

Abstract
Studies from the 1950s and 1960s already recognize the fact that osteocytes, although long living cells, die, as evidenced by accumulation of osteocytic lacunae devoid of cells. More recently, it was demonstrated that these cells die by apoptosis. The rate of osteocyte apoptosis is regulated by the age of the bone, as well as by systemic hormones, local growth factors, cytokines, pharmacological agents, and mechanical forces. Apoptotic osteocytes, in turn, recruit osteoclasts to initiate targeted bone resorption. This results in the removal of “dead” bone and may improve the mechanical properties of the skeleton. However, the molecular regulators of osteocyte survival and targeted bone remodeling are not completely known. In this review, the current knowledge on the molecular mechanism that lead to osteocyte death or survival, and the signals that mediate targeted bone resorption is discussed.
Introduction: osteocytes, long living cells that can undergo premature death
Osteocytes were considered to be inactive bone residents for long time. They are now believed to be the master regulators of bone remodeling, via the production of soluble factors that reach the bone marrow and influence the differentiation and behavior of osteoblasts and osteoclasts [1]. Part of this regulatory mechanism is exerted following osteocyte death. In particular, osteocyte apoptosis is associated with local bone resorption in several conditions. This could be due either to the absence of restraining signals or the release of pro-osteoclastogenic signals by dying osteocytes or their neighbors. In the 1950s it was shown that radiation damage was associated with osteocyte death, and that osteocytes located near fractures also died [2]. Later, Harold Frost reported increased osteocyte death with aging, evidenced by accumulation of empty lacunae [3]; and Dunstan and colleagues showed increased osteocyte apoptosis in patients with fractures of the femoral neck and osteoarthritis [2]. The accumulation of dead osteocytes in older individuals was reproduced more recently in mice, by measuring the prevalence of apoptotic TUNEL positive osteocytes [4]. In addition, increased osteocyte apoptosis was found in the absence of sex steroids [5,6], with glucocorticoid excess [7], due to immobilization [8,9], or induced by fatigue loading [10]. Conversely, agents that preserve bone mass and strength reduce the prevalence of osteocyte apoptosis. This is the case for intermittent parathyroid hormone (PTH) administration [11], bisphosphonates [12], and sex steroids [6,13]. In this review, the current knowledge on the molecular mechanism that lead to osteocyte death and the signals that mediate targeted bone resorption will be discussed.
Why osteocytes die?
Parfitt and colleagues have estimated that approximately 2.5% osteocytes die each year, and that the lifespan of osteocytes can vary from 1 to 50 years [14]. The cause of this spontaneous apoptosis remains unknown. It is believed that apoptosis is the default fate for all nucleated cells and only cells that have active survival signals remain alive [15]. For example, inhibition of osteocyte apoptosis by deletion of the pro-apoptotic proteins Bak and Bax generates dysfunctional osteocytes [16]. This evidence suggests that a normal rate of osteocyte death is required to maintain bone health; and, that artificial prolongation of the life span of osteocytes results in accumulation of cell damage as the age of the bone increases.
Aging accelerates osteocyte death, by a mechanism associated with increased oxidative stress, as evidenced by a reduction in anti-oxidant enzymes, and phosphorylation of the markers of oxidative stress p53 and of p66Shc in bone from aging mice [4]. A similar mechanism has been proposed for the accumulation of apoptotic osteocytes induced by removal of sex steroids [5]. Thus, administration of the anti-oxidant N-acetyl Cysteine prevents the accumulation of reactive oxygen species, the decrease activity of anti-oxidant enzymes, the increase in phosphorylated p53 and p66shc, and, ultimately, osteocyte apoptosis in ovariectomized and orchidectomized mice [4].
