

Abstract
The 231-residue capsid (CA) protein of HIV-1 spontaneously self-assembles into tubes with a hexagonal lattice that is believed to mimic the surface lattice of conical capsid cores within intact virions. We report the results of solid state nuclear magnetic resonance (NMR) measurements on HIV-1 CA tubes that provide new information regarding changes in molecular structure that accompany CA self-assembly, local dynamics within CA tubes, and possible mechanisms for the generation of lattice curvature. This information is contained in site-specific assignments of signals in two- and three-dimensional solid state NMR spectra, conformation-dependent 15N and 13C NMR chemical shifts, detection of highly dynamic residues under solution NMR conditions, measurements of local variations in transverse spin relaxation rates of amide 1H nuclei, and quantitative measurements of site-specific 15N-15N dipole-dipole couplings. Our data show that most of the CA sequence is conformationally ordered and relatively rigid in tubular assemblies and that the structures of N-terminal and C-terminal domains (NTD and CTD) observed in solution are largely retained. However, specific segments, including the N-terminal β-hairpin, the cyclophilin A binding loop, the inter-domain linker, segments involved in intermolecular NTD-CTD interactions, and the C-terminal tail, have substantial static or dynamical disorder in tubular assemblies. Other segments, including the 310-helical segment in CTD, undergo clear conformational changes. Structural variations associated with curvature of the CA lattice appear to be localized in the inter-domain linker and intermolecular NTD-CTD interface, while structural variations within NTD hexamers, around local three-fold symmetry axes, and in CTD-CTD dimerization interfaces are less significant.
Keywords: human immunodeficiency virus, AIDS, solid state NMR, electron microscopy, hexagonal lattice
Introduction
The capsid core of the human immunodeficiency virus type 1 (HIV-1) encapsulates the viral genome and enables its transfer into host cells as part of the viral life cycle1,2. The mature form of the capsid has an outer shell formed by self-assembly of the capsid protein (CA). According to studies by X-ray crystallography3–8 and multidimensional solution nuclear magnetic resonance9–17 (NMR), the CA monomer is comprised of independently folding N-terminal (NTD, residues 1–143) and C-terminal (CTD, residues 148–231) domains, joined by a four-residue inter-domain linker. NTD and CTD are primarily α-helical, with seven α-helices (H1–H7) in NTD and four α-helices (H8–H11) in CTD. In solution under typical experimental conditions, both full-length CA and CTD dimerize through intermolecular interactions involving H9 and other sites5,6,12,18. Mutations that disrupt the CTD dimerization interface have facilitated solution NMR studies of CTD and full-length CA in monomeric form11,13,14,16, as well as the crystallization of hexamers7 and pentamers8 of cross-linked CA. NTD is compact and presents similar conformations in various solution and crystal structures3,4,7,8,15,16, except for flexibility in the cyclophilin A binding loop (residues 84–100) and in the extreme N-terminus, which forms a β-hairpin under certain conditions9,15,16. CTD may be more malleable, as indicated by conformational differences among structures determined from different constructs13, particularly at the dimer interface12. The C-terminal tail (residues 220–231) is not observed in crystal structures5,6 and is dynamically disordered in solution12,16. In solution, the two domains sample a variety of conformations relative to one another in both monomeric and dimeric states, facilitated by flexibility of the inter-domain linker16,17.
Capsid cores within mature HIV-1 are apparently closed shells with diameters of roughly 50–100 nm and shapes that are often conical, but are also variable19. At high ionic strength (as in experiments described below), full-length CA spontaneously self-assembles in vitro into tubes with diameters similar to those of capsid cores20–22. Cryo-electron microscopy (cryoEM) studies show that the walls of CA tubes have structures in which CA hexamers form a regular hexagonal lattice12,20,23, analogous to the structure of a carbon nanotube24 (with a CA hexamer substituting for the center of each six-carbon ring of the nanotube). CA hexamers are held together by intermolecular NTD-NTD interactions around local six-fold symmetry axes, primarily involving H1, H2, and H3, and by intermolecular NTD-CTD interactions, primarily involving the N-terminal end of H4 and H8. Hexamers are joined by the CTD-CTD dimerization interactions involving H9 at local two-fold symmetry axes. Stabilizing interactions involving intermolecular H10-H10 contacts at local three-fold symmetry axes have also been identified12,23. Closed CA assemblies, as observed in mature HIV-1 and under certain in vitro assembly conditions22,25, are proposed to result from inclusion of precisely twelve CA pentamers in this lattice26, which introduces points of high surface curvature in analogy to the curvature introduced by the twelve five-carbon rings in all-carbon fullerene molecules27.
Crystal structures of full-length CA hexamers and pentamers have been solved, using double cysteine substitutions in NTD to promote intermolecular cross-linking at NTD-NTD contacts and mutations in H9 to abrogate CTD-CTD dimerization interactions7,8. In crystal structures of CA hexamers, as also shown by cryoEM studies of CA tubes and planar CA assemblies12,20,23,25, NTDs of six CA proteins form a ring around the six-fold axis, mediated by interactions among H1, H2, and H3 segments, while CTDs form a surrounding belt and interact with NTDs of neighboring CA molecules within the hexamer.
Variable lattice curvature, a key feature of both native capsids and tubular CA assemblies, eliminates the perfect symmetry of the ideal hexagonal lattice, necessitating variations in the time-averaged structures and/or structural environments of individual CA molecules (in addition to the time-dependent structural fluctuations that are inevitably present at normal temperatures). Curvature has been attributed to various complementary mechanisms, including variations in relative orientations of NTD and CTD within individual molecules allowed by flexibility of the inter-domain linker7,12,28, variations in intermolecular NTD-CTD orientations within hexamers that are also facilitated by the flexible linker7,12,28, variations in contacts or crossing angles between H9 segments in the CTD-CTD dimerization interface12,23, and variations in interactions among H10 segments around local three-fold symmetry axes12,23. Detailed models for conical HIV-1 CA shells, based on the fullerene analogy described above, have been constructed, using information from cryoEM measurements on CA tubes20, high-resolution structural information from crystallography and solution NMR8, or a combination of structural information from crystallography, solution NMR, and cryoEM data with molecular dynamics simulations23. The latter models include structural variations in the dimerization interface, around local three-fold axes, and in the inter-domain linker.
A comprehensive atomic-level description of noncrystalline CA assemblies is necessary for a full understanding of important properties of native capsids, including their stability, variable curvature, and pleomorphism. While many aspects of CA structures have been firmly established by the ingenious studies cited above, certain important features remain unsettled due to inherent limitations of the experimental techniques that have been applied. Solid state NMR techniques can provide unique information about structural and dynamical properties of noncrystalline supramolecular assemblies, as demonstrated in recent studies of amyloid fibrils29,30, prion fibrils31,32, viruses33,34, membrane proteins35,36, and other supramolecular assemblies37–39. In systems such as HIV-1 CA assemblies, where the monomer molecular weight is relatively high but where basic aspects of the structure are already known from other techniques, solid state NMR measurements can provide high-resolution information about local structure and dynamics that complements information from other sources, thereby contributing to the comprehensive description. Initial results from solid state NMR measurements on HIV-1 CA assemblies have been reported in earlier publications from our laboratory40 and from Polenova and coworkers41,42.
Here we describe solid state NMR measurements on tubular HIV-1 capsid protein assemblies that address the following questions: (i) Can site-specific assignments of NMR signals from CA assemblies be obtained from solid state NMR data alone, independent of results for unassembled CA from solution NMR? (ii) How ordered and rigid is CA in tubular assemblies? (iii) Which regions of CA undergo structural changes upon tube formation, compared with CA structures observed in solution or crystalline states? (iv) Which segments exhibit structural variations that may be associated with lattice curvature within tubular CA assemblies?
Specifically, using improved sample preparation protocols that produce homogeneous assemblies resistant to degradation during lengthy solid state NMR experiments, we obtain high-quality two- and three-dimensional (2D and 3D) magic-angle spinning (MAS) NMR spectra, from which we obtain unambiguous site-specific 15N and 13C resonance assignments for the majority of the CA sequence. From the resonance assignments, we distinguish segments of the CA sequence that are structurally ordered in tubular assemblies from segments that are dynamically or statically disordered. Additional 2D solid state NMR experiments allow us to identify protein backbone sites that are primarily ordered but also exhibit partial mobility. Quantitative measurements of 15N-15N dipole-dipole couplings among backbone amide sites provide information about backbone conformations in the CA tubes. We use the assigned 15N and 13C chemical shifts and the measured 15N-15N couplings as restraints in structure calculations that reveal specific sites in CA that undergo significant conformational changes upon tube formation.
Results
Resonance assignments from 2D and 3D solid state NMR data
Full-length, wild type HIV-1 CA assemblies with tubular morphologies were prepared by spontaneous self-assembly following addition of NaCl (1 M) to CA solutions at pH 8.0. Prior to solid state NMR measurements, the NaCl concentration was reduced to 0.1 M NaCl and polyethylene glycol (PEG-8000) was added to a concentration of 17.5% (w/w). CA tubes (~15 mg) were then pelleted and loaded into 3.2 mm MAS NMR rotors. Samples were prepared with uniform 15N and 13C labeling of all residues (U-CA) and with uniform 15N labeling but partial 13C labeling, by expression on media containing either 1,3-13C2-glycerol or 2-13C-glycerol as the carbon source43,44 (1,3-CA or 2-CA). Details of sample preparation and measurement conditions are given in Materials and Methods.
