of a class I transgene is elevated on CD4+CD25+ T cells in thymus and lymph node. Thymocytes and lymph nodes from mice transgenic for the MHC class I gene, PD1, were stained for endogenous H2-Kb (upper panels), transgenic PD1 (lower panels), CD4 and CD25, as described in Materials and Methods. FACS profiles of class I expression on the CD4+CD25− (shaded) and CD4+CD25+ (black line) populations are shown. Mean fluorescence intensity (MFI) is indicated for CD25+/CD25−. The data are representative of three experiments.
D. Levels of MHC class I expression correlate with levels of Foxp3 expression. Double positive (DP) thymocytes were isolated from B6 mice and two lines of Foxp3 transgenics, T3 and A10. Expression of Foxp3 (upper panels) and H-2Kb (lower panels) were monitored by FACS. Foxp3 staining (solid line) and negative control (dotted line) are shown; MFI of Foxp3 and H-2Kb staining are indicated. The data are representative of three experiments.
E. Class I expression is proportional to Foxp3 levels in native Tregs. CD4 single positive thymocytes were stained for Foxp3 (left) and H-2Kb (right). The expression of H-2Kb on Foxp3 high (right, lower panel) and Foxp3 low (right, upper panel) cells is compared to Foxp3 negative cells. The data are representative of three experiments.
F. MHC class I expression is elevated on CD4+Foxp3+ thymocytes relative to CD4+Foxp3− thymocytes. Thymocytes from B6 and BALB/c were surface stained for H2-Kb, H-2Db, H-2Kd, H-2Dd, CD4 and CD8, fixed and stained for intracellular Foxp3. FACS profiles of class I expression on the CD4+Foxp3+ (black line) and CD4+Foxp3- (shaded) populations are shown. The data are representative of three experiments.
To assess the mechanisms that result in elevated MHC class I expression in Tregs, we first asked if increased expression was intrinsic to the MHC class I gene or an indirect positional effect linked to overall regulation of H-2 locus. To distinguish these two possibilities, we examined the expression in Tregs of an exogenous MHC class I transgene (PD1) that is not linked to the H-2 locus but whose pattern of expression fully recapitulates that of endogenous MHC class I genes (13). We reasoned if the elevated H-2 class I levels in Tregs result from a direct effect, expression of the PD1 transgene also would be increased in Tregs. Indeed, both thymic and peripheral CD4+ Tregs expressed higher levels of both surface PD1 and H-2Kb than did CD4+Tconv cells (Figure 1C). Thus, increased class I expression is independent of chromosomal location, consistent with a direct effect on the MHC class I gene in Tregs. Furthermore, CD4+ T regs also express higher levels of β2microglobulin RNA than conventional CD4+ T cells (Supplementary Figure 1). Interestingly, Tregs from β2m-heterozygous mice express nearly wild-type levels of H-2Kb (Supplementary Figure 1.) Since Tregs differ from Tconv cells in their expression of Foxp3, we next considered the possibility that the elevated MHC class I expression in Tregs was mediated by Foxp3.
Over-expression of Foxp3 correlates with increased MHC class I expression
To determine whether elevated MHC class I expression was due to Foxp3, we examined surface MHC class I levels on immature DP thymocytes from two lines of transgenic Foxp3 mice that both express transgenic Foxp3 protein but at different levels (14). Immature DP thymocytes from wild type mice do not express Foxp3 protein and expressed the low levels of surface class I that are normally observed on DP thymocytes (Figure 1D). In contrast, DP thymocytes from the Foxp3 transgenic mice express high levels of Foxp3 protein (Figure 1D, upper panels). Importantly, cell surface H-2Kb class I levels were directly proportional to the amount of Foxp3 protein (Figure 1D, lower panels). Thus, DP thymocytes from the A10 transgenic line, which contain higher levels of Foxp3 than the T3 line, also expressed about two-fold higher levels of class I. Importantly, MHC class I expression also correlated with endogenous Foxp3 protein levels. Among Foxp3-expressing CD4+SP cells, class I levels were higher on the subset of Foxp3-high positive cells, relative to the Foxp3-low subset (Figure 1E). MHC class I expression also was increased on newly generated Foxp3+ Tregs compared with that on CD4+CD25+Foxp3− Treg precursors in the same culture following in vitro induction with IL2 (data not shown). Thus, class I expression is directly correlated with Foxp3 protein levels and increased in a dose-dependent manner with Foxp3. Taken together, these results demonstrate a direct correlation between Foxp3 protein levels and class I expression in Tregs.
The increased class I expression on Tregs, relative to conventional CD4+ T cells, is not restricted to H-2Kb as H-2Db is also elevated on Tregs. Similarly, Tregs from BALB/c mice also have increased H-2Kd and H-2Dd surface expression (Fig. 1F)
Foxp3 enhances MHC class I promoter activity in Jurkat T cells
Since only Tregs contain Foxp3 protein, we next asked whether introduction of Foxp3 into any T cell would result in increased class I expression. Indeed, transient transfection of a Foxp3 expression vector into Jurkat T cells, which express the components of the T cell enhanceosome (TCE) and other T cell specific transcription factors, such as NFAT (12), resulted in increased cell surface expression of HLA class I (Table I). Furthermore, transient cotransfection of the Foxp3 expression vector with a class I promoter reporter construct enhanced class I promoter activity (Figure 2A). Although modest, the effect was significant and reproducible and was within the range of increase that is observed on Tregs in vivo. Therefore, the enhanced class I expression in Tregs is transcriptional and a function of Foxp3.
Table I. Foxp3 overexpression increases endogenous MHC Class I expression in Jurkat cells.
Jurkat cells were transfected with Foxp3 cDNA (2.0 ug/ 106 cells); after 24 hrs, cells were surface stained either with anti-class I antibody and secondary antibody or secondary alone, as a control. Foxp3+ transfectants were distinguished from non-transfectants by intracellular staining with anti-Foxp3. Both cell surface and intracellular staining levels were assayed by flow cytometry. Transfection efficiency was 10%.
MFI
Figure 2. Foxp3 activation of MHC class I promoter activity in Jurkat cells maps to the IRE and core promoter.
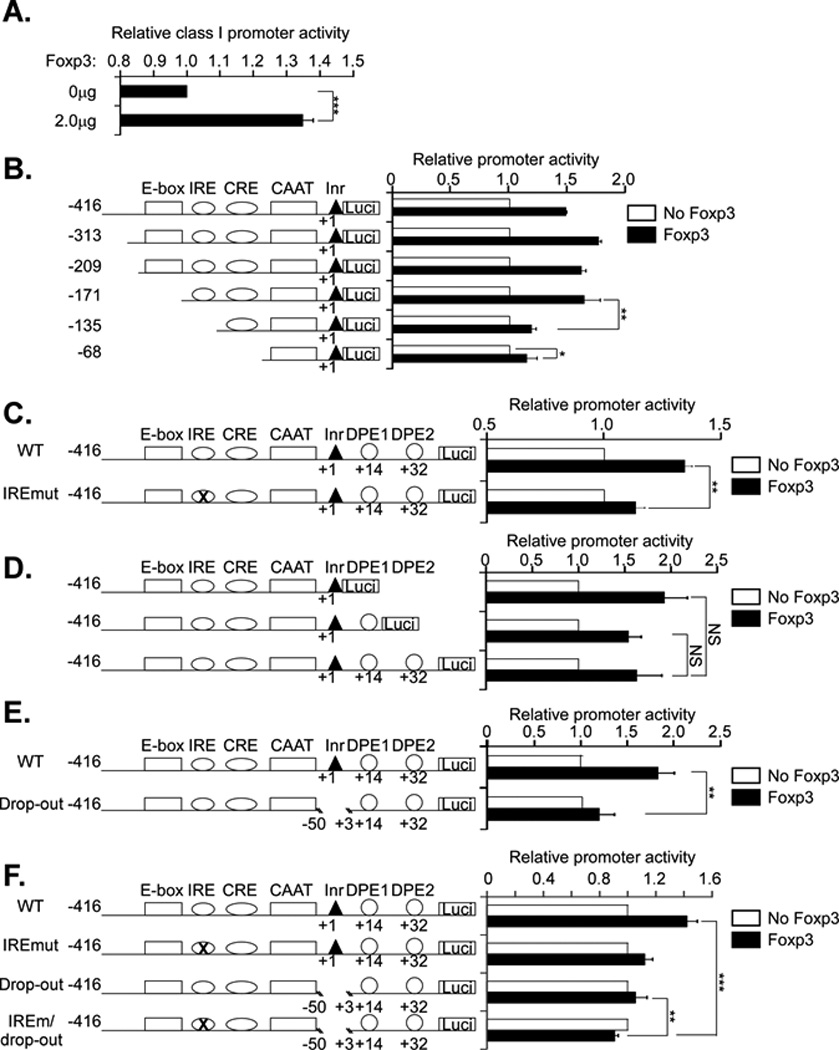
A. Jurkat T cells were cotransfected with a 1.0 µg of the MHC class I PD1 promoter construct containing DNA sequences from −416 to +32 bp (WT), ligated to a luciferase reporter gene and 2.0 µg of a Foxp3 expression vector or control vector. Results are expressed relative to class I promoter activity of the control co-transfection. Results are the average ± SEM of 6 experiments. *** p<0.0004.