Accumulation of apoptotic osteocytes is also a feature of glucocorticoid-induced bone disease in mice and humans [7,12]. In particular, glucocorticoid-induced osteonecrosis and the consequent collapse of the femoral head are associated with apoptotic osteocytes localized adjacent to the sub-chondral region [17]. The pro-apoptotic effect of the steroids is mediated via the glucocorticoid receptor, is independent of gene transcription and results from the activation of the focal adhesion related kinase Pyk2/JNK signaling pathway [18]. This leads to cell detachment-induced apoptosis (also known as anoikis). Actions of glucocorticoid on osteoblasts might also contribute to the induction of osteocyte apoptosis. This includes the local suppression of the synthesis of survival factors such as IGF-1, IL-6, MMPs [19], and the induction of the Wnt antagonist SFRP-1 [20]. The effect of glucocorticoids has also been associated with increased oxidative stress [21,22]; and elevated glucocorticoid levels can contribute to the increase in reactive oxygen species and osteocyte apoptosis during aging. This was demonstrated by a decrease in osteocyte apoptosis in 21-month-old mice in which the osteoblasts and osteocytes expressed an enzyme to locally inactivate glucocorticoids [23].
Mechanical forces also affect osteocyte viability. Lack of mechanical stimulation [8,9] and excessive loading [10,24] both increase the prevalence of osteocyte apoptosis in mice and rats. The lack of mechanical forces could decrease survival signals induced by normal physical activity. In particular, disuse increases the expression of the Wnt inhibitor sclerostin [25]. By interfering with the survival effect of Wnt signaling activation in osteocytes, sclerostin also contributes to increasing osteocyte death in the absence of loading [26]. In osteocytes in the vicinity of microdamage, elevated osteocyte apoptosis is associated with increased expression of the pro-apoptotic protein Bax, and, coincides with increased TUNEL staining. Conversely, the anti-apoptotic protein Bcl-2 increases farther away from the damage focus [27], suggesting the activation of protective signals that avoid widespread cell death.
How osteocytes are maintained alive?
Most therapeutic interventions that increase or maintain bone mass and strength also preserve osteocyte viability. This was first demonstrated following daily injections of PTH, the only FDA-approved bone anabolic treatment [11]. Although the mechanism by which PTH prevents osteocyte apoptosis has not been investigated, we have shown using osteoblastic cells that the hormone prevents glucocorticoid- and etoposide-induced cell death via activating the PKA/cAMP/CREB signaling pathway [28]. PTH-induced cell survival also requires the inactivation of the pro-apoptotic protein BAD and increased synthesis of the anti-apoptotic protein Bcl-2. Interestingly, the transcription factor Runx2 is required for the survival effect of PTH, but the hormone also induces Runx2 proteosomal degradation, which limits the duration of the survival effect of PTH. The anti-apoptotic effect of PTH also requires the expression of connexin43 (Cx43), which sequesters β-arrestin thereby allowing PTH-induced cAMP production and cell survival [29].
Bisphosphonates are anti-resorptive agents that block osteoclast activity by inhibiting enzymes of the mevalonate pathway [30]. We showed that bisphosphonates also prevent osteocyte apoptosis in vitro and in vivo [12]. The drugs exert this effect by opening Cx43 hemichannels, leading to activation of the Src/MEK/ERK pathway [12,31]. The requirement of Cx43 expression in osteocytes for the anti-apoptotic effect of bisphosphonates, and the induction of ERK activation in bone, has been demonstrated in vivo [32,33]. Interestingly, unlike most agents that induce ERK activation, bisphosphonates do not induce the nuclear translocation of the kinase [34]. Instead, they promote the phosphorylation of the cytoplasmic target of ERKs p90RSK, followed by phosphorylation of CEBPβ and BAD, without requiring new gene transcription. The survival effect of bisphosphonates on osteocytes (and osteoblasts) is distinct from the effect of the drugs on osteoclasts. Thus, it is exerted at much lower concentrations than the ones required for inhibiting osteoclast function, it is mediated by activation of the extracellular signal-activated kinases (ERKs), independently of the mevalonate pathway, and it is reproduced by IG9402, a bisphosphonate analog that does not affect osteoclasts [33,35].