Figures 1a and 1b show representative transmission electron microscopy (TEM) images of the CA tubes, with negative staining. The overall tube dimensions and appearance are similar to those in previous reports12,20,23,40. Apparent tube diameters vary from about 35 nm to about 60 nm (see Fig. S1 of supplemental information), indicating a mixture of helical symmetries12,20,23 for the cylindrical surface lattice. Figures 1c–1f show examples of 2D MAS NMR spectra. The uniform tubular morphology and good overall sample homogeneity give rise to well-resolved solid state NMR spectra, comparable to those reported for other well-ordered, non-crystalline biological solids29,31,32,37, indicating that the molecular structure within the tubes is largely uniform despite variations in tube diameters. In spectra acquired at 14.1 T (599.2 MHz 1H NMR frequency), 13C and 15N NMR linewidths (full width at half maximum) are typically 0.7 ppm and 1.3 ppm for U-CA tubes (Figs. 1c and 1d), and typically 0.5 ppm and 1.3 ppm for 2-CA and 1,3-CA tubes (Figs. 1e and 1f).
Figure 1.

Characterization of HIV-1 CA tubes by electron microscopy and solid state NMR. (a,b) TEM images of negatively stained CA tubes. (c) 2D NCACX solid state NMR spectrum of U-CA tubes, showing only the region that contains 15N-13Cα crosspeaks. (d,e,f) Aliphatic regions of 2D 13C-13C MAS NMR spectra of U-CA, 2-CA, and 1,3-CA tubes, respectively, with 4.0 ms RFDR mixing periods. All 2D spectra were obtained at 14.1 T with MAS at 12.00 kHz. Contour levels increase by successive factors of 1.5.
2D spectra in Figure 1 are better resolved than our earlier spectra of CA tubes40, primarily due to the reduced ionic strength and the addition of PEG-8000 to the solid state NMR samples. These changes reduced sample heating from radio-frequency (rf) pulses and stabilized the tubular morphologies at the high CA concentrations and high centrifugal forces of MAS NMR measurements.
Site-specific assignments of 15N and 13C chemical shifts are a prerequisite for the extraction of useful structural and dynamical information from solid state NMR data. In our previous study of CA tubes40, tentative assignments were based on comparisons with chemical shifts reported for monomeric CA and NTD in solution11,15. In a study of conical CA assemblies by Han et al.41, assignments were based in part (but not entirely) on comparisons with solution NMR data. In light of the conformational changes that accompany CA self-assembly and effects of intermolecular contacts on NMR chemical shifts, it is important to obtain chemical shift assignments from solid state NMR data alone. For this purpose, we obtained 3D NCACX and NCOCX spectra of U-CA tubes, 3D NCACX, NCOCX, and CONCA spectra of 1,3-CA tubes, and a 3D CONCA spectrum of 2-CA tubes. Examples of 2D planes from these 3D spectra are shown in Figure 2 (see also Fig. S2). 3D spectra of U-CA and 1,3-CA tubes were obtained both at 14.1 T and at 21.1 T (900.1 MHz 1H NMR frequency). Spectra obtained at 21.1 T exhibited typical 13C and 15N NMR linewidths of 0.5 ppm and 0.9 ppm, and were particularly useful for resolving crosspeaks involving carbonyl 13C sites and for detecting crosspeaks involving proline 15N sites.
Figure 2.

Representative 2D planes from 3D solid state NMR spectra. (a) 15N-13CO region of a f1/f3 plane from a 3D NCACX spectrum of U-CA tubes, recorded at 21.1 T, with f2 =57.1 ppm. (b) Part of the aliphatic region of a f1/f3 plane from a 3D NCACX spectrum of U-CA tubes, recorded at 14.1 T, with f2 = 57.1 ppm. (c,d) Parts of the aliphatic regions of f1/f3 planes from a 3D NCOCX spectrum of U-CA tubes, recorded at 21.1 T, with f2 = 175.2 ppm and f2 = 177.3 ppm. (e,f) Parts of the aliphatic regions of f1/f3 planes from a 3D CONCA spectrum of 2-CA tubes, recorded at 14.1 T, with f2 = 175.2 ppm and f2 = 177.3 ppm. (Here f1, f2, and f3 refer to NMR frequencies in the first, second, and third dimensions of a 3D spectrum.)
In order to obtain nearly complete backbone chemical shift assignments for CA tubes, we combined conventional, manual spectral analysis with the resonance assignment program MCASSIGN245 as described below in Material and Methods. The final stage of this procedure was the execution of 50 independent MCASSIGN2 runs, using NCACX and NCOCX signal tables (see Tables S1 and S2 of supplemental information) as input. Results of the final runs are summarized in Figure 3a, which plots assignment consistency, defined as the fraction of times that each residue is assigned with the same NCACX signal set (the most likely assignment) in the final runs. Only residues with an assignment consistency of 100% are considered to have unambiguous signal assignments. As shown in Figure 3a, 159 out of 231 residues have unambiguous assignments. An additional 6 residues with somewhat lower consistencies (as low as 80%) can be considered to have tentative assignments. In addition, sidechain signals were assigned for many residues during the manual analysis of the 2D and 3D spectra. Final 15N and 13C chemical shift assignments for CA tubes are given in Table S3.
Figure 3.

Summary of signal assignments from solid state NMR spectra of CA tubes. (a) Assignment consistency in the 50 final MCASSIGN2 runs, with 100% consistency indicating unambiguous assignment of signals to a given residue and 0% consistency indicating a complete absence of signal assignments to a given residue. (b) Secondary structure predictions from TALOS+ based on solid state NMR chemical shifts, where negative values indicate helical backbone conformations and positive values indicate extended conformations. (c) Secondary structure elements in CA structures from solution NMR and X-ray crystallography. Rectangular bars indicate the 11 α-helical segments. The small rounded bar indicates the 310-helix (residues 149–152) between NTD and CTD.
Differences between chemical shifts in CA tubes from solid state NMR and chemical shifts for CA dimers in solution17 are plotted in Figure S3. While many residues have similar 15N and 13C chemical shifts in solution and in tubular assemblies, differences greater than 0.5 ppm (for 13Cα) and 1.0 ppm (for 15N) occur at many sites, attributable to differences in intermolecular interactions, protein conformation, and motional amplitudes. These differences highlight the importance of deriving signal assignments for CA tubes solely from solid state NMR data.
Chemical shifts in Table S3 are in reasonable agreement with a partial set of 15N and 13C chemical shifts for CA tubes reported recently by Han et al.46 It should be noted that CA sequences examined by Han et al. and by us are not identical; Cys198 and Cys218 were also apparently reduced in the CA tubes examined by Han et al., but form the native intramolecular disulfide linkage within CTD in our CA tubes.
Secondary structure in CA tubes
Having obtained resonance assignments for the majority of the protein backbone, we analyzed the chemical shifts with the program TALOS+ to generate torsion angle and secondary structure predictions47, which are plotted in Figure 3b (see also Table S4). In this plot, negative values indicate helical conformations, while positive values indicate extended conformations. Values close to zero indicate either a non-helical, non-extended prediction or the absence of a reliable prediction. Residues with predicted helical conformations in CA tubes were found in all eleven α-helical segments that are observed in high-resolution solution NMR and crystal structures of full-length CA, CTD, and NTD. The positions of helices in the CA monomer in solution (PDB code 2LF4) are shown as bars in Figure 3c. Thus, in general, solid state NMR spectra of CA tubes indicate that the secondary structure of CA in these assemblies is similar to that found in soluble and crystalline states. Interestingly, residues 149–151, following the inter-domain linker, display strand-like chemical shifts consistent with an extended conformation, rather than the 310-helical conformation observed in the CA monomer in solution (PDB code 2LF4), crystalline CTD dimers (PDB code 1A43), and the cross-linked CA hexamer crystals (PDB code 3MGE).
Dynamics and disorder in CA tubes
Most residues in CA tubes display strong, sharp solid state NMR signals characteristic of well-ordered conformations with only small-amplitude motions (plus methyl group rotations and aromatic ring flips). We refer to these residues as “rigid”. Other residues fall into three categories: (i) “Disordered” residues whose signals are absent from the 3D spectra, due to either the presence of large-amplitude motions that attenuate 1H-15N, 1H-13C, 13C-13C, and/or 15N-13C dipole-dipole couplings (i.e., dynamic disorder) or the presence of local static disorder that produces large inhomogeneous broadening of the NMR lines; (ii) “Partially mobile” residues whose signals are present and assignable in 3D spectra, but which undergo motions on the sub-millisecond time scale with sufficient amplitudes to attenuate local 1H-1H dipole-dipole couplings. Partially mobile residues are identified by the behavior of their signals in 1H T2-filtered NCA spectra as described below; (iii) Residues to which signals are assigned with consistencies below 100%. These residues may contribute to the solid state NMR signals, and thus be either rigid or partially mobile, but their signals are insufficiently resolved or insufficiently unique to allow unambiguous assignment. Alternatively, these residues may be disordered.