B. Jurkat T cells were cotransfected with 2.0 µg of Foxp3 or control vector (no Foxp3) and 1.0µg of a series of 5’ truncation deletion mutants of the class I promoter, as diagrammed. The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active (12).The results are the average ± SEM of 4 separate experiments. *, p<0.2; ** p=0.016.
C. Jurkat T cells were cotransfected with 2.0 µg Foxp3 or control vector (no Foxp3) and either WT (1.0 µg) or a class I promoter construct containing a mutation in the interferon response element (IRE). The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the two constructs differed, both were transcriptionally active. The results are the average ± SEM of 4 separate experiments. ** p=0.019.
D. Jurkat T cells were cotransfected with 2.0 µg Foxp3 or control vector and 1 µg of a series of 3’ truncation deletion mutants of the class I promoter. The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active (16). The results are the average ± SEM of 3 separate experiments.
E. Jurkat T cells were cotransfected with 2.0 µg Foxp3 or control vector and 1.0 µg of either WT or a MHC class I promoter construct containing a −50 to +3 deletion (drop out). The results are average ± SEM of 4 separate experiments. ** p=0.018.
F. Jurkat T cells were cotransfected with 2.0 µg Foxp3 or control vector and 1.0 µg of either WT, IRE mutant, the drop-out or a mutant containing both the IREmut and the drop-out (IREm/drop-out). The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active (16).The results are the average ± SEM of 5 separate experiments.**, p<0.01; ***,p<0.004
Since Foxp3 activated the class I promoter construct in transfected Jurkat T cells, we next mapped the DNA sequence element(s) targeted by Foxp3. Jurkat T cells were contransfected with Foxp3 and a 5’ promoter truncation series cotransfected with Foxp3 into Jurkat T cells (Figure 2B). All of the promoter constructs had a common 3’ terminus at +1, but differed in their 5’ extension. All of the constructs that terminated between −416 bp and −171 bp were activated by Foxp3 (Figure 2B). In contrast, further deletion to −135 bp and beyond significantly reduced, but did not eliminate completely, Foxp3 activation of class I promoter activity. Thus, a Foxp3 response element maps to the class I promoter segment between −171 bp and −135 bp. Interestingly, contained within this segment is the conserved class I interferon response element (IRE) (Supplementary Figure 2). Inspection of the IRE sequence (AGTTTCACTTCT) revealed a consensus Foxp3 binding site (AGTTTCA) imbedded within it. Indeed, mutation of the IRE sequence significantly reduces the ability of Foxp3 to affect class I promoter activity (Figure 2C). (Attempts to demonstrate direct binding of Foxp3 to either this element or the canonical one, as reported by others (20, 21), were not successful. It is likely that the interaction of Foxp3 with DNA is relatively weak and needs to be stabilized by co-factors.)
The fact that the promoter activity of the IRE mutant is still enhanced by Foxp3, albeit to a reduced degree, suggested the presence of an additional Foxp3 response element. Indeed as noted above, the construct containing only 68bp of core promoter sequences maintained a small, but reproducible, response to Foxp3, also suggesting the presence of another response element. To map this second element, a series of 3’ promoter truncations were examined for their ability to respond to Foxp3. As shown in Figure 2D, Figure 3’ truncation of the class I promoter from +32 bp to +1 bp does not affect the Foxp3 response, mapping the second Foxp3 response element between −68 and +1 bp. To verify the presence of a response element in this region, a deletion between −50 bp and +3 bp was introduced into the class I promoter construct (Figure 2E, drop-out). Although, as previously reported, this construct retains promoter activity (16), the Foxp3 response is significantly reduced (Figure 2E). Since no consensus Foxp3 site occurs in the −50 bp to +3 bp region, the response to Foxp3 may be mediated by an interaction with another transcription factor(s). To determine whether the residual response to Foxp3 was mediated by the intact IRE remaining in the drop-out construct, we generated a construct with both the IRE mutation and the drop-out (IREm/drop-out). As shown in Figure 2F, the IREm/drop-out no longer responded to Foxp3 activation. Taken together, these experiments map two Foxp3 response elements to the MHC class I promoter region: one contained within the IRE and one mapping between −50 bp and +3bp.
Figure 3. Foxp3 regulation of class I expression in Tregs depends on the class I core promoter.
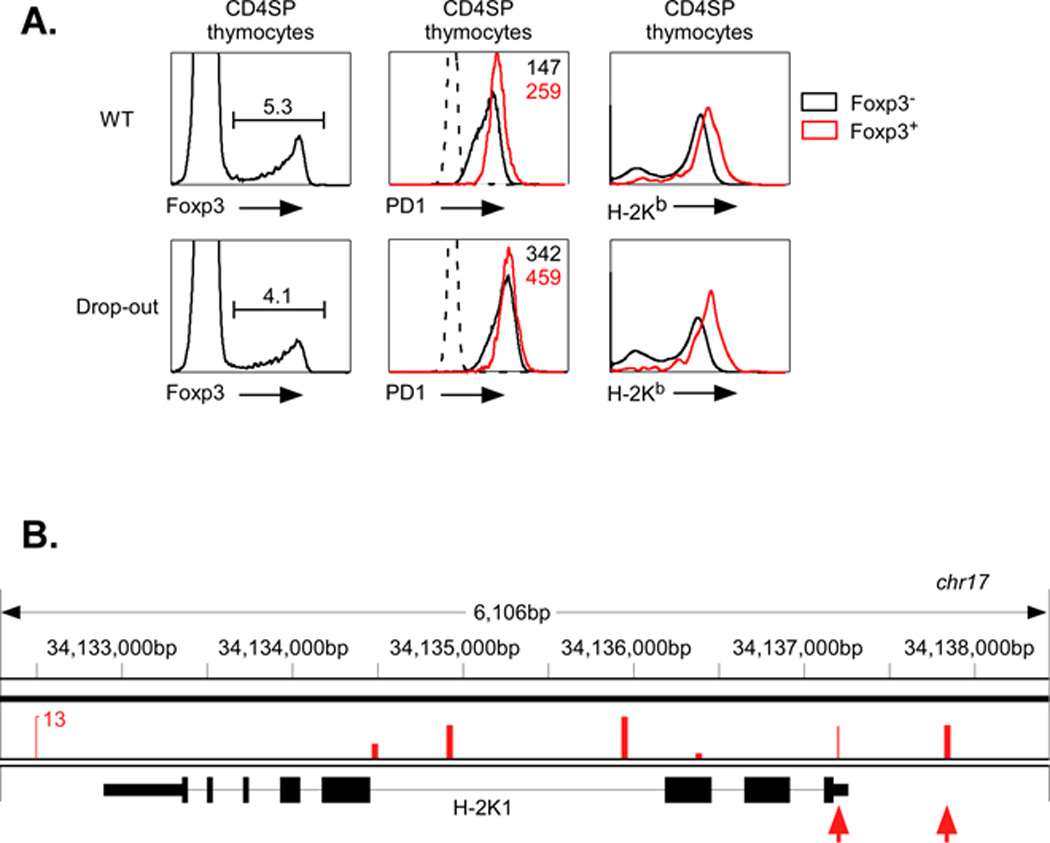
A. Foxp3 (left panels), PD1 (middle panels) and H-2Kb (right panels) levels were measured by FACS on CD4+ SP thymocytes from mice transgenic for either the WT PD1 class I gene (upper panels) or a mutant PD1 class I gene with the −50bp to +3bp drop-out (lower panels). The levels of class I expression of Foxp3+ (red line) and Foxp3− (black line) were compared. The levels of expression (MFI) of Foxp3+ (red) and Foxp3− (black) CD4+ SP thymocytes from wild type and drop-out mutant PD1 transgenes are indicated. Data are representative of three separate experiments.