Both estrogens and androgens prevent osteocyte apoptosis [36,37]. Similar effect has been demonstrated with the selective estrogen receptor modulator (SERM) LY 117018 [38], but not with raloxifene [39,40]. The survival effect of the sex steroids can be exerted via activation of either estrogen or androgen receptor [6], is independent of gene transcription mediated by the receptors, and can be mimicked by estren, a non-genomic activator of estrogen receptor [6,37]. Like bisphosphonates, the survival effect of sex steroids depends on ERK activation, but, unlike bisphosphonates, nuclear accumulation of the kinase and ERK-activated transcription factors are required [34,39]. The survival effect of sex steroids also requires the activation of PI3K and JNK, inactivation of the pro-apoptotic protein BAD and the transcriptional activity of AP-1 [39].
Physiological levels of mechanical stimulation reduce osteocyte apoptosis in rats [24]. We showed in vitro that mechanical stimulation by biaxial stretching prevents osteocyte apoptosis by inducing the engagement of integrins and activation of the focal adhesion kinase FAK [41]. This is followed by Src/MEK/ERK activation, nuclear ERK accumulation and ERK-mediated gene transcription. In addition, the estrogen receptors ERα and ERβ (but not the androgen receptor), and their presence in the cell membrane within caveolae, are required for the survival effect of mechanical loading, albeit in a ligand-independent manner [42]. Others have shown that membrane shear stress induced by fluid flow in vitro leads to the opening of Cx43 hemichannels, the release of prostaglandin E2, and osteocytic cell survival [43]. In this context, prostaglandin E2 induces the activation of both cAMP/PKA and PI3K/Akt/βcatenin signaling pathways to preserve osteocyte viability.
Osteocyte apoptosis and osteoclast recruitment: lack of signals from absent osteocytes versus signals from dying cells
The connection between disruption of the osteocyte network and bone remodeling was first proposed by Frost (reviewed in [44]). Later, Noble and colleagues observed signs of apoptosis in osteocytes located in bones with high rates of remodeling in human bone samples [45]. This led the authors to speculate the existence of a link between osteocyte apoptosis and bone replacement. This hypothesis is supported by microscopy studies showing apoptotic osteocytes engulfed by osteoclasts [46–48]. More recently, Schaffler’s group showed that apoptotic osteocytes in rats subjected to fatigue loading accumulate in areas where bone resorption occurs [10]. Our group also demonstrated co-localization of apoptotic osteocytes and osteoclasts in a murine model of reduced mechanical stimulation [8]. Accumulation of apoptotic osteocytes preceded the increase in osteoclasts, suggesting a cause-effect relationship between dead osteocytes and bone resorption [8,10]. This relationship was confirmed in studies in which osteocyte apoptosis was directly blocked using caspase inhibitors, resulting in a substantial decrease in bone resorption following fatigue loading and ovariectomy [49,50].
Genetically modified mouse models also help to support the hypothesis that accumulation of apoptotic osteocytes induces the recruitment of osteoclasts to initiate bone resorption. Using transgenic mice, expressing the diphtheria toxin receptor in osteocytes, Tatsumi et al showed that administration of diphtheria toxin not only induces a dramatic increase in apoptotic osteocytes and empty lacunae, but also accumulation of osteoclasts and cortical bone porosity [51].
We showed accumulation of apoptotic osteocytes in cortical bone from mice lacking Cx43 in osteoblasts and osteocytes, or in osteocytes only (Figure 1) [52]. This was later confirmed by Lloyd et al. in mice lacking Cx43 in osteoblasts and osteocytes [53]. In bones from mice lacking osteocytic Cx43, anatomical mapping of cells showed that apoptotic osteocytes are preferentially located near endocortical surfaces where osteoclasts accumulate [52]. Interestingly, although deletion of Cx43 results in accumulation of empty lacunae, these did not localize in any particular region of the cortical bone. This suggests that signals from dying osteocytes, rather than absence of signals released by living cells, are required to recruit osteoclasts. However, although some candidate molecules have been proposed [54], the molecular nature of these signals is not known.
Figure 1.