Most disordered residues occur within or adjacent to segments that are expected to be dynamically or statically disordered, including the C-terminal tail (residues 220–231), the N-terminal segment (residues 7–13), which was found to form a β-hairpin in solution15,16 but is disordered in some crystal structures7,48, and the inter-domain linker (residues 141–145), which is highly dynamic in solution, allowing a wide range of relative orientations between the two domains16,17. The C-terminal tail is expected to be unconstrained at its C-terminal end, and is therefore likely to be dynamically disordered (see below). However, the linker may be either statically or dynamically disordered, as it is constrained at either end by the fixed positions of the NTD and CTD domains. Similarly, since residues 1–6 of the N-terminal segment are observed in our solid state NMR spectra, the rest of this segment (residues 7–13) is also constrained at either end. Absence of disorder in residues 1–6 may reflect the Pro1-Asp51 salt bridge that is observed in solution and crystal structures.7,15
An additional stretch with missing signals is residues 179–182, which are at the N-terminal end of H9, adjacent to residues whose sidechains are essential for dimerization (Trp184 and Met185)11,13. Interestingly, this region presents variable conformations in different CA constructs, depending on the arrangement of the dimer interface, and is missing from the CA hexamer crystal structure (PDB code 3MGE) but present in the hexamer structure derived from cryoEM of CA tubes (PDB code 3J4F). Signals from residues 172–175, at the C-terminal end of H8, are also missing, although these residues are included in both the crystal structure and the cryoEM-based structure.
When local molecular motions are sufficiently rapid and large in amplitude, dynamic disorder within an immobilized protein assembly can be distinguished from static disorder by recording 2D NMR spectra under conditions similar to those used in solution NMR measurements, i.e., by using scalar couplings (rather than dipole-dipole couplings) to drive spin polarization transfers and relatively weak rf fields for 1H decoupling. A 2D 13C-13C TOBSY49 spectrum of U-CA tubes (see Fig. S4) shows crosspeaks from dynamically disordered residues that can be assigned to Gly, Val, Pro, Lys, Ala, Arg, and Leu, all of which are attributable to the C-terminal tail. In addition, crosspeaks from Ile, Thr, Glu, Met, Ser, and Asp/Asn are observed. Ile signals could arise from Ile91 or Ile141. Thr signals most likely arise from Thr171, while Glu signals could arise from Glu175 or Glu180. Met signals could arise from Met10 or Met144. Ser signals are attributable to Ser146. (Note that all other Thr, Glu, and Met residues that lack unambiguous signal assignments are contained in helical segments, which are unlikely to contribute to a 2D 13C-13C TOBSY spectrum. All other Ser residues have unambiguous signal assignments.) These results indicate that at least part of the inter-domain linker (including Ser146, and possibly Ile141 and Met144) is dynamically disordered in CA tubes, as is part of the segment that connects H8 and H9 (including Thr171 and Glu175 or Glu180). Residues 7–13 may also contain dynamic disorder, but existing data are not definitive.
All Asp and Asn residues in CA tubes have unambiguous signal assignments. The observation of Asp/Asn signals in the 2D 13C-13C TOBSY spectrum therefore suggests dynamical heterogeneity, with certain Asp or Asn residues being sufficiently rigid in some CA molecules to contribute to spectra acquired under standard solid state NMR conditions, but also sufficiently dynamic in other CA molecules to contribute to spectra acquired under solution NMR conditions. Asn139 is a likely site of dynamical heterogeneity, if the length of the dynamically disordered part of the inter-helical linker varies among structurally inequivalent molecules in CA tubes (see below).
Figure 4 shows 1H T2-filtered NCA spectra of U-CA tubes. In these measurements, a 1H spin echo period τecho is inserted at the beginning of the rf pulse sequence, during which signals decay according to the strength of local 1H-1H dipole-dipole couplings. At τecho = 168 μs, crosspeak signal volumes from rigid residues are reduced to roughly 20–30% of their values at τecho = 0. In contrast, partially mobile residues retain more than 40% of their crosspeak signal volumes. Table S5 lists the signal reductions for residues whose signals were sufficiently well resolved in the NCA spectra for reliable crosspeak volume measurements. Partially mobile residues are located almost exclusively in non-α-helical segments, indicating that the interhelical segments have greater motional amplitudes. Specifically, partially mobile residues are identified in segments between H2 and H3, H4 and H5, H6 and H7, H8 and H9, and H10 and H11, as well as in the N-terminal segment before H1. In contrast, certain residues in segments between H5 and H6, H7 and H8, and H9 and H10 are apparently rigid, indicating that different interhelical segments have different degrees of mobility.
Figure 4.

Evidence for partial mobility at specific residues in HIV-1 CA tubes. (a) Reference 2D NCA spectrum, with zero 1H spin echo time. Crosspeaks are labeled with their residue-specific assignments. (b) 1H T2-filtered NCA spectrum, with a 168 μs 1H spin echo time. Crosspeaks are labeled with their amplitudes relative to the reference spectrum. Dark blue, light blue, green, and magenta labels corresponding to values of 15–30%, 31–40%, 41–55%, and 56–85%, respectively, with higher percentages corresponding to greater mobility.
The greatest mobility (i.e., smallest signal reduction) is observed for residues 88–94, in the cyclophilin A binding loop between H4 and H5, which is a highly flexible segment in solution9. The least mobility is observed in or proximal to H3, H5, H6, H7, and H8. For the most part, partially mobile residues are located on the solvent-exposed face of CA hexamers, based on comparisons with structural models from cryoEM (PDB code 3J34). Partially mobile residues between H10 and H11 (Gly206, Pro207, Gly208, and Thr210, with signal reductions = 41–65%) are not in the dimerization interface. Residues whose sidechains participate in the dimerization interface or which are adjacent to such residues (according to PDB code 2KOD), including Ile150, Ala177, Thr188, and Asn193, have signal reductions approximately equal to 35%.
Only one partially mobile residue is tentatively identified in a nominal α-helical segment, namely Thr171, at the C-terminal end of H8 (see Table S5). As noted above, signals from residues 172–175 are missing, and signals attributable to Thr171 are also observed in the 2D 13C-13C TOBSY spectrum (Fig. S4). These observations again point to dynamical heterogeneity, with molecule-to-molecule variations in the dynamical properties of Thr171, and to dynamical disorder in residues 172–175. Partial mobility of Ala177 also appears to be consistent with elevated dynamics in a segment that runs from the nominal C-terminal end of H8 to the nominal N-terminal end of H9.
Figure 5 summarizes information about site-specific structural order and dynamics from the data discussed above. In Figure 5a, residues within a CA monomer are color-coded according to the certainty of their contributions to 3D solid state NMR spectra. Gray residues are either disordered (i.e., have no signal assignments in the final MCASSIGN2 runs) or have assignment consistencies below 80%. In Figure 5c, residues within a CA monomer are color-coded according to their partial mobility. Gray residues do not have resolved signals in 1H T2-filtered NCA spectra, precluding measurements of partial mobility. CA hexamers in Figures 5b and 5d are colored as in Figures 5a and 5c, respectively.
Figure 5.

Summary of information concerning local order and dynamics. (a) Cartoon representation of a CA monomer (from PDB code 2LF4) with unambiguously assigned residues in brown and tentatively assigned residues in yellow. Residues in gray have assignment consistencies below 80%, including residues to which solid state NMR signals are never assigned. Helical segments of CA are labeled in red. According to solid state NMR data, the 310-helix, between helices 7 and 8, is not present in our CA tubes. (b) CA hexamer (from PDB code 3GV2) with the same coloring as in panel a, except that dark brown and light brown are used for alternate CA molecules. The hexamer is viewed from outside a CA tube (top) and from inside a CA tube (bottom). (c) CA monomer with residues colored according to percentages in Figure 4b, with dark blue, light blue, green, and magenta corresponding to values of 15–30%, 31–40%, 41–55%, and 56–85%, respectively. Residues whose crosspeak signals are insufficiently resolved in the 1H T2-filtered NCA spectra are gray. (d) CA hexamer with the same coloring as in panel c, except that gray and light brown are used for alternate CA molecules. The hexamer is viewed from outside a CA tube (top) and from inside a CA tube (bottom).
Quantitative measurements of backbone conformation
To obtain quantitative experimental restraints on the CA backbone conformation in tubular assemblies, we used the 15N-15N backbone recoupling (15N-BARE) technique of Hu et al.50 to probe sequential amide 15N-15N distances. In these measurements, residue-specific 15N magnetization decay curves under 15N-15N dipole-dipole couplings are detected through crosspeak volumes in a series of 2D NCACX spectra with increasing dipolar evolution periods (see Fig. S5). The decay curve for residue i is sensitive to the backbone torsion angles ψi and ψi−1, which determine distances from the amide 15N site of residue i to the amide 15N sites of residues i+1 and i−1. The decay curve is also somewhat sensitive to ϕi, which affects the angle between these internuclear displacement vectors. Figure 6 presents examples of 15N-BARE decay curves. 15N-BARE curves for other residues are shown in Figure S6.
Figure 6.
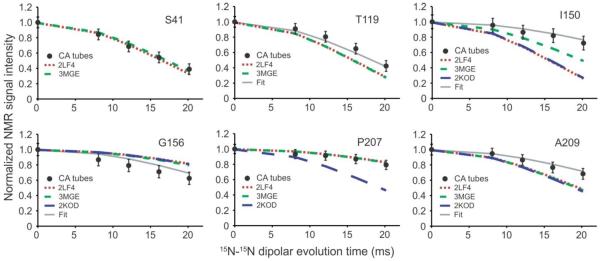
Quantitative restraints on backbone conformation in HIV-1 CA tubes. 15N-BARE signal decay curves for the indicated residues are shown as filled circles, normalized to the signal amplitude at zero evolution time and with error bars representing root-mean-squared uncertainty due to noise in the experimental solid state NMR spectra. These curves are measurements of sequential amide 15N-15N distances, which are conformation-dependent. Dashed and dotted lines are simulations based on a CA monomer structure from solution NMR (red dots, PDB code 2LF4), a crystal structure of cross-linked CA hexamers (short green dashes, PDB code 3MGE), and a solution NMR structure of CTD dimers (long blue dashes, PDB code 2KOD). Simulations based on the refined structure in Figure 7a and 7b are shown in solid gray lines (“Fit”). See Figure S5 for examples of 2D spectra from which the 15N-BARE curves are extracted and Figure S6 for additional 15N-BARE curves.