B. Published Chip-seq data were analyzed as described in Methods. Significant Foxp3 binding was observed at multiple sites along the H-2K gene (veritical bars). Two sites were in the H-2K promoter region (arrows); the proximal site is located at −86bp. Additional peaks were detected within the large intron 3.
The above conclusions were based on results from transient transfection assays, which clearly showed that Foxp3 targeted a promoter region between −50 bp and +3 bp. To determine whether endogenous Foxp3 activation of the class I promoter also depended on this same region in Tregs in vivo, we generated a transgenic mouse containing a full-length PD1 MHC class I gene from which the −50 bp to +3 bp segment had been deleted (PD1drop-out). In contrast to the control, wild type PD1 transgene, PD1 expression of the PD1drop-out transgenic mice was not significantly different on Tregs compared with CD4+ Tconv cells (Figure 3A). This difference was not due to differences in overall transgene expression since the PD1drop-out is expressed at levels at least as high as the wild type PD1 transgene (Figure 3A). Furthermore, endogenous H-2Kb levels in Tregs of the two transgenic lines were identical. Thus, the promoter region between −50 bp and +3 bp contains a Foxp3 response element that functions to regulate class I expression in Tregs in vivo.
These findings suggest that Foxp3 activation of the endogenous the H-2K gene results from a direct interaction. Indeed, analysis of published ChipSeq data (17) reveals a significant peak of Foxp3 binding to the H-2K promoter at −86 bp (Fig. 3B). This is within approximately the same region of the promoter as the Foxp3 target site on the PD1 promoter. Thus, Foxp3 binds to MHC class I promoters to modulate transcription.
Foxp3 reduces MHC class I promoter activity in Hela and MCF-7 cells
Foxp3 is known to interact with a variety of different transcription and chromatin remodeling factors, including many that are not expressed exclusively in T cells. Among the factors known to bind Foxp3 are AP1 and Runx1, both of which regulate class I transcription in HeLa cells (12). Therefore, we next asked whether Foxp3 affects class I expression in non-lymphoid cells. We first examined the effect of Foxp3 on class I promoter activity in the HeLa epithelial tumor cell line. Surprisingly, co-transfection of Foxp3 with the −416/+32 bp PD1 promoter reporter construct resulted in a dramatic reduction in promoter activity (Figure 4A). Using the same promoter deletion series as in Figure 2, no discrete Foxp3 response elements were mapped between −416 bp and −68 bp (Figure 4B). Indeed, deletion of upstream sequences to −209 bp actually augmented Foxp3-mediated repression. Further deletion to −68 bp partially restored promoter activity in the presence of Foxp3, but only to level of the −416 bp. In contrast, truncation of the region between +1 and +32 bp had no effect on Foxp3 repression (Figure 4C).
Figure 4. Foxp3 suppression of class I expression in HeLa cells maps to the IRE and the core promoter.
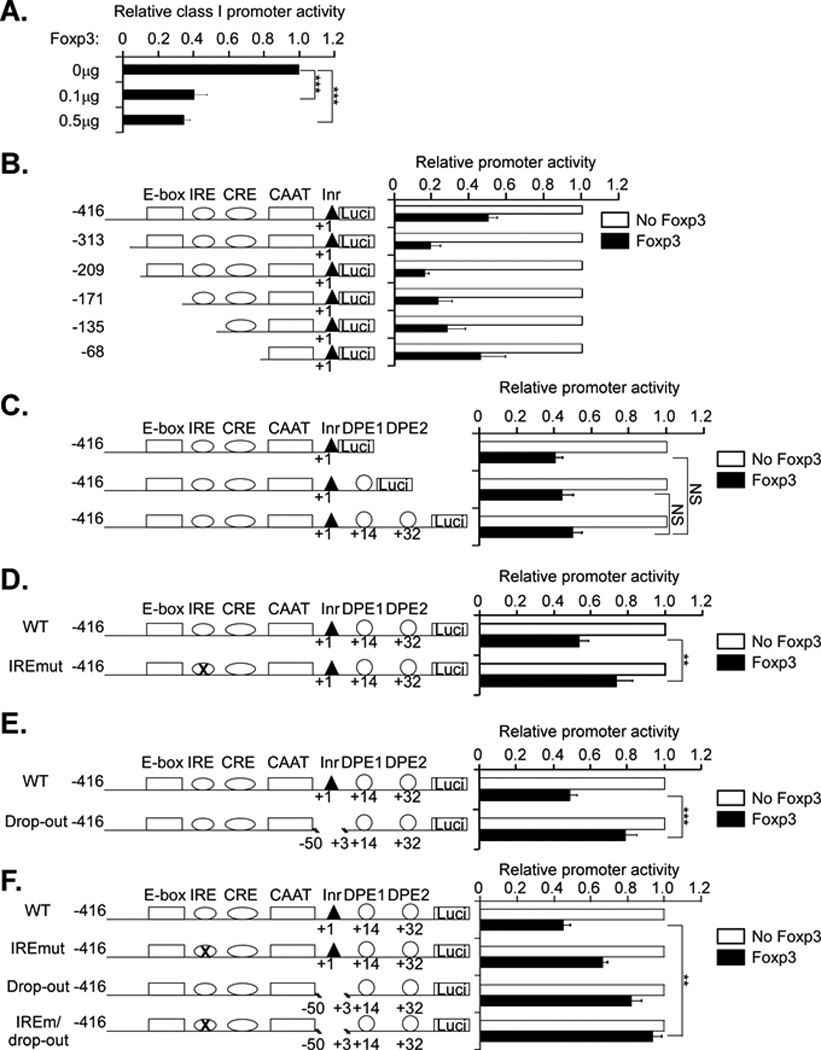
A. Hela cells were cotransfected with WT class I promoter (2.0 µg) and Foxp3 (0 µg, 0.1 µg or 0.5 µg) or equal amounts of control vector. Results are expressed relative to class I promoter activity of the control co-transfection. Results are the average ± SEM of 3 experiments. ***p< 0.001.
B. Hela cells were cotransfected with 1.0 µg Foxp3 or control vector and 2.0 µg each of a series of 5’ truncation deletion mutants of the class I promoter, as diagrammed. The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active (12). The results are the average ± SEM of 4 separate experiments.
C. Hela cells were cotransfected with 1.0 µg Foxp3 or control vector and 2.0 µg each of a series 3’ truncation deletion mutants of the class I promoter. The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active. The results are the average ± SEM of 4 separate experiments.
D. Hela cells were cotransfected with 1.0 µg Foxp3 or control vector and 2.0 µg WT class I promoter or IRE mutation constructs. The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the two constructs differed, both were transcriptionally active. The results are the average ± SEM of 4 separate experiments. **p=0.002.
E. HeLa cells were cotransfected with 1.0 µg Foxp3 or control vector and 2.0 µg of either WT or a MHC class I promoter construct containing a −50 to +3 bp deletion (drop-out). Although the absolute activity of the two constructs differed both were transcriptionally active. The results are the average ± SEM of 4 separate experiments. ***p<0.0001.
F. HeLa cells were cotransfected with 1.0 µg Foxp3 or control vector and 2.0 µg of either WT, IRE mutant, the drop-out or a mutant containing both the IREmut and the drop-out (IREm/drop-out). The results for each construct are normalized to the control transfection with empty vector. Although the absolute activity of the constructs differed, all of them were transcriptionally active (16).The results are the average ± SEM of 4 separate experiments. **p=0.004.