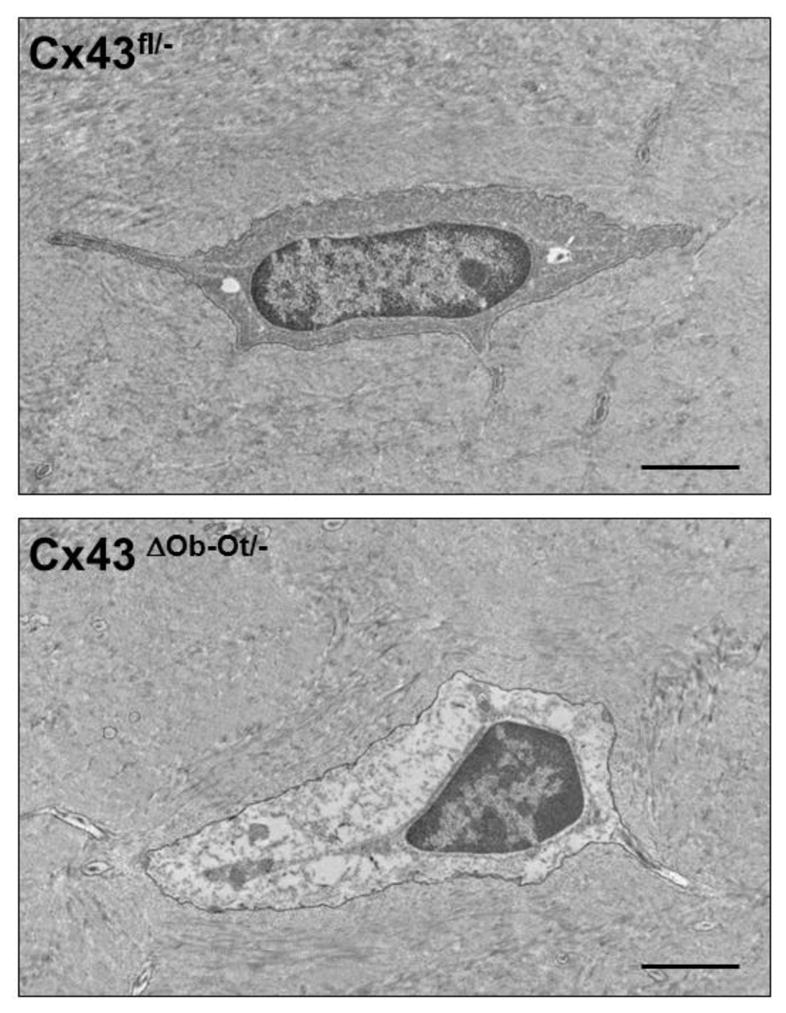
Transmission electron microscopy images of femoral bone showing a Cx43-expressing osteocyte from control Cx43fl/− mice (top panel), and a Cx43-deficient osteocyte from mice lacking Cx43 in osteoblasts and osteocytes (Cx43 ΔOb−Ot/−) (bottom panel). Whereas the cell on the top contains a nucleus with normal appearance, the one on the bottom exhibit typical features of a cell undergoing apoptosis, including nuclear condensation and detachment from the extracellular matrix. Bar indicates 2 μm. Images were taken at the Electron Microscopy Center of the Department of Anatomy and Cell Biology (Indiana University School of Medicine). Digital images were taken with an Advanced Microscope Techniques (Danvers, MA, USA) CCD camera. See [52] for more details.
Evidence for some candidate molecules linking osteoclastogenesis to osteocyte apoptosis are now emerging. For example, viable osteocytes in the vicinity of apoptotic cells express high RANKL/OPG ratio and elevated VEGF expression in bones subjected to fatigue loading [55]. Monocyte chemoattractant protein-1 (CCL2) is also specifically regulated at this site [56], contributing to the localized elevation of osteoclastogenesis. An increase in the RANKL/OPG ratio is also observed in Cx43 deficient osteocytes [52,57], suggesting that disruption of cell-to-cell communication between the dying osteocyte and the neighbor osteocyte might cause the elevation of the pro-osteoclastogenic cytokines in the latter cell.