In α-helices, sequential 15N-15N distances are short (~2.7 Å), leading to relatively rapid decays of 15N-BARE signals. This is indeed observed for most residues in α-helical segments of CA (e.g., Ser41, Met66, Gly116, Thr186). Similar rapid dephasing is also exhibited by several residues in tight turns, where bending of the backbone brings sequential amide nitrogen atoms closer together (e.g., Ala105, Gly106, Thr107). On the other hand, several residues in well-ordered loop regions display slower dephasing due to their more extended backbone conformations, with 15N-15N distances up to ~3.8 Å (e.g., Gly46, Thr110, Asn121, Pro160).
At a more detailed level, the 15N-BARE data can be used to reveal changes in the local structure of CA in tubes compared with the structure in soluble and crystalline states, through comparisons between experimental decay curves and curves calculated from the backbone torsion angles in solution NMR and crystal structures. Thus, Figures 6 and S6 include calculated 15N-BARE curves for several high-resolution structures. Good agreement between experimental and calculated curves is typically found within α-helices, while some residues at the ends of α-helices (e.g., Thr119, Phe161) appear to adopt more extended conformations in CA tubes. A major discrepancy between experimental and calculated curves occurs at Ile150, located at the beginning of CTD, which participates in a 310-helix in certain solution NMR and crystal structures but apparently adopts an extended conformation in CA tubes. Absence of the 310-helix in CA tubes is also indicated by chemical shifts, as discussed above.
Structure refinement based on solid state NMR data
To visualize the extent to which the conformation of CA in tubular assemblies differs from that in soluble and crystalline forms, we used our solid state NMR data as restraints in structure calculations with the program Xplor-NIH51. Deviations from TALOS+ torsion angle predictions (see Table S4) and from 15N-BARE data were used as potential energy terms to drive the simulated annealing process in Xplor-NIH, as previously described50. Atomic coordinates for CA derived from the solution NMR monomer (PDB code 2LF4) and the crystalline hexamer (PDB code 3MGE) were used as initial structures in different sets of calculations. The refRMSD potential of Xplor-NIH was applied separately to NTD (residues 1–144) and CTD (residues 149–231) to maintain the two domains close to their initial structures, aside from conformational changes within each domain and changes in their relative orientation required to accomodate the solid state NMR data. Full details of these calculations are given in Materials and Methods.
As most of the solid state NMR restraints generally agree with the initial NTD and CTD structures, the initial and final structures from these Xplor-NIH calculations are very similar. Values of the backbone root-mean-squared deviations between initial and final structures are less than 0.7 Å for both NTD and CTD in all calculations. However, there are a number of adjustments that reflect local structural changes associated with CA assembly into tubes. Figure 7a shows a superposition of initial and final structures from calculations that started with the solution NMR structure, displayed with optimal alignment of the CTD regions. The 310-helix at the beginning of CTD in the solution NMR structure clearly adopts a more extended conformation, reflecting the solid state NMR data for CA tubes described above. The relative orientations of NTD and CTD are also very different in initial and final structures, but this difference is attributable to flexibility of the inter-domain linker in solution and to the absence of structural restraints for the linker from solid state NMR (since signals from residues 141–145 are not observed).
Figure 7.

Changes in published CA structures required to fit solid state NMR restraints on the CA structure within tubular assemblies. (a) Comparison of initial (red) and final (blue) structures, with optimal superposition of CTD segments, from a restrained molecular dynamics/simulated annealing calculation that used a CA monomer structure from solid state NMR (PDB code 2LF4) as the initial structure. The 310-helix near the NTD-CTD linker in the initial structure, indicated by the green arrow, changes to an extended conformation in order to accommodate solid state NMR chemical shift and 15N-BARE data. (b,c) Comparison of initial (orange) and final (blue) structures from a calculation that used a CA hexamer structure from X-ray crystallography (PDB code 3MGE) as the initial structure. Either NTD (helices 1–7) or CTD (helices 8–11) segments are optimally superposed. Green arrows indicate conformational changes between helices 3 and 4 (near the intermolecular NTD-CTD interface in a CA hexamer) and helices 10 and 11 (near the local three-fold axis in a CA lattice). Helical segments of CA are labeled in red. (d) CA hexamer (from PDB code 3GV2) with segments that exhibit conformational changes colored red. The hexamer is viewed from outside a CA tube (top) and from inside a CA tube (bottom). Segments in which conformational changes are not identified by our data are gray and light brown in alternate CA molecules.
Figures 7b and 7c show superpositions of initial and final NTD and CTD structures from calculations that started with the CA hexamer crystal structure. In this case, calculations were performed separately for each domain and residues disordered in the crystal structure were omitted. Again, most α-helices are unchanged because the original structure agrees well with the solid state NMR restraints. However, significant conformational changes are observed for residues 58–62, which comprise the C-terminal end of H3 and the loop between H3 and H4 (Fig. 7b). The root-mean-squared angular deviations in this segment between initial and final structures are 71° and 90° for ϕ and ψ angles, respectively. A different conformation in this region may be the result of the intermolecular NTD-NTD and NTD-CTD interfaces adapting to the tubular lattice, since residues 58–62 make contacts to both H2 and H8 of a neighboring molecule within a hexamer in the crystal structure.
Finally, significant changes in backbone torsion angles are observed for residues 206–210, in the loop between H10 and H11 (Fig. 7c). The root-mean-squared angular deviations in this segment between initial and final structures are 47° and 49° for ϕ and ψ angles, respectively. These changes are driven by 15N-BARE data for Gly208, Ala209, and Thr210, which do not agree with simulations that use the crystal structure coordinates (Figs. 6 and S6). It is worth noting that 15N-BARE data for Pro207 do agree well with simulations that use the crystal structure coordinates, but not with simulations that use CTD dimer coordinates from solution NMR (PDB code 3KOD). 15N-BARE data for Gly208 also disagree strongly with simulations that use the CTD dimer coordinates from solution NMR. Interestingly, residues 206–210 are near the local three-fold symmetry axes observed in cryoEM reconstructions of tubular CA assemblies12,23. Intermolecular interactions around three-fold symmetry axes are a property of the hexagonal lattice and hence are absent from both CA hexamer crystals and CTD dimers in solution. Therefore, it is plausible that the structure in this region differs between tubular CA assemblies and the crystalline and soluble states.
Ordered aromatic sidechains
Sidechains of certain aromatic residues are located in the cores of NTD and CTD and are therefore expected to be conformationally ordered, while others are partially exposed on the protein surface and may be more flexible. In particular, four of the five tryptophan residues in CA (residues 23, 80, 117, and 133) are buried in the hydrophobic pockets of NTD, but Trp184 points outward from H9 of CTD. Due to their characteristic chemical shifts, we were able to identify aromatic sidechains of certain tryptophan and tyrosine residues in solid state NMR spectra of CA tubes, and assign them to specific residues via their backbone signals. Figure 8a shows the aromatic-aliphatic region of a 2D 13C-13C DARR spectrum of 2-CA tubes, in which tryptophan and tyrosine spin systems are labeled. Interestingly, we observe all five tryptophan backbone and sidechain spin systems in solid state NMR spectra of CA tubes, showing that these residues adopt rigid, well-defined conformations. According to crystal and solution structures of CTD dimers5,6,12, Trp184 forms key intermolecular interactions across the CTD-CTD dimer interface, rather than being buried within CTD or exposed to the solvent. The observation of clear sidechain signals from Trp184 indicates that Trp184 establishes specific intermolecular contacts in CA tubes, as observed in structures of the CTD dimer (see Fig. 8b). From available data, we can not determine whether the dimer interface structure in CA tubes more closely resembles structures identified in solution or in crystalline states.
Figure 8.
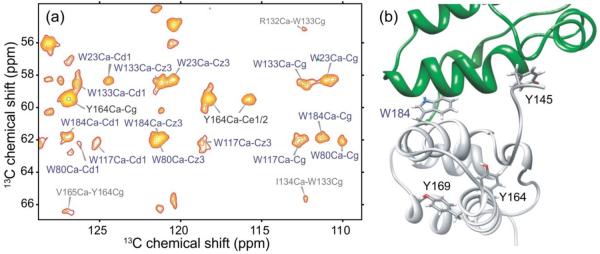
Conformationally ordered aromatic sidechains in HIV-1 CA tubes. (a) Aromatic-aliphatic region of 2D 13C-13C spectrum of 2-CA tubes, acquired at 14.1 T with a 500 ms DARR mixing period and MAS at 12.00 kHz. Crosspeaks arising from Trp and Tyr residues are labeled in blue and black, respectively. Additional peaks are labeled in gray. (b) CTD dimer structure (PDB code 2KOD), highlighting the positions of tryptophan and tyrosine residues in one of the subunits. Trp184 adopts a well-defined, relatively rigid conformation in CA tubes, as indicated by its strong, sharp crosspeak signals, while Tyr164 is the only tyrosine residue with observable sidechain crosspeaks.