These findings suggest a series of DNA sequence elements upstream of +1 bp that contribute to Foxp3 regulation of class I promoter activity in HeLa cells. To determine whether the IRE is among those elements, the IRE promoter mutant was co-transfected with Foxp3 into HeLa cells. The IRE mutation significantly reduced (p<0.002), but did not ablate, Foxp3-mediated repression (Figure 4D). Similarly, deletion of the segment between −50 bp and +3 bp significantly reduced (p<0.0001), but did not eliminate, Foxp3 repression (Figure 4E). However, deletion of both the IRE and the −50bp/+3bp segment eliminated the response to Foxp3 (Figure 4F).
The disparate effect of Foxp3 on class I promoter activity in Jurkat T cells and HeLa epithelial tumor cells led to the question of whether the HeLa response was anomalous. To address this, the effect of Foxp3 on class I transcription was assessed in a breast cancer cell line, MCF-7. As shown in Figure 5A (left panel), Foxp3 represses class I promoter activity in MCF-7 cells and to a similar degree to that observed in HeLa cells.
Figure 5. Foxp3 and Runx1 independently regulate class I promoter activity in Hela and MCF-7 cells.
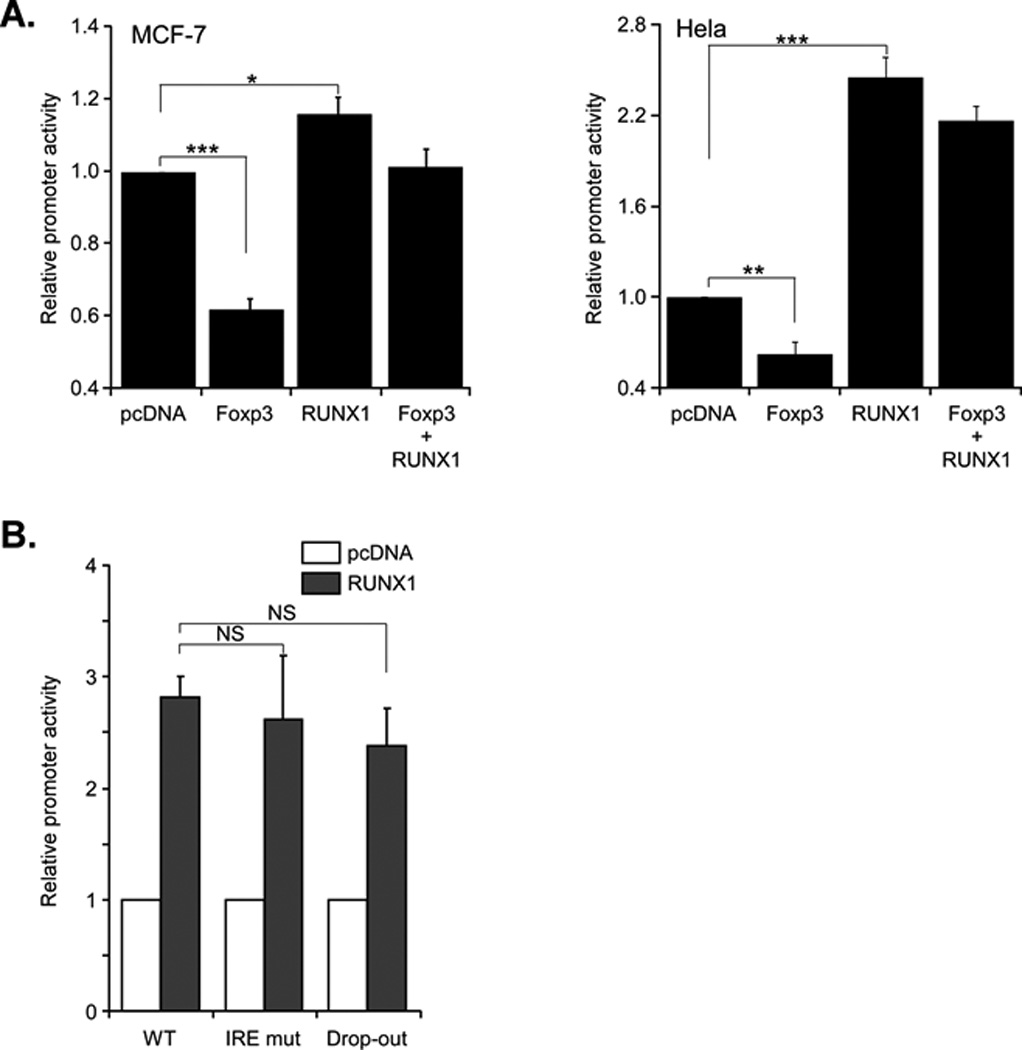
A. MCF-7 cells (left panel) and Hela cells (right panel) were cotransfected with WT class I promoter (2.0 µg) and Foxp3 (1.0 µg) or Runx1 (1.0 µg) alone or Foxp3 and Runx1 together. The results are the average ± SEM of 4 separate experiments for MCF-7 cells. ***p<0.001; *p<0.05, and four experiment for Hela cells. ***p<0.001; **p<0.01.
B. Hela cells were cotransfected with Runx1 (1.0 µg) or control vector and either WT class I promoter (2.0 µg) or IREmutant (2.0 µg) or drop-out (2.0 µg). The results are the average ± SEM of 2 separate experiments.
Taken together, these findings demonstrate the effect of Foxp3 on class I promoter activity is cell type dependent: Foxp3 activates the promoter in T cells but represses it in epithelial HeLa and MCF-7 cells. However, the response elements targeted by Foxp3 are common to both cell types, mapping to the IRE and a region between −50bp and +3 bp.
Runx1 and Foxp3 independently regulate class I promoter activity in HeLa and MCF-7 cells
Depending on its interactions with other transcription factors, Foxp3 functions as either an activator or repressor of different target gene promoters (22, 23). Therefore, we speculated that Foxp3 enhancement of class I promoter activity in T cells, but not in non-T cells, resulted from a T cell-specific transcription factor that modulated the effect of Foxp3 on the class I promoter. Aside from Foxp3, the major transcription factor expressed in Jurkat and Treg cells, but not in HeLa or MCF-7 cells, is Runx1. Runx1, in complex with CBFβ and LEF1, forms the TCE that activates transcription of the TCR genes. Runx1 also interacts with Foxp3 (24). As we have shown previously, introduction of Runx1 into HeLa cells strongly enhance MHC class I promoter activity (12). Therefore, we asked whether in the presence of Runx1, Foxp3 would enhance class I promoter activity in HeLa and MCF-7 cells, neither of which expresses Runx1 constitutively (data not shown). As expected, cotransfection of the class I promoter with only Runx1 into either MCF-7 or HeLa cells increased class I promoter activity, whereas cotransfection of Foxp3 alone repressed it (Fig 5A,B). Importantly, co-transfection of the class I promoter with both Foxp3 and Runx1 did not activate class I promoter activity above that observed by Runx1 alone. Rather, the net promoter activity was intermediate to each alone. Therefore, Runx1 expression alone is not sufficient to generate Foxp3 activation of class I expression.
If Foxp3 and Runx1 are independently regulating class I promoter activity, then their target DNA sequence elements might also be distinct. Since Foxp3 targets the class I IRE and the −50bp to +3bp sequence, we assessed the ability of Runx1 to activate these two promoter constructs. As shown in Fig. 5B, Runx1 efficiently activated both promoters, demonstrating that Runx1 targets a DNA sequence element distinct from that targeted by Foxp3.
Taken together, these results indicate that the differential expression of Runx1 in T lymphocytes and epithelial cells does not determine the effect of Foxp3 on class I promoter activity. Rather, Foxp3 acts independently of Runx1, suggesting that it does not require Runx1/TCE for the enhancement of class I promoter activity observed in T cells.