Still, the signals driving from dying cells to neighboring osteocytes or to the bone marrow have not been identified. In MLO-Y4 osteocytic cells and primary osteocytes, the apoptotic osteocyte bodies released by dying cells induce bone resorption in quiescent surfaces of calvariae [58]. Further, they also stimulate osteoclast differentiation in co-cultures with bone marrow cells, even in the absence of RANKL. Another candidate to mediate the recruitment of osteoclasts is the high mobility group box protein 1 (HMGB1), which is released by apoptotic MLO-Y4 osteocytic cells [59]. HMGB1 is a pro-inflammatory cytokine that can activate the receptor for advanced glycation end products (RAGE) and the Toll-like receptor [60]. HMGB1 induces osteoclastogenesis by activating RAGE [61]. In addition, HMGB1 is chemotactic for osteoclasts [62], increases RANKL/OPG mRNA ratio, and decreases OPG release to the cell culture media [59]. Further studies are required to identify the molecules responsible for the association of apoptotic osteocytes and osteoclast recruitment in targeted bone resorption in vivo.
Conclusion
Studies from the past 2 decades have underscored the relevance of osteocyte viability for the maintenance of bone structure and strength. Targeted osteoclast recruitment and localized bone resorption results in the removal of the “dead” bone, allowing for renewal of the skeleton and maintenance of bone strength. Conversely, both increased osteocyte apoptosis and its inhibition throughout life result in defective bone, with increased porosity, altered bone geometry and deficient mechanical properties. Although advances have been made in understanding the relationship between osteocyte death and the recruitment of osteoclasts, the main effectors of this association remain unknown. Complete understanding of the relationship between osteocyte apoptosis and bone remodelling may lead to the development of improved therapies to maintain bone health.
Acknowledgments
This research was supported by National Institutes of Health (R01-AR053643) and by a Biomedical Research Grant and a Developing Diverse Researchers with InVestigative Expertise (DRIVE) Grant from Indiana University School of Medicine.
Footnotes
Conflict of Interest
LI Plotkin declares no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by Lilian Plotkin involving murine samples were performed after approval by the Institutional Animal Care and Use Committees of University of Arkansas for Medical Sciences and Indiana University School of Medicine.
References
Papers of particular interest, published recently have been highlighted as:
* Of importance
** Of major importance
- 1.Bellido T. Osteocyte-Driven Bone Remodeling. Calcif Tissue Int. 2013 doi: 10.1007/s00223-013-9774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noble BS, Reeve J. Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol. 2000;159:7–13. doi: 10.1016/s0303-7207(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 3.Frost HM. In vivo osteocyte death. J Bone Joint Surg Am. 1960;42-A:138–143. [PubMed] [Google Scholar]
- 4.Almeida M, Han L, Martin-Millan M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285–27297. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomkinson A, Reeve J, Shaw RW, et al. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab. 1997;82:3128–3135. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- 6.Kousteni S, Bellido T, Plotkin LI, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- 7.Weinstein RS, Jilka RL, Parfitt AM, et al. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguirre JI, Plotkin LI, Stewart SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Min Res. 2006;21:605–615. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 9.Morse LR, Xu Y, Solomon B, et al. Severe Spinal Cord Injury Causes Immediate Multicellular Dysfunction at the Chondro-Osseous Junction. Transl Stroke Res. 2011;2:643–650. doi: 10.1007/s12975-011-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verborgt O, Gibson G, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Min Res. 2000;15:60–67. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 11.Jilka RL, Weinstein RS, Bellido T, et al. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plotkin LI, Weinstein RS, Parfitt AM, et al. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–1374. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology. 1999;140:5339–5347. doi: 10.1210/endo.140.11.7135. [DOI] [PubMed] [Google Scholar]