In contrast, only one of the four tyrosine residues in CA, namely Tyr164, shows detectable sidechain signals in our solid state NMR spectra, although backbone signals for Tyr130, Tyr164, and Tyr169 (but not Tyr145) are assigned. The Tyr164 sidechain is buried in the hydrophobic core of CTD and is thus apparently rigid enough to generate strong signals. Tyr169, which is partially exposed to the solvent, and Tyr145, located near the inter-domain linker and also solvent-exposed, are likely to be more flexible and may adopt disordered or dynamic states in CA tubes. This is an interesting finding because Tyr145 is critical for capsid formation and HIV-1 infectivity, according to mutational studies12,52. In addition, in the solution structure of a CTD construct that includes this residue (PDB code 2KOD), Tyr145 participates in specific intermolecular interactions across the dimer interface and adopts an ordered conformation. The absence of Tyr145 sidechain signals from our 2D and 3D solid state NMR spectra may result from librational motions or ring flips on time scales in the 10−5−10−3 s range.
In solid state NMR studies of conical CA assemblies, Byeon et al. also found that signals from Tyr145 were absent from 2D spectra, although the backbone amide 15N signal was detected in 1D spectra42. From a variety of solid state NMR measurements, Byeon et al. concluded that Tyr145 underwent motions on the millisecond timescale, consistent with our observations for CA tubes.
Discussion
Solid state NMR signal assignments
Previous studies of HIV-1 CA assemblies by solid state NMR have relied wholly40 or partially41 on solution NMR data for site-specific assignment of solid state NMR signals. In contrast, we have used only solid state NMR data to obtain the assignments in Table S3. We find significant differences between 13C and 15N chemical shifts determined from solution NMR measurements on soluble constructs and chemical shifts in CA tubes (Fig. S3), highlighting the importance of using only solid state NMR data to make assignments for CA assemblies. Nonetheless, secondary structure predictions based on assignments from solid state NMR indicate that the α-helical segments of NTD and CTD in soluble and crystalline constructs are largely maintained in CA tubes.
From the standpoint of methodology, our approach to signal assignments for HIV-1 CA tubes demonstrates the utility of computational methods such as MCASSIGN245 as a means of identifying all assignments that are consistent with available solid state NMR data. By executing multiple independent MCASSIGN2 runs, we identify segments that have unambiguous signal assignments, segments that have tentative assignments, and segments that have no signal assignments (i.e., that definitely do not contribute to 3D solid state NMR spectra). This information is difficult to extract reliably from solid state NMR spectra with traditional manual assignment approaches, especially for relatively large proteins such as CA and especially when a significant fraction of the amino acid sequence does not contribute to the spectra.
Local disorder and dynamics
Segments that definitely do not contribute to our 3D solid state NMR spectra include residues 7–13, 96–97, 141–145, 172–175, 179–182, and 220–231. Absence of detectable solid state NMR signals can be due to dynamic disorder or static disorder. Residues within CA tubes that undergo rapid, large-amplitude motions can be observed in 2D spectra obtained under solution NMR conditions, e.g., in the 2D 13C-13C TOBSY spectrum in Figure S4. Most signals in this spectrum can be attributed to the C-terminal tail (residues 220–231). However, signals arising from Ile, Thr, Glu, Met, and Ser residues must come from other segments, most likely the inter-domain linker (residues 141–146) and the segment that connects H8 with H9 (residues 171–182). Thus, it appears that most missing signals are associated with localized dynamic disorder, rather than purely static disorder.
Residues that are partially mobile, revealed by the 1H T2-filtered NCA spectra (Fig. 4 and Table S5), are limited to inter-helical segments, especially the cyclophilin A binding loop between H4 and H5. Mobility of the cyclophilin A binding loop is supported by the absence of detectable solid state NMR signals from residues 96–97. Thus, the cyclophilin A binding loop is dynamic in CA tubes, but not so dynamic (or disordered) that signals from residues 84–95 are absent from solid state NMR spectra, as most of these residues have unambiguous signal assignments.
The N-terminal β-hairpin seen in solution NMR structures of NTD and monomeric CA (PDB codes 1GWP and 2LF4) contains both rigid (residues 1–6 and 14–16) and disordered (residues 7–13) segments in CA tubes. Solution NMR measurements indicate significant dynamic disorder in the β-hairpin9. Residues 4–9 are missing from the CA hexamer crystal structure (PDB code 3MGE). Residues 1–10 are missing from a NTD/cyclophilin A crystal structure (PDB code 1M9C). Residues 1–16 are present in the recent cryoEM-based model for CA tubes (PDB code 3J34), but have significantly different conformations in different CA molecules within this model. Thus, available data suggest either that a stable, hydrogen-bonded N-terminal β-hairpin does not exist in tubular CA assemblies or that the β-turn and flanking residues undergo large-amplitude motions.
Conformational changes that accompany CA self-assembly
Residues 141–145, at the C-terminal end of H7, are α-helical in solution NMR and crystal structures of NTD (PDB codes 1GWP, 1M9C, 2JPR), monomeric CA (PDB code 2LF4), and CA hexamers (PDB code 3MGE), as well as in the cryoEM-based model for CA tubes (PDB code 3J34). As discussed above, these residues are disordered and dynamic in CA tubes. The precise time-averaged conformation of residues 141–145 and the precise length of H7 may vary from molecule to molecule within CA tubes. The segment that follows, residues 149–153, contains a 310-helix in crystal structures7 and solution NMR structures12,16. This 310-helix is not present in CA tubes, as shown by both chemical shifts and 15N-15N dipole-dipole couplings (Figs. 3c, 6, and 7a). Thus, it appears that residues 141–153 constitute a segment where major conformational changes accompany CA self-assembly.
In crystal and solution NMR structures of CTD dimers (PDB codes 2KOD and 1A43), H8 extends to residue 173 or 174, while H9 begins at residue 178 or 179. The disorder in residues 172–175 and 179–182 discussed above indicates that these α-helices are shorter or frayed at their ends in CA tubes. According to our data, residues 172–182 may undergo “flapping” motions in CA tubes, while the rest of H8 and H9 remains rigid. According to the cryoEM-based model (PDB code 3J34), this segment is exposed on the outer surface of CA tubes, without intermolecular or intramolecular contacts that would preclude such motions.
Conformational changes also occur in residues 58–62, which connect H3 with H4, and 206–210, which connect H10 with H11. As discussed above, these conformational changes may be driven by intermolecular interactions that exist in the CA lattice but not in solution or crystalline states, especially interactions between neighboring NTD units within the same CA hexamer and interactions among hexamers around local three-fold symmetry axes.
Conformational variations within CA tubes
Curvature of the hexagonal lattice necessarily introduces asymmetries that should be reflected in molecule-to-molecule variations in CA structure and intermolecular interactions. The precise structural basis for lattice curvature is one of the central unresolved questions in the current understanding of CA assemblies, including both tubular and conical assemblies. In addition to the curvature of individual CA tubes, structural variations in our samples can arise from the coexistence of tubes with different diameters and helicities (see Figs. 1 and S1).
Structural heterogeneity generally produces inhomogeneous broadening of solid state NMR lines. Given the expected structural heterogeneity of CA tubes, it is remarkable that our 2D and 3D solid state NMR spectra show sharp crosspeaks that can be assigned to most residues from the N-terminal end of H1 to the C-terminal end of H11 (see Fig. 5a). The fact that 15N and 13C NMR linewidths (measured in ppm) are smaller in spectra acquired at 21.1 T than in spectra acquired at 14.1 T indicates that inhomogeneous broadening is not a major contribution to the linewidths of signals from most residues. In other words, very little structural heterogeneity actually exists at most sites, including residues that are believed to play important roles in intermolecular interactions. For example, all residues in H1, H2, H3 have unambiguous signal assignments. These α-helical segments participate in the intermolecular NTD-NTD interactions in the center of each hexamer7,12,20,23,25. Residues 183–187 in H9, which are believed to participate in CTD-CTD dimerization interactions5,6,11–13,16, have unambiguous signal assignments, including assignments for sidechain signals of Trp184, Met185, Thr186, and Glu187. Residues 200–207, which are believed to participate in intermolecular interactions around local three-fold symmetry axes12,23, also have unambiguous signal assignments.
In contrast, a number of residues in the vicinity of intermolecular NTD-CTD contacts between neighboring CA molecules within the same hexamer have no signal assignments or are assigned with low consistency, including residues 166, 167, 172–174, and 212–214. As discussed above, residues 179–182, in the vicinity of the CTD-CTD dimerization interface, also have no signal assignments. Thus, the most likely regions of conformational variations within CA that may be associated with lattice curvature are the inter-domain linker segment, residues that participate in intermolecular NTD-CTD contacts, and the N-terminal end of H9.
Finally, what are the implications of our solid state NMR data for previous proposals regarding structural variations within CA assemblies? Byeon et al. proposed that the arrangement of CA molecules around local three-fold symmetry axes within CA tubes was significantly asymmetric, with variations in intermolecular distances and in angles among H10 segments12. Similar asymmetries are apparent in the cryoEM-based model for CA tubes developed by Zhao et al.23. However, since residues around local three-fold symmetry axes have sharp solid state NMR signals with unambiguous assignments, it appears that pronounced asymmetries in intermolecular distances and H10-H10 angles are not present in our CA tubes. Byeon et al. also suggested that structural variations in CTD-CTD dimerization interfaces might contribute to lattice curvature12, and such variations are also apparent in the model of Zhao et al., which exhibits multiple H9-H9 angles23. In light of our solid state NMR data, especially the sharp sidechain signals of Trp184 (Fig. 8a), pronounced variations in H9-H9 angles appear not to be present in our CA tubes, although the N-terminal end of H9 is likely to be significantly disordered.
