

Abstract
AGAP2 [Arf (ADP-ribosylation factor) GAP (GTPase-activating protein) with GTP-binding-protein-like, ankyrin repeat and PH (pleckstrin homology) domains] is a multidomain Arf GAP that was shown to promote the fast recycling of transferrin receptors. In the present study we tested the hypothesis that AGAP2 regulates the trafficking of β2-adrenergic receptors. We found that AGAP2 formed a complex with