- 14.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010;21:369–374. doi: 10.1016/j.tem.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jilka RL, Bellido T, Almeida M, Plotkin LI, O’Brien CA, Weinstein RS, Manolagas SC. Apoptosis in bone cells. In: Bilezikian JP, Raisz LG, Martin TJ, editors. Principles of Bone Biology. Academic Press; San Diego, San Francisco, New York, London, Sydney, Tokyo: 2008. pp. 237–261. [Google Scholar]
- 16**.Jilka RL, O’Brien CA, Roberson PK, et al. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates bone porosity with age. J Bone Miner Res. 2014;29:103–117. doi: 10.1002/jbmr.2007. Reports the effect of preventing osteocyte apoptosis on gene expression and bone remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85:2907–2912. doi: 10.1210/jcem.85.8.6714. [DOI] [PubMed] [Google Scholar]
- 18.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: evidence for inside-out signaling leading to anoikis. J Biol Chem. 2007;282:24120–24130. doi: 10.1074/jbc.M611435200. [DOI] [PubMed] [Google Scholar]
- 19.Canalis E, Delany AM. Mechanisms of glucocorticoid action in bone. Ann N Y Acad Sci. 2002;966:73–81. doi: 10.1111/j.1749-6632.2002.tb04204.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang FS, Lin CL, Chen YJ, et al. Secreted frizzled-related protein 1 (SFRP1) modulates glucocorticoid attenuation of osteogenic activities and bone mass. Endocrinology. 2005;146:2415–2423. doi: 10.1210/en.2004-1050. [DOI] [PubMed] [Google Scholar]
- 21.Almeida M, Han L, Ambrogini E, et al. Glucocorticoids and tumor necrosis factor (TNF) alpha increase oxidative stress and suppress WNT signaling in osteoblasts. J Biol Chem. 2011;286:44326–44335. doi: 10.1074/jbc.M111.283481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia J, Yao W, Guan M, et al. Glucocorticoid dose determines osteocyte cell fate. FASEB J. 2011;25:3366–3376. doi: 10.1096/fj.11-182519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinstein RS, Wan C, Liu Q, et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in 21-month-old mice. Aging Cell. 2009;9:147–161. doi: 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noble BS, Peet N, Stevens HY, et al. Mechanical loading: biphasic osteocyte survival and the targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol. 2003;284:C934–C943. doi: 10.1152/ajpcell.00234.2002. [DOI] [PubMed] [Google Scholar]
- 25.Lin C, Jiang X, Dai Z, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–1661. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 26.Lin C, Jiang X, Dai Z, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–1661. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 27.Verborgt O, Tatton NA, Majeska RJ, et al. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res. 2002;17:907–914. doi: 10.1359/jbmr.2002.17.5.907. [DOI] [PubMed] [Google Scholar]
- 28.Bellido T, Ali AA, Plotkin LI, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–50272. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- 29.Bivi N, Lezcano V, Romanello M, et al. Connexin43 interacts with arrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem. 2011;112:2920–2930. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers MJ, Crockett JC, Coxon FP, et al. Biochemical and molecular mechanisms of action of bisphosphonates. Bone. 2011;49:34–41. doi: 10.1016/j.bone.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277:8648–8657. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 32.Plotkin LI, Lezcano V, Thostenson J, et al. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res. 2008;23:1712–1721. doi: 10.1359/JBMR.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plotkin LI, Bivi N, Bellido T. A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone. 2011;49:122–127. doi: 10.1016/j.bone.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plotkin LI, Aguirre JI, Kousteni S, et al. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of ERK activation. J Biol Chem. 2005;280:7317–7325. doi: 10.1074/jbc.M412817200. [DOI] [PubMed] [Google Scholar]
- 35.Plotkin LI, Manolagas SC, Bellido T. Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone. 2006;39:443–452. doi: 10.1016/j.bone.2006.02.060. [DOI] [PubMed] [Google Scholar]
- 36.Tomkinson A, Gevers EF, Wit JM, et al. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Min Res. 1998;13:1243–1250. doi: 10.1359/jbmr.1998.13.8.1243. [DOI] [PubMed] [Google Scholar]