Both Pornillos et al.7,8 and Byeon et al.12 suggested that CA lattice curvature in tubular assemblies involves variations in the orientations of CTD units around hexamer cores formed by NTD units, permitted by variability in the conformation of the linker segment and in conformations of sidechains involved in intermolecular NTD-CTD interactions. From crystallographic studies of the Rous sarcoma virus (RSV) capsid protein, Bailey et al. developed a similar proposal for the generation of RSV lattice curvature based on pivoting of CTD units relative to a NTD hexamers28. Our solid state NMR data support this curvature mechanism, since our data indicate disorder in residues 141–145 (the C-terminal end of NTD), conformational changes in residues 149–153 (the N-terminal end of CTD), and disorder in residues of CTD that are involved in NTD-CTD interactions. Disorder at the N-terminal end of H9 may then be linked to the same variations in the orientations of CTD units around NTD hexamers.
Materials and Methods
Protein expression and purification
Wild-type HIV-1 CA was prepared by modification of the protocol described earlier.40 Competent E. coli BL21(DE3) cells carrying a pET-11a vector (HIV-1 CA plasmid pNL4–3) were added into 1.0 ml of 2xYT microbial medium with 100 μg/ml ampicillin for 3 hr, then transferred into 50.0 ml of minimal medium and grown at 37° C overnight to OD600 of 4–5. Each 500 ml of minimal medium contained 16.6 ml of 30× salt solution (1 M Na2HPO4·7H2O, 1 M KH2PO4, and 100 mM Na2SO4), 1.5 g of glucose, 0.75 g of NH4Cl, 0.5 ml of 2 M MgCl2, 25 mg of thiamine, 15 mg of CaCl2·2H2O and 5.0 ml of MEM vitamin solution (100×), and 100 μg/ml ampicillin. 500 ml of this medium was inoculated with the overnight culture to OD600 of 0.20–0.25. The cells were grown to OD600 of 1.5–2.0 at 37° C with 250 rpm shaking and spun down by 4,000 × g centrifugation for 20 min. Cell pellets were immediately transferred to warm fresh medium with 13C6-D-glucose and 15NH4Cl to produce a uniformly labeled sample (U-CA). Protein expression was initiated by addition of 1 mM IPTG after one hr of growth in the fresh medium. The cells were harvested by centrifugation at 3700 rpm for 60 min after 4 hr of induction to OD600 of 3.0–3.5. The pellets were stored at −80° C or purified immediately. The protein yield was about 65–70 mg/l.
The harvested cells were resuspended in 40 ml of lysis solution [25 mM Tris, 50 mM NaCl, 5mM β-mercaptoethanol (βME), one tablet of Complete protease inhibitor cocktail (Roche), and 200 μl of lysozyme (20 mg/ml), pH 8.0] on ice. The solution was then sonicated (Branson Sonifier model 250) at duty cycle 40, amplitude 4, for 60 min on ice. The cell debris was spun down by centrifugation at 9000 × g for 60 min. The protein was precipitated from the supernatant by addition of (NH4)2SO4 to a final concentration of 25% with stirring on ice for 2 hr. The precipitate was collected by centrifugation at 9000 × g for 1 hr. Pellets were redissolved in 20 ml Sepharose buffer A (25 mM KMOPS at pH 6.9 with 5 mM βME). The protein solution was then desalted through a Sephadex G-25 column before being loaded onto a SP Sepharose size exclusion column (GE Healthcare). After washing with two columns of Sepharose buffer A, the protein was eluted at around 45 ml by running a linear gradient of Sepharose buffer B (25 mM KMOPS at pH 6.9 with 1 M NaCl and 5 mM βME) over 300 ml.
Partially 13C-labeled CA samples (1,3-CA and 2-CA) were prepared in the same manner, but with 1,3-13C2-glycerol or 2-13C-glycerol instead of 13C6-D-glucose. After growth to OD600 of 0.8–0.85 at 37° C with 250 rpm shaking, the cells were spun down by centrifugation and the cell pellets were resuspended in fresh medium with 1,3-13C2-glycerol (1,3-CA) or 2-13C1-glycerol (2-CA) and with 15NH4Cl. Protein expression was initiated when the cells were grown to OD600 of 0.9–1.0. The cells were harvested after 4 hr of induction to OD600 of 1.5–1.9. The pellets were stored at −80° C or purified immediately. The protein yield for glycerol-labeled CA was about 35–45 mg/l.
CA assembly
Purified protein was concentrated to 30 mg/ml in Tris buffer (50 mM, pH 8.0) and 50 mM NaCl. To initiate CA self-assembly, Tris buffer with 5.0 M NaCl was added to this solution for a final NaCl concentration of 1.0 M and final protein concentration of 24 mg/ml. This assembly solution was incubated at 37° C for 15 min and then incubated at 4° C for 12–16 hr. Pre-incubation at 37° C appears to encourage nucleation, while assembly at low temperature allows elongation into long, uniform tubes. The formation of tubes was verified by TEM. The solution was then centrifuged gently and resuspended in 1.0 M NaCl multiple times to wash off incompletely assembled CA. Finally, the 1.0 M NaCl solution was replaced by 0.1 M NaCl and 17.5% PEG-8000 (w/w). Lower NaCl concentrations reduce sample heating due to rf irradiation during NMR experiments, while PEG-8000 aids in preserving the integrity of the tubes. Approximately 30 mg of sample, containing around 15 mg of protein, were packed into each MAS rotor for solid state NMR measurements.
Transmission electron microscopy
TEM images were recorded with an FEI Morgagni microscope, operating at 80 keV. Negatively stained samples were prepared on glow-discharged carbon films, supported by lacey carbon on 300 mesh copper grids. A 5 μl aliquot of 10-fold-diluted CA assembly solution (in 1 M NaCl) was adsorbed for 30 s, blotted with filter paper, rinsed with deionized water and immediately blotted, stained for 20 s with 5 μl of 2% uranyl acetate, blotted, and dried in air.
Solid state NMR
Solid state NMR experiments were recorded in our laboratory with a Varian InfinityPlus spectrometer operating at a 1H NMR frequency of 599.2 MHz, equipped with a Varian BioMAS probe with a 3.2 mm MAS module, which had been modified to increase the flow of cooling gas to the sample and to increase the rf efficiency of the scroll coil. Sample temperature was maintained at 5–10° C with cooled nitrogen gas and monitored through the water 1H NMR line. 2D and 3D experiments for signal assignments were recorded at 12.00 kHz MAS frequency, with frequency-selective cross-polarization for 15N-13C polarization transfer and radio frequency-driven recoupling (RFDR)53,54 and dipolar assisted rotary resonance (DARR)55,56 for 13C-13C mixing. 2D 13C-13C RFDR correlation experiments were recorded with a 4.0 ms mixing period, during which 15.0 μs 13C π pulses and 90 kHz 1H decoupling were applied. Heteronuclear 15N-13C transfer was achieved in all 2D and 3D experiments with 3.0 ms cross-polarization periods with rf fields of 30 kHz on 15N and 42 kHz on 13C for NCO/CON transfer steps and 30 kHz on 15N and 18 kHz on 13C for NCA transfer steps, all with 90 kHz 1H decoupling. With the U-CA sample, two 3D NCACX experiments were recorded, with 20 and 50 ms of DARR mixing. One 3D NCOCX experiment was recorded with 70 ms of DARR mixing. Similar experiments were recorded with the 1,3-CA sample. These 3D spectra were recorded with 28 and 32 complex points and 5.38 kHz and 5.38 kHz spectral widths in the indirect 15N and 13C dimensions, respectively, with 32 scans per free-induction decay (FID) and a recycle delay of 2.2 s. Finally, 3D CONCA experiments were recorded for the 1,3-CA and 2-CA samples. These spectra were recorded with 20 and 24 complex points and 5.76 kHz and 5.76 kHz spectral widths in the indirect 15N and 13C dimensions, respectively, with 64 scans per FID and a recycle delay of 2.2 s. Total measurement times were approximately 3 days per 3D spectrum.
1H T2-filtered NCA spectra of the U-CA sample were recorded by inserting a 1H Hahn echo period (0 or 168 μs) between the initial 1H π/2 pulse and the 1H-15N cross-polarization step. These spectra were recorded with 80 real points and a 8.96 kHz spectral width in the indirect dimension, with 512 scans per FID, for a total measurement time of approximately 2 days per 2D spectrum.
15N-BARE measurements were performed as previously described50, but at an MAS frequency of 11.905 kHz. Effective 15N-15N dipolar evolution periods were 0 ms, 8.064 ms, 12.096 ms, 16.128 ms, and 20.16 ms, using a constant-time recoupling period of 20.16 ms. Each 2D spectrum was acquired with 80 complex points and a 8.96 kHz spectral width in the indirect dimension and 256 scans per FID. Simulations of 15N-BARE data were performed as previously described50.
A 2D 13C-13C TOBSY spectrum was recorded with preparation of initial 13C polarization by refocused 1H-13C INEPT57 (insensitive nuclei enhanced polarization transfer) and with a 5.04 ms 13C-13C TOBSY49 (total through-bond correlation spectroscopy) mixing period. The MAS frequency was set to 5.952 kHz to allow for a TOBSY matching condition with the 13C rf field of 71.4 kHz. The 1H decoupling amplitude was 50 kHz. The spectrum was recorded with 512 complex points and 45.5 kHz spectral width in the indirect dimension, with eight scans per FID.