The DNA-binding forkhead domain of Foxp3 is required for its effect on class I promoter activity in Jurkat and HeLa cells
Because Foxp3 has multiple structural domains that allow it to interact with both DNA and a variety of different transcription factors, our next questions were 1) which Foxp3 domain(s) is required for class I regulation and 2) is the same domain required in Jurkat and HeLa cells? To address these questions, we examined the effects of two different Foxp3 deletion mutants on class I promoter activity in the Jurkat and HeLa cell lines. Foxp3-ΔFKH is a C-terminal truncation of the DNA forkhead binding domain (FKH) and Foxp3-ΔN is an N-terminal deletion of the proline rich domain. Both constructs contain the central zinc finger (ZF) and leucine zipper (LZ) dimerization domains (14, 20). In both Jurkat and HeLa cells, the Foxp3-ΔN was as effective as the Foxp3-WT in modulating class I promoter activity. In contrast, Foxp3-ΔFKH was markedly impaired in its ability to affect class I promoter activity in either cell type, although the three Foxp3 constructs were expressed comparable levels of protein (Figure 6, lower panel). Thus, class I promoter activation by Foxp3 depends on the Foxp3 DNA binding FKH domain. Interestingly, in HeLa cells, expression of Foxp3-ΔFKH not only reversed the effect but actually increased class I promoter activity. Whether the Foxp3-ΔFKH is acting as a dominant negative of an endogenous repressor remains to be examined. We conclude that the forkhead domain is necessary for Foxp3 effects in both Jurkat and HeLa cells and that the ZF, LZ and FKH domains are sufficient for activity.
Figure 6. Foxp3 modulation of class I promoter activity depends on its DNA binding domain.
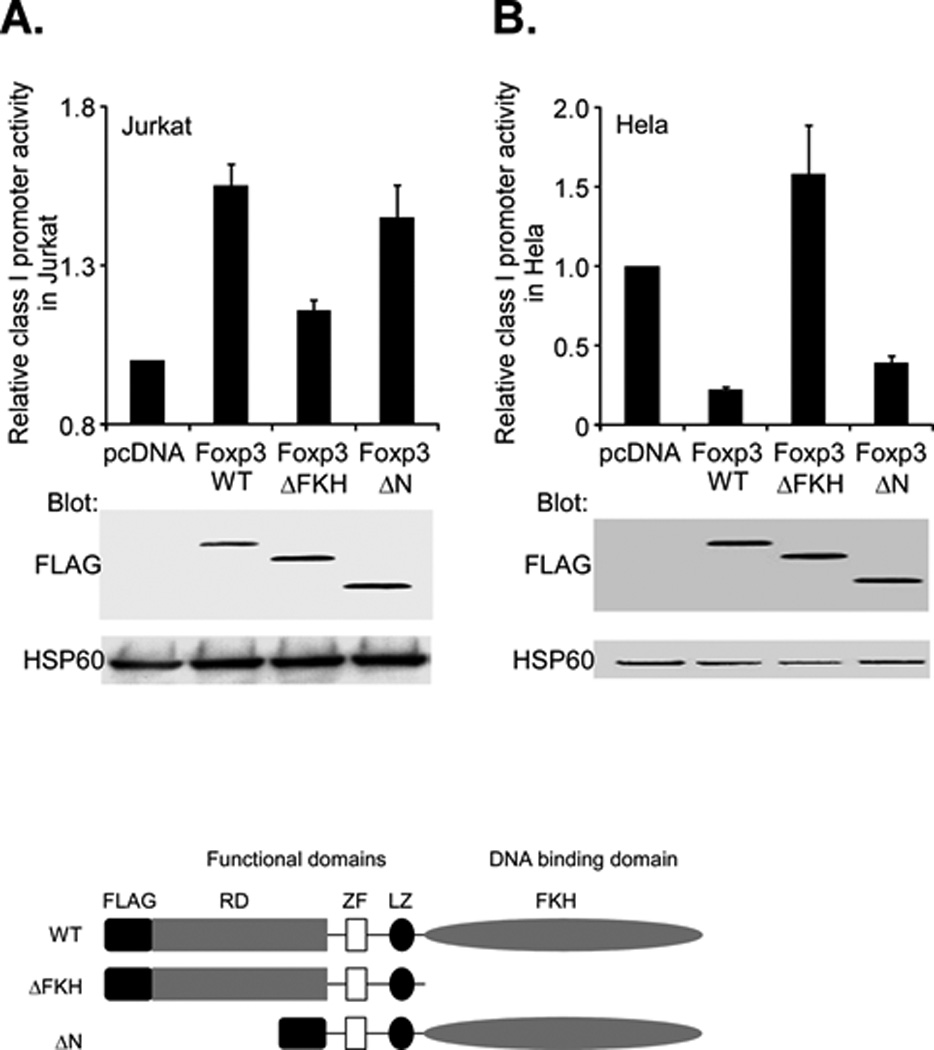
A. Jurkat T cells were cotransfected with the class I WT promoter (1.0µg) and either 2.0 µg of Foxp3 full length (Foxp3-WT), a C-terminal truncation of FKH domain (ΔFKH) or an N-terminal deletion of the proline rich domain (ΔN). Relative promoter activity, as assessed by luciferase activity (upper histogram) was determined. The activity of the ΔFKH construct was significantly less (p<4×10-5) than WT. The activity of the ΔN construct was not significantly different than WT (p=0.13) Expression of Foxp3 constructs was measured by western blotting with an anti-Flag antibody (middle) and compared with the internal control, HSP60 (bottom). Schematic diagrams of the wild type (WT) Foxp3 protein and the derivative truncations are shown below.
B. Hela cells were cotransfected with the class I WT promoter (2.0µg) and either 1.0 µg of Foxp3 full length (Foxp3-WT), a C-terminal truncation of FKH domain (ΔFKH) or an N-terminal deletion of the proline rich domain (ΔN). Relative promoter activity, as assessed by luciferase activity (upper histogram) was determined. Expression of Foxp3 constructs was measured by western blotting with an anti-Flag antibody (middle) and compared with the internal control, HSP60 (bottom).
Class I expression contributes to optimal Treg function
The finding that Foxp3 modulates class I promoter activity raises the question of whether there is a functional consequence to class I overexpression on Tregs. Since Foxp3 is essential for Treg generation, it is not possible to test directly the effect of depleting Foxp3 on class I expression or Treg function. Therefore, to address this question, we examined the effect of the complete elimination of class I on Treg development and function. Treg development does not require MHC class I expression since β2m-deficient mice that do not express surface class I develop normal numbers of CD4+CD25+ Tregs in both lymph node and thymus (Figure 7A). Consequently, we determined the impact of class I-deficiency on various aspects of Treg function. We first assessed the ability of class I deficient Tregs to suppress in vitro proliferative responses of CD4+CD25− B6 T cells (Tconv) stimulated by anti-CD3 mAb and antigen presenting cells (APC). B6 Tconv cells were stimulated with anti-TCR and APCs in the presence of increasing numbers of Tregs from either wild type B6 or β2m-deficient mice. Interestingly, β2m-deficient Tregs were detectably less efficient than B6 Tregs in that greater numbers of β2m-deficient Tregs than B6 Tregs were required to achieve the same level of suppression. The same was true whether the Tconv responders were from B6 or β2m-deficient mice (Fig. 7B, C; Supplementary Figure 3). Although these effects were modest, they were reproducible over multiple experiments.These results indicate that class I expression contributes to efficient Treg cell suppressive function in vitro.
Figure 7. Class I deficiency reduces Treg function in vitro and in vivo.