- 37.Kousteni S, Chen JR, Bellido T, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 38.Huber C, Collishaw S, Mosley JR, et al. Selective estrogen receptor modulator inhibits osteocyte apoptosis during abrupt estrogen withdrawal: implications for bone quality maintenance. Calcif Tissue Int. 2007;81:139–144. doi: 10.1007/s00223-007-9049-6. [DOI] [PubMed] [Google Scholar]
- 39.Kousteni S, Han L, Chen JR, et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Essen HW, Holzmann PJ, Blankenstein MA, et al. Effect of raloxifene treatment on osteocyte apoptosis in postmenopausal women. Calcif Tissue Int. 2007;81:183–190. doi: 10.1007/s00223-007-9050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plotkin LI, Mathov I, Aguirre JI, et al. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases and ERKs. Am J Physiol Cell Physiol. 2005;289:C633–C643. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- 42.Aguirre JI, Plotkin LI, Gortazar AR, et al. A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem. 2007;282:25501–25508. doi: 10.1074/jbc.M702231200. [DOI] [PubMed] [Google Scholar]
- 43.Kitase Y, Barragan L, Jiang JX, et al. Mechanical induction of PGE(2) in osteocytes blocks glucocorticoid induced apoptosis through both the beta-catenin and PKA pathways. J Bone Miner Res. 2010;25:2657–2668. doi: 10.1002/jbmr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frost HM. Bone’s mechanostat: A 2003 update. Anat Rec. 2003;275A:1081–1101. doi: 10.1002/ar.a.10119. [DOI] [PubMed] [Google Scholar]
- 45.Noble BS, Stevens H, Loveridge N, et al. Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone. 1997;20:273–282. doi: 10.1016/s8756-3282(96)00365-1. [DOI] [PubMed] [Google Scholar]
- 46.Elmardi AS, Katchburian MV, Katchburian E. Electron microscopy of developing calvaria reveals images that suggest that osteoclasts engulf and destroy osteocytes during bone resorption. Calcif Tissue Int. 1990;46:239–245. doi: 10.1007/BF02555002. [DOI] [PubMed] [Google Scholar]
- 47.Boabaid F, Cerri PS, Katchburian E. Apoptotic bone cells may be engulfed by osteoclasts during alveolar bone resorption in young rats. Tissue Cell. 2001;33:318–325. doi: 10.1054/tice.2001.0179. [DOI] [PubMed] [Google Scholar]
- 48.Cerri PS, Boabaid F, Katchburian E. Combined TUNEL and TRAP methods suggest that apoptotic bone cells are inside vacuoles of alveolar bone osteoclasts in young rats. J Periodontal Res. 2003;38:223–226. doi: 10.1034/j.1600-0765.2003.02006.x. [DOI] [PubMed] [Google Scholar]
- 49.Cardoso L, Herman BC, Verborgt O, et al. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emerton KB, Hu B, Woo AA, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2009;46:577–583. doi: 10.1016/j.bone.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Bivi N, Condon KW, Allen MR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Min Res. 2012;27:374–389. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lloyd SA, Loiselle AE, Zhang Y, et al. Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone. 2013;57:76–83. doi: 10.1016/j.bone.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaffler MB, Cheung WY, Majeska R, et al. Osteocytes: Master Orchestrators of Bone. Calcif Tissue Int. 2013 doi: 10.1007/s00223-013-9790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Kennedy OD, Herman BC, Laudier DM, et al. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–1122. doi: 10.1016/j.bone.2012.01.025. Reports the temporal relationships between injury, osteocyte apoptosis and pro-osteoclastogenic signaling following excess loading. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu AC, Morrison NA, Kelly WL, et al. MCP-1 expression is specifically regulated during activation of skeletal repair and remodeling. Calcif Tissue Int. 2013;92:566–575. doi: 10.1007/s00223-013-9718-6. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Paul EM, Sathyendra V, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localised bone destruction. J Bone Miner Res. 2008;23:915–927. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 59.Yang J, Shah R, Robling AG, et al. HMGB1 is a bone-active cytokine. J Cell Physiol. 2008;214:730–739. doi: 10.1002/jcp.21268. [DOI] [PubMed] [Google Scholar]
- 60.Klune JR, Dhupar R, Cardinal J, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Z, Han JY, Xi CX, et al. HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J Bone Miner Res. 2008;23:1084–1096. doi: 10.1359/JBMR.080234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taniguchi N, Yoshida K, Ito T, et al. Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol. 2007;27:5650–5663. doi: 10.1128/MCB.00130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]