In addition, solid state NMR spectra were recorded with a Bruker Avance spectrometer operating at a 1H NMR frequency of 900.1 MHz at the MIT-Harvard Center for Magnetic Resonance. This spectrometer was equipped with a Bruker E-free probe containing a 3.2 mm MAS module. For U-CA and 1,3-CA samples, 3D NCACX and NCOCX spectra were obtained simultaneously using concurrent 15N-13Cα and 15N-13CO transfers via transferred echo double resonance (TEDOR)58 followed by 13C-13C mixing via RFDR.59 The MAS frequency was set to 20.0 kHz. Mixing times were 1.6 ms and 3.2 ms for TEDOR and RFDR, respectively. The RFDR period consisted of 6.0 Μs 13C π pulses, with 83 kHz 1H decoupling applied during both RFDR and TEDOR periods. Indirect dimensions consisted of 122 complex points with a 20.00 kHz spectral width for the 13C dimension and 64 complex points with a 6.67 kHz spectral width for the 15N dimension. The carbonyl region of the indirect 13C dimension was aliased to allow for a smaller spectral width. Four scans were averaged per FID with a 2.5 s recycle delay, for a total acquisition time of 3.5 days per 3D experiment. All spectra were processed with NMRPipe60 and analyzed with Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco; available from http://www.cgl.ucsf.edu/home/sparky/).
Computer-aided signal assignments
Our procedure for assignment of solid state NMR signals relied on the program MCASSIGN2 (available upon request from robertty@mail.nih.gov), which uses a Monte Carlo/simulated annealing algorithm to assign signal sets from 2D or 3D chemical shift correlation spectra to individual residues in a manner that maximizes a simple score function45. The score function includes terms that favor correct connections between signal sets in different signal tables, disfavor incorrect connections, favor the use of all available signal sets, and disfavor gaps between amino acid segments to which signals are assigned. An important advantage of this computer-aided approach, compared with traditional manual approaches to sequential assignment, is that large numbers of independent MCASSIGN2 runs can easily be performed, allowing the identification of all assignments that are consistent with available data. Comparisons of assignments from multiple independent runs allow residues with unambiguous assignments to be distinguished from residues with multiple possible assignments. Residues with no signal assignments in any successful MCASSIGN2 runs are considered not to contribute to the solid state NMR data, due to either static or dynamic disorder.
The initial step in the assignment process was to identify the most intense crosspeaks in the 3D spectra of U-CA tubes. These peaks were grouped with interconnecting signals in 2D CC, 2D NCA, and 3D NCACX spectra of U-CA tubes to build a signal table comprised of sets of N(i), Cα(i), CO(i), and Cβ(i) chemical shifts for each resolved spin system i. Each signal set (i.e., each row in the signal table) was given an array of possible amino acid type assignments based on Cα and Cβ chemical shifts. In cases where sidechain resonances were observed and unambiguously assigned to a signal set, they were used to narrow down the possible amino acid type assignments for the signal set. In the case of certain amino acid spin systems with characteristic resonances (i.e., Ala, Pro, Ser, Thr), it was possible to assign a single amino acid type to the given signal set. A second signal table was generated by applying the same procedure to 3D NCOCX spectra, resulting in signal sets comprised of N(j+1), Cα(j), CO(j), and Cα(j) chemical shifts for each resolved spin system j.
The initial NCACX and NCOCX signal tables (containing 150 and 138 signal sets, respectively) served as the starting input for the MCASSIGN2 program. In multiple independent runs, MCASSIGN2 generated unique assignments for some of the signal sets, while other signal sets where assigned with lower consistency. The spectra were then examined manually to verify the unique assignments from MCASSIGN2, to check for peak-picking errors, and to shorten the lists of possible amino acid types for certain signal sets. CONCA spectra of 1,3-CA and 2-CA samples were used to corroborate assignments from MCASSIGN2 and refine amino acid types, using the dependences of 13CO and 13Cα labeling on amino acid type in these samples. New MCASSIGN2 runs were performed using the updated signal tables as input, while keeping the signal sets that were assigned unambiguously in the previous runs fixed to their assigned values. In these runs, new signal sets were assigned uniquely, while others were assigned less consistently. This process of manual analysis and computer-aided assignment was iterated several times until no new unique assignments were made by MCASSIGN2. In this way, unique sequential assignments were determined for 122 signal sets from the NCACX signal table.
Weaker signals and incomplete spin systems in the spectra were then analyzed manually and included in new signal tables for a second set of iterative MCASSIGN2 and manual analysis. At the end of this process, the final signal tables, shown in Tables S1 and S2, were used as input for the final 50 independent MCASSIGN2 runs, with no fixed assignments of any signal sets.
In the final runs, coefficients in the score function increased from initial values of zero to final values of 10, 20, 6, and 0 for w1, w2 , w3, and w4, respectively, in the notation of reference45. Each run consisted of 20 annealing steps, with 500 million Monte Carlo attempts in each step. Typically, 10 runs were performed simultaneously with a Linux computer equipped with a 12-core 2.8 GHz Xeon processor. Each run took approximately 1.5 hr to complete.
Final assignments in Table S3 have been deposited in the BioMagResBank with accession number 19575.
Structural modeling
Calculations to visualize differences between CA structures determined by solution NMR or crystallography and the structure of CA in tubes were performed with Xplor-NIH51. Solid state NMR restraints were applied as energy potentials to drive the simulated annealing process, which started from atomic coordinates in PDB files 2LF4 and 3MGE. Torsion angle restraints based on TALOS+ predictions used the CDIH potential of Xplor-NIH. Restraints from 15N-BARE data were converted to three-dimensional torsion angle potential surfaces as previously described45 and implemented with the TorsionInterpolPot potential. Calculated structures were kept close to their starting structures with the refRMSD potential, with scaling factors adjusted to allow localized structural changes to satisfy the solid state NMR restraints. Separate refRMSD potentials were applied to residues 1–144 and 149–231 to maintain the CTD and NTD structures, while allowing the relative orientation of CTD and NTD and the conformation of residues 145–148 to vary freely.
Simulated annealing was performed from 3500 K to 25 K in 12.5 K steps, with 0.2 ps of torsion angle dynamics at each temperature step. The CDIH scale factor was set to 400 kcal/mol-rad2. The refRMSD scale factor was set to 80 kcal/mol-Å2. Scale factors for 15N-BARE potentials increased geometrically from 0.001 to 400 kcal/mol. The scale factor for van der Waals-like repulsions increased from 0.004 to 4 kcal/mol-Å4, with a radius scale factor decreasing from 0.9 to 0.8. Standard bond length, bond angle, and improper angle potentials were also used. After annealing, energy minimization was performed, first in torsion angle space, then in Cartesian coordinate space. Final structures in Fig. 7 are lowest-energy structures from 100 independent Xplor-NIH calculations. In each set of calculations, no significant differences were found in the 10 lowest-energy final structures.