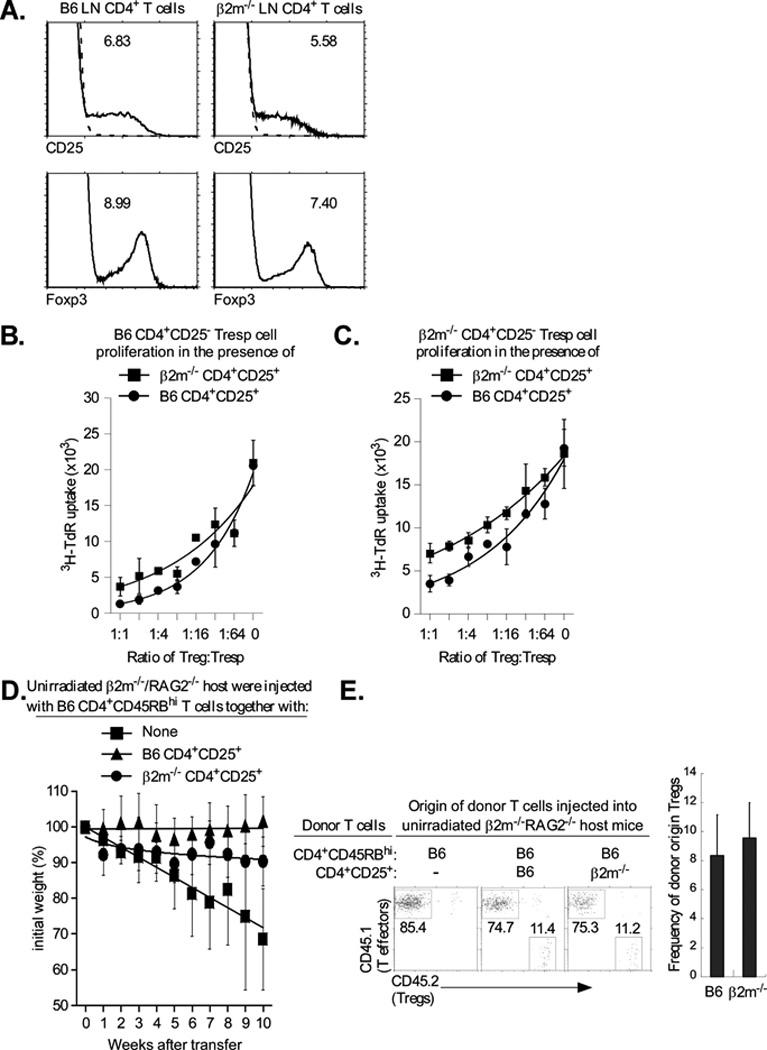
A. MHC class I is not required to generate Tregs. Lymph node T cells were stained for CD25 and Foxp3, as described in Material and Methods. Top panels display CD25 surface staining of gated CD4+ LNT cells and the numbers indicate the percentage of CD4+ T cells that are CD4+CD25+ (dashed lines represent negative control staining). Bottom panels display intracellular Foxp3 staining of gated CD4+ LNT cells and the numbers indicate the percentage of CD4+ T cells that are Foxp3+. Data are representative of three independent experiments.
B., C. MHC class I expression is required for efficient Treg function. CD4+CD25− LN Tconv cells from normal B6 mice (B.) or β2m−/− mice (C.) were cultured with purified CD4+CD25+ Tregs of mice from the two different origins and stimulated to proliferate by anti-CD3 (1µg/ml) and APC. Proliferation was measured by 3H-thymidine incorporation and mean cpm ± SD of triplicate wells are shown. The proliferation levels of B6 and β2m−/− Tregs, alone, were 108±23 and 110±7.2. Data are representative of 3 independent experiments and graphed with best fit curves. The IC50 for the β2m−/− and B6 Tregs was 1:6 and 1:21, respectively with β2m−/− responders.
D. Five week old β2m−/−/RAG-2−/− mice were injected either with 4×105 CD4+CD45RBhigh (CD45.1+) T cells alone, or together with 4×105 purified CD4+CD25+ (CD45.2+) Tregs from either B6 or β2m−/− mice. Tregs of B6 origin were used as a positive control. Percentage change from initial body weight of the recipients was monitored over time. β 2m−/−/RAG-2−/− mice were used as host instead of RAG-2−/− mice which rejected the transferred β 2m−/− Tregs, possibly by NK-mediated killing of class I deficient T cells (data not shown). Each data point represents between 4 and 5 β 2m−/−/RAG-2−/− recipient mice. Data are graphed with best fit curves.
E. β2m−/−/RAG-2−/− mice were injected either with 4×105 CD4+CD45RBhigh (CD45.1+) T cells alone, or together with 4×105 purified CD4+CD25+ (CD45.2+) Tregs from either B6 or β 2m−/− mice. Five weeks later, mice were sacrificed and their mesenteric LNs were stained for the relative proportions of CD4+CD45RBhigh effectors and CD4+CD25+ Tregs. Four or five β2m−/−/RAG-2−/− recipient mice per group were used.
To determine if class I expression on Tregs similarly contributes to Treg suppression in vivo, we compared the ability of Tregs from B6 and β2m-deficient mice to suppress the induction of inflammatory bowel disease (IBD) in vivo. Transfer of B6 CD4+CD45RBhi T-effector cells into unirradiated β2m−/−/RAG-2−/− mice induced severe diarrhea that resulted in weight loss and death (Fig. 7D). However, IBD was suppressed and mice maintained their weight when Tregs of B6 were co-transferred with the effector T cells. In sharp contrast, class I-deficient Tregs from β2m−/− mice did not provide any protection during the first 5 weeks post-transfer as indicated by progressive weight loss. This weight loss occurred even though both Treg populations were equally efficient in engrafting and populating the mesenteric lymph nodes of host mice (Fig. 7E). Interestingly, after five weeks, host mice transferred with class I-deficient Tregs gradually regained weight and showed reduced signs of the IBD. Thus, these in vitro and in vivo results reveal that MHC class I expression contributes to optimal Treg suppressor function.
To further characterize the underlying defects in class I-deficient Tregs, we focused on the well described Treg cell signature genes, Ctla4, Foxp3, IL-10 and TGFβ, which are differentially expressed between Treg cells and conventional CD4+ T cells. There were no significant differences in the expression of Foxp3, Ctla4 or TGFβ between class I-deficient and wild type Tregs (Fig. 8). (Note that relative to B6, the class I-deficient Tconv did express reduced levels of TGFβ. In contrast, we found that class I deficient Tregs expressed significantly reduced IL-10 mRNA levels than wild type, consistent with the finding that class I on Tregs is necessary to upregulate IL-10 expression (25).
Figure 8. MHC class I expression enhances IL-10 but not CTLA-4 and TGF-β.
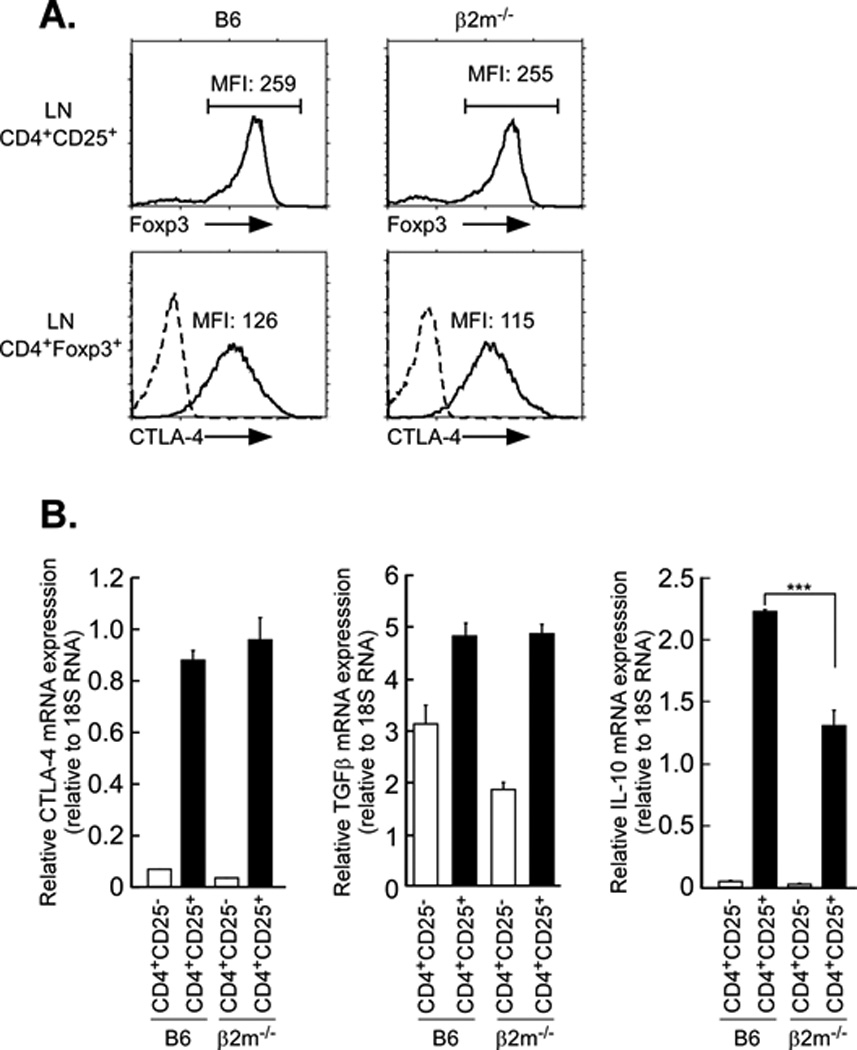
A. MHC class I deficiency does not affect Foxp3 and CTLA-4 expression in Tregs. Lymph node T cells were first stained for surface CD25, fixed and then stained for Foxp3 and CTLA-4. Top panels display Foxp3 intracellular staining of gated CD4+CD25+ LNT cells and the numbers indicate the MFI of Foxp3. Bottom panels display intracellular CTLA-4 staining of gated CD4+Foxp3+ LNT cells and the numbers indicate the MFI of CTLA-4. Dashed lines represent negative control staining. Data are representative of three independent experiments.
IL-10 RNA is reduced in β2m−/− CD4+CD25+ T cells relative to that in B6 CD4+CD25+ T cells. Total RNA was isolated from purified CD4+CD25− Tconv and CD4+CD25+ Tregs and subjected to quantitative RT-PCR for CTLA-4, TGF-β and IL-10. 18S rRNA was used as an internal control. Mean ± SEM of triplicate samples in three experiments. ***p<0.001.
Discussion
Foxp3, a member of the winged helix/forkhead family of transcription factors, is a master regulator of Treg development and function and is also induced in a variety of cancer cells. Consistent with Treg’s role as a suppressor of immune responses, most of the genes regulated by Foxp3 are in the TCR signaling pathway and are repressed upon Treg stimulation; the small proportion that are activated are involved in immune suppression (26). In the present study, we provide evidence that Foxp3 also regulates MHC class I transcription in vivo and its effect is cell-type dependent. In Treg cells, Foxp3 enhances expression of MHC class I genes. In contrast, in epithelial tumor cells, Foxp3 represses MHC class I expression. Whereas Foxp3 has been known to act either as an activator or repressor of different genes, this is the first example of it differentially affecting a single gene in a tissue-specific fashion. Importantly, the enhanced class I expression mediated by Foxp3 contributes to the IL10 content and suppressor function of Tregs.
Regulation of MHC class I gene expression is tissue specific and mediated by cell-type specific factors that interact with DNA sequence elements in the MHC class I gene promoter. For example, a B lymphocyte-specific enhanceosome consisting of the coactivator CIITA and DNA bound transcription factors RFX, CREB/ATF, and NF-Y leads to high cell surface class I and II expression in B lymphocytes (8–11). In conventional T cells, the high levels of class I transcription are regulated by the T cell enhanceosome, consisting of RUNX1, CBFβ and LEF1(12). In Tregs, Foxp3 further enhances class I expression above that in conventional T cells. This enhancement is observed in T cells both in vivo and in vitro. In contrast, in epithelial tumor cells, which do not express the T cell enhanceosome, Foxp3 represses class I transcription, presumably as a result of interactions with cell-type specific factors. Foxp3 is known to interact with a number of transcriptional activators such as AP1, NFκB, NFAT, RORγt, RORα and Runx1, as well as chromatin modifying enzymes such at the acetylase Tip60 and the deacetylase HDAC7 (27). We speculate that the different effects of Foxp3 on class I transcription reflect its interactions with different co-factors present in the different cell types.
Although the effect of Foxp3 binding to the class I promoter differs between T cells and epithelial tumor cells, two common DNA sequence elements are targeted. In both T cells and epithelial tumor cells, two distinct elements contribute to Foxp3 regulation. Interestingly, one of the elements resides within the class I interferon response element, which is the binding site for IRFs (28). The co-localization of Foxp3 and IRF1 at the IRE suggests that they may synergize to activate transcription. The other element resides within the core promoter of the class I gene, between −50 and +1 bp, the region where transcription initiates. Previous studies also mapped the Runx target site to this segment. Nevertheless, Runx and Foxp3 seem to act independently on their regulation of MHC class I transcription. Both elements contributed to Foxp3 activity in both cell types, their relative importance differed. In HeLa cells, repression of class I transcription by Foxp3 was mediated primarily by the response element within the core promoter. In contrast, in Jurkat T cells, the IRE and the core promoter both contributed equally to the increased transcription. The importance of the core promoter elements in Foxp3-mediated activation was demonstrated by deleting the core promoter from a class I transgene. The deletion did not abrogate its expression (29), but did eliminate Foxp3-dependent overexpression in Tregs. Thus, Foxp3 targets two major elements in the class I promoter, although the effect on transcription is cell-type specific.
In addition to these two major Foxp3 response elements, a series of additional response elements, spanning the segment from −416 to −171 bp, were active in HeLa cells. In particular, an element(s) between −416 and −313 bp enhanced Foxp3 mediated repression. Contained within this region is an AP-1 binding site. Since that site negatively regulates class I transcription in HeLa cells, it is unlikely it mitigates the effect of Foxp3 (30, 31). The finding that the IRE/dropout mutation, which still contains all of the upstream sequences is not repressed by Foxp3 suggests that any upstream regulatory elements are only active in the context of the IRE and core promoter.
Foxp3 activity in both Jurkat T cells and Hela epithelial cells depends on its forkhead (FKH) DNA binding domain. A mutant deleted of the FKH domain loses its ability to modulate class I transcription. In contrast, deletion of the N-terminal activation domain (ΔN) does not abrogate activity. The ΔN protein retains the zinc finger and leucine zipper domains of the wild type Foxp3, which would maintain interactions with a variety of co-factors and thus function. We propose that Foxp3 modulates class I transcription through its direct binding to the forkhead consensus sequence within the IRE and through its interactions with cell-type specific co-factors, resulting in either the activation or repression of class I transcription.
Class I molecules are receptors for intracellular derived peptides and provide immune surveillance by presenting them on the cell surface. Although class I molecules are ubiquitously expressed, their level of expression is tightly regulated in every tissue. Aberrant expression is associated with autoimmunity; lack of expression is associated with cancer. Accordingly, the expression of class I in Tregs had a functional correlate: both in vitro and in vivo, the absence of class I resulted in impaired Treg function. Thus, class I expression contributes to optimal Treg function. The mechanism(s) by which class I affects Treg function remain to be delineated. It is known that class I interaction with CD8 is required for optimal Treg activation and that Treg-specific inactivation of IL10 results in spontaneous colitis (25, 32). Thus, it is possible to speculate that class I deficiency results in impaired signaling, reduced IL10 content and consequently inefficient Treg function. Recent studies that class I molecules signal in NK cells and macrophages support the model that class I molecules signal in Tregs (33, 34).
The repression of class I expression in both Hela epithelial cells derived from an ovarian cancer and in MCF7 breast cancer cells is consistent with the down-regulation of class I commonly observed in tumors that allows them to escape immune surveillance. Indeed, Foxp3 expression has been detected in a variety of human and mouse tumors, including breast, ovarian and prostate (35–38). For example, single cell analysis of circulating tumor cells from prostate cancer patients (35) detected Foxp3 expression in the large majority of cases. Foxp3 was also observed in epithelial cells in breast DCIS and invasive carcinoma (39). The present findings that Foxp3 represses class I expression in epithelial tumor cells lead to the speculation that this is a mechanism whereby the tumor evades immune surveillance, enabling tumor progression.