Supplementary Material
Highlights
Structure and dynamics of self-assembled HIV-1 CA are not yet fully understood
Solid state NMR spectroscopy distinguishes ordered and disordered residues in CA tubes
Solid state NMR data identify conformational changes accompanying CA self-assembly
Structural variations localized to NTD-CTD linker and intermolecular NTD-CTD contacts
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, a component of the National Institutes of Health, and by the NIH Intramural AIDS Targeted Antiviral Program. Bo Chen acknowledges support from the Air Force Office of Scientific Research, award number FA9550-13-1-0150. NMR spectra at 21.1 T were acquired at the MIT/Harvard Center for Magnetic Resonance, which supported by NIH grant EB-002026. We thank Lalit Deshmukh and Guillermo Bermejo for helpful discussions regarding solution NMR spectroscopy of CA and Xplor-NIH calculations, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Briggs JAG, Krausslich HG. The molecular architecture of HIV. J. Mol. Biol. 2011;410:491–500. doi: 10.1016/j.jmb.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 2.Engelman A, Cherepanov P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012;10:279–290. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gamble TR, Vajdos FF, Yoo SH, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 4.Momany C, Kovari LC, Prongay AJ, Keller W, Gitti RK, Lee BM, Gorbalenya AE, Tong L, McClure J, Ehrlich LS, Summers MF, Carter C, Rossmann MG. Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 1996;3:763–770. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- 5.Gamble TR, Yoo SH, Vajdos FF, vonSchwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 6.Worthylake DK, Wang H, Yoo SH, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 angstrom resolution. Acta Crystallogr. Sect. D-Biol. Crystallogr. 1999;55:85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- 7.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua YZ, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pornillos O, Ganser-Pornillos BK, Yeager M. Atomic-level modelling of the HIV capsid. Nature. 2011;469:424–428. doi: 10.1038/nature09640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campos-Olivas R, Summers MF. Backbone dynamics of the N-terminal domain of the HIV-1 capsid protein and comparison with the G94D mutant conferring cyclosporin resistance/dependence. Biochemistry. 1999;38:10262–10271. doi: 10.1021/bi990991x. [DOI] [PubMed] [Google Scholar]
- 10.Tang C, Ndassa Y, Summers MF. Structure of the N-terminal 283-residue fragment of the immature HIV-1 gag polyprotein. Nat. Struct. Biol. 2002;9:537–543. doi: 10.1038/nsb806. [DOI] [PubMed] [Google Scholar]
- 11.Wong HC, Shin R, Krishna NR. Solution structure of a double mutant of the carboxy-terminal dimerization domain of the HIV-1 capsid proteint. Biochemistry. 2008;47:2289–2297. doi: 10.1021/bi7022128. [DOI] [PubMed] [Google Scholar]
- 12.Byeon IJL, Meng X, Jung JW, Zhao GP, Yang RF, Ahn JW, Shi J, Concel J, Aiken C, Zhang PJ, Gronenborn AM. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell. 2009;139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcaraz LA, del Alamo M, Barrera FN, Mateu MG, Neira JL. Flexibility in HIV-1 assembly subunits: Solution structure of the monomeric C-terminal domain of the capsid protein. Biophys. J. 2007;93:1264–1276. doi: 10.1529/biophysj.106.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcaraz LA, del Alamo M, Mateu MG, Neira JL. Structural mobility of the monomeric C-terminal domain of the HIV-1 capsid protein. FEBS J. 2008;275:3299–3311. doi: 10.1111/j.1742-4658.2008.06478.x. [DOI] [PubMed] [Google Scholar]
- 15.Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 16.Shin R, Tzou YM, Krishna NR. Structure of a monomeric mutant of the HIV-1 capsid protein. Biochemistry. 2011;50:9457–9467. doi: 10.1021/bi2011493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshmukh L, Schwieters CD, Grishaev A, Ghirlando R, Baber JL, Clore GM. Structure and dynamics of full length HIV-1 capsid protein in solution. J. Am. Chem. Soc. 2013;135:16133–16147. doi: 10.1021/ja406246z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen B, Tycko R. Simulated self-assembly of the HIV-1 capsid: Protein shape and native contacts are sufficient for two-dimensional lattice formation. Biophys. J. 2011;100:3035–3044. doi: 10.1016/j.bpj.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J. Mol. Biol. 2005;346:577–588. doi: 10.1016/j.jmb.2004.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- 21.Ehrlich LS, Liu TB, Scarlata S, Chu B, Carter CA. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: Structure switching by pH-induced conformational changes. Biophys. J. 2001;81:586–594. doi: 10.1016/S0006-3495(01)75725-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 2004;78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao GP, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning JY, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang PJ. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ajayan PM. Nanotubes from carbon. Chem. Rev. 1999;99:1787–1799. doi: 10.1021/cr970102g. [DOI] [PubMed] [Google Scholar]
- 25.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: A model for the mature capsid lattice. Cell. 2007;131:70–79. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 27.Manolopoulos DE, Fowler PW. Molecular graphs, point groups, and fullerenes. J. Chem. Phys. 1992;96:7603–7614. [Google Scholar]
- 28.Bailey GD, Hyun JK, Mitra AK, Kingston RL. A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J. Mol. Biol. 2012;417:212–223. doi: 10.1016/j.jmb.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 29.Lu J-X, Qiang W, Yau W-M, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang LC, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TPJ, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Molecular conformation and dynamics of the Y145stop variant of human prion protein. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6284–6289. doi: 10.1073/pnas.0711716105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Melckebeke H, Wasmer C, Lange A, Ab E, Loquet A, Bockmann A, Meier BH. Atomic-resolution three-dimensional structure of HET-s(218–289) amyloid fibrils by solid state nmr spectroscopy. J. Am. Chem. Soc. 2010;132:13765–13775. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- 33.Goldbourt A, Day LA, McDermott AE. Intersubunit hydrophobic interactions in Pf1 filamentous phage. J. Biol. Chem. 2010;285:37051–37059. doi: 10.1074/jbc.M110.119339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu TY, Schaefer J. REDOR NMR characterization of DNA packaging in bacteriophage t4. J. Mol. Biol. 2008;382:1031–1042. doi: 10.1016/j.jmb.2008.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doherty T, Su YC, Hong M. High-resolution orientation and depth of insertion of the voltage-sensing S4 helix of a potassium channel in lipid bilayers. J. Mol. Biol. 2010;401:642–652. doi: 10.1016/j.jmb.2010.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang M, Nesbitt AE, Sperling LJ, Berthold DA, Schwieters CD, Gennis RB, Rienstra CM. Structure of the disulfide bond generating membrane protein DSBB in the lipid bilayer. J. Mol. Biol. 2013;425:1670–1682. doi: 10.1016/j.jmb.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bayro MJ, Daviso E, Belenky M, Griffin RG, Herzfeld J. An amyloid organelle: solid state NMR evidence for cross-β assembly of gas vesicles. J. Biol. Chem. 2012;287:3479–3484. doi: 10.1074/jbc.M111.313049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. Atomic model of the type III secretion system needle. Nature. 2012;486:276–279. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jehle S, van Rossum B, Stout JR, Noguchi SM, Falber K, Rehbein K, Oschkinat H, Klevit RE, Rajagopal P. Alpha B-crystallin: A hybrid solid state/solution state NMR investigation reveals structural aspects of the heterogeneous oligomer. J. Mol. Biol. 2009;385:1481–1497. doi: 10.1016/j.jmb.2008.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen B, Tycko R. Structural and dynamical characterization of tubular HIV-1 capsid protein assemblies by solid state nuclear magnetic resonance and electron microscopy. Protein Sci. 2010;19:716–730. doi: 10.1002/pro.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han Y, Ahn J, Concel J, Byeon IJL, Gronenborn AM, Yang J, Polenova T. Solid state NMR studies of HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 2010;132:1976–1987. doi: 10.1021/ja908687k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byeon IJL, Hou GJ, Han Y, Suiter CL, Ahn J, Jung J, Byeon CH, Gronenborn AM, Polenova T. Motions on the millisecond time scale and multiple conformations of HIV-1 capsid protein: Implications for structural polymorphism of CA assemblies. J. Am. Chem. Soc. 2012;134:6455–6466. doi: 10.1021/ja300937v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, van Rossum BJ, Oschkinat H. Assigning large proteins in the solid state: A MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J. Biomol. NMR. 2009;44:245–260. doi: 10.1007/s10858-009-9338-7. [DOI] [PubMed] [Google Scholar]
- 44.Hong M. Determination of multiple ϕ torsion angles in proteins by selective and extensive 13C labeling and two-dimensional solid state NMR. J. Magn. Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 45.Hu KN, Qiang W, Tycko R. A general Monte Carlo/simulated annealing algorithm for resonance assignment in NMR of uniformly labeled biopolymers. J. Biomol. NMR. 2011;50:267–276. doi: 10.1007/s10858-011-9517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han Y, Hou G, Suiter CL, Ahn J, Byeon IL, Lipton AS, Burton S, Hung I, Gor'kov PL, Gan Z, Brey W, Rice D, Gronenborn AM, Polenova T. Magic angle spinning NMR reveals sequence-dependent structural plasticity, dynamics, and the spacer peptide 1 conformation in HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 2013;135:17793–17803. doi: 10.1021/ja406907h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP. Structural insights into the catalytic mechanism of cyclophilin A. Nature Struct. Biol. 2003;10:475–481. doi: 10.1038/nsb927. [DOI] [PubMed] [Google Scholar]
- 49.Hardy EH, Verel R, Meier BH. Fast MAS total through-bond correlation spectroscopy. J. Magn. Reson. 2001;148:459–464. doi: 10.1006/jmre.2000.2258. [DOI] [PubMed] [Google Scholar]
- 50.Hu KN, Qiang W, Bermejo GA, Schwieters CD, Tycko R. Restraints on backbone conformations in solid state NMR studies of uniformly labeled proteins from quantitative amide 15N-15N and carbonyl 13C-13C dipolar recoupling data. J. Magn. Reson. 2012;218:115–127. doi: 10.1016/j.jmr.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:47–62. [Google Scholar]
- 52.Jiang JY, Ablan SD, Derebail S, Hercik K, Soheilian F, Thomas JA, Tang SX, Hewlett I, Nagashima K, Gorelick RJ, Freed EO, Levin JG. The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology. 2011;421:253–265. doi: 10.1016/j.virol.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett AE, Rienstra CM, Griffiths JM, Zhen WG, Lansbury PT, Griffin RG. Homonuclear radio frequency-driven recoupling in rotating solids. J. Chem. Phys. 1998;108:9463–9479. [Google Scholar]
- 54.Ishii Y. 13C-13C dipolar recoupling under very fast magic angle spinning in solid state nuclear magnetic resonance: Applications to distance measurements, spectral assignments, and high-throughput secondary-structure determination. J. Chem. Phys. 2001;114:8473–8483. [Google Scholar]
- 55.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- 56.Morcombe CR, Gaponenko V, Byrd RA, Zilm KW. Diluting abundant spins by isotope edited radio frequency field assisted diffusion. J. Am. Chem. Soc. 2004;126:7196–7197. doi: 10.1021/ja047919t. [DOI] [PubMed] [Google Scholar]
- 57.Morris GA, Freeman R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979;101:760–772. [Google Scholar]
- 58.Hing AW, Vega S, Schaefer J. Transferred-echo double-resonance NMR. J. Magn. Reson. 1992;96:205–209. [Google Scholar]
- 59.Daviso E, Eddy MT, Adreas LB, Griffin RG, Herzfeld J. Efficient resonance assignment of proteins in MAS NMR by simultaneous intra- and inter-residue 3D correlation spectroscopy. J. Biomol. NMR. 2013;55:257–265. doi: 10.1007/s10858-013-9707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe: a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.