Supplementary Material
Acknowledgements
The authors thank Drs. Richard Hodes and Hyun Park for their critical reading of the manuscript. We also thank the members of the D. Singer lab for helpful discussions during the course of this work. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Mozes E, Kohn LD, Hakim F, Singer DS. Resistance of MHC class I-deficient mice to experimental systemic lupus erythematosus. Science. 1993;261:91–93. doi: 10.1126/science.8316860. [DOI] [PubMed] [Google Scholar]
- 2.Mozes E, Lovchik J, Zinger H, Singer DS. MHC class I expression regulates susceptibility to spontaneous autoimmune disease in (NZBxNZW)F1 mice. Lupus. 2005;14:308–314. doi: 10.1191/0961203305lu2079oa. [DOI] [PubMed] [Google Scholar]
- 3.Singer DS, Mozes E, Kirshner S, Kohn LD. Role of MHC class I molecules in autoimmune disease. Critical reviews in immunology. 1997;17:463–468. [PubMed] [Google Scholar]
- 4.Singer DS, Zinger H, Kohn LD, Mozes E. Differing MHC class I requirements for induction and propagation of experimental systemic lupus erythematosus. European journal of immunology. 1999;29:2259–2268. doi: 10.1002/(SICI)1521-4141(199907)29:07<2259::AID-IMMU2259>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Girdlestone J. Transcriptional regulation of MHC class I genes. European journal of immunogenetics : official journal of the British Society for Histocompatibility and Immunogenetics. 1996;23:395–413. doi: 10.1111/j.1744-313x.1996.tb00015.x. [DOI] [PubMed] [Google Scholar]
- 6.Howcroft T, Singer D. Expression of nonclassical MHC class Ib genes: comparison of regulatory elements. Immunologic research. 2003;27:1–30. doi: 10.1385/IR:27:1:1. [DOI] [PubMed] [Google Scholar]
- 7.Singer DS, Ehrlich R. Identification of regulatory elements associated with a class I MHC gene. Current topics in microbiology and immunology. 1988;137:148–154. doi: 10.1007/978-3-642-50059-6_22. [DOI] [PubMed] [Google Scholar]
- 8.Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman L, Peterlin BM. MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators? Int Immunol. 2003;15:467–475. doi: 10.1093/intimm/dxg048. [DOI] [PubMed] [Google Scholar]
- 9.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Gene Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 10.Masternak K, Reith W. Promoter-specific functions of CIITA and the MHC class II enhanceosome in transcriptional activation. Embo J. 2002;21:1379–1388. doi: 10.1093/emboj/21.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J. CIITA regulates transcription onset viaSer5-phosphorylation of RNA pol II. Embo J. 2003;22:5125–5136. doi: 10.1093/emboj/cdg496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howcroft TK, Weissman JD, Gegonne A, Singer DS. A T lymphocyte-specific transcription complex containing RUNX1 activates MHC class I expression. J Immunol. 2005;174:2106–2115. doi: 10.4049/jimmunol.174.4.2106. [DOI] [PubMed] [Google Scholar]
- 13.Frels WI, Bluestone JA, Hodes RJ, Capecchi MR, Singer DS. Expression of a microinjected porcine class I major histocompatibility complex gene in transgenic mice. Science. 1985;228:577–580. doi: 10.1126/science.3885396. [DOI] [PubMed] [Google Scholar]
- 14.Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, Guinter T, McCaughtry T, Etzensperger R, Feigenbaum L, Singer DS, Singer A. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013;38:1116–1128. doi: 10.1016/j.immuni.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer DS, Camerini-Otero RD, Satz ML, Osborne B, Sachs D, Rudikoff S. Characterization of a porcine genomic clone encoding a major histocompatibility antigen: expression in mouse L cells. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:1403–1407. doi: 10.1073/pnas.79.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee N, Iyer SS, Mu J, Weissman JD, Ohali A, Howcroft TK, Lewis BA, Singer DS. Three novel downstream promoter elements regulate MHC class I promoter activity in mammalian cells. PloS one. 2010;5:e15278. doi: 10.1371/journal.pone.0015278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, Li MO, Leslie C, Stamatoyannopoulos JA, Rudensky AY. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh KP, Sundrud MS, Rao A. Domain requirements and sequence specificity of DNA binding for the forkhead transcription factor FOXP3. PloS one. 2009;4:e8109. doi: 10.1371/journal.pone.0008109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, Roncador G, Banham AH, Riley JL, Wang Q, Shen Y, Saouaf SJ, Greene MI. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. 2007;19:825–835. doi: 10.1093/intimm/dxm043. [DOI] [PubMed] [Google Scholar]
- 22.Hu H, Djuretic I, Sundrud MS, Rao A. Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends in immunology. 2007;28:329–332. doi: 10.1016/j.it.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 24.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 25.Joetham A, Takeda K, Miyahara N, Matsubara S, Ohnishi H, Koya T, Dakhama A, Gelfand EW. Activation of naturally occurring lung CD4(+)CD25(+) regulatory T cells requires CD8 and MHC I interaction. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15057–15062. doi: 10.1073/pnas.0706765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, Young RA. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Z, Song X, Li B, Greene MI. FOXP3 and its partners: structural and biochemical insights into the regulation of FOXP3 activity. Immunologic research. 2008;42:19–28. doi: 10.1007/s12026-008-8029-x. [DOI] [PubMed] [Google Scholar]
- 28.Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. The Journal of biological chemistry. 2007;282:20065–20069. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- 29.Barbash ZS, Weissman JD, Campbell JA, Jr., Mu J, Singer DS. MHC class I core promoter elements are not essential for transcription in vivo. Molecular and cellular biology. 2013 doi: 10.1128/MCB.00553-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howcroft TK, Richardson JC, Singer DS. MHC class I gene expression is negatively regulated by the proto-oncogene, c-jun. Embo J. 1993;12:3163–3169. doi: 10.1002/j.1460-2075.1993.tb05985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SM, Gao B, Fang D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood. 2008;111:3599–3606. doi: 10.1182/blood-2007-09-115014. [DOI] [PubMed] [Google Scholar]
- 32.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr., Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Rubio G, Ferez X, Sanchez-Campillo M, Galvez J, Marti S, Verdu R, Hernandez-Caselles T, Garcia-Penarrubia P. Cross-linking of MHC class I molecules on human NK cells inhibits NK cell function, segregates MHC I from the NK cell synapse, and induces intracellular phosphotyrosines. Journal of leukocyte biology. 2004;76:116–124. doi: 10.1189/jlb.1103597. [DOI] [PubMed] [Google Scholar]
- 34.Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, Zhang X, Li W, Cao X. Constitutive MHC class I molecules negatively regulate TLR-triggered inflammatory responses via the Fps-SHP-2 pathway. Nature immunology. 2012;13:551–559. doi: 10.1038/ni.2283. [DOI] [PubMed] [Google Scholar]
- 35.Chen CL, Mahalingam D, Osmulski P, Jadhav RR, Wang CM, Leach RJ, Chang TC, Weitman SD, Kumar AP, Sun L, Gaczynska ME, Thompson IM, Huang TH. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. The Prostate. 2013;73:813–826. doi: 10.1002/pros.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W, Wang L, Katoh H, Liu R, Zheng P, Liu Y. Identification of a tumor suppressor relay between the FOXP3 and the Hippo pathways in breast and prostate cancers. Cancer research. 2011;71:2162–2171. doi: 10.1158/0008-5472.CAN-10-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, McNally B, Lin L, Zhou P, Zuo T, Cooney KA, Liu Y, Zheng P. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer cell. 2009;16:336–346. doi: 10.1016/j.ccr.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, Liu Y, Wang Y, Liu X, Chan MW, Liu JQ, Love R, Liu CG, Godfrey V, Shen R, Huang TH, Yang T, Park BK, Wang CY, Zheng P. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129:1275–1286. doi: 10.1016/j.cell.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lal A, Chan L, Devries S, Chin K, Scott GK, Benz CC, Chen YY, Waldman FM, Hwang ES. FOXP3-positive regulatory T lymphocytes and epithelial FOXP3 expression in synchronous normal, ductal carcinoma in situ, and invasive cancer of the breast. Breast cancer research and treatment. 2013;139:381–390. doi: 10.1007/s10549-013-2556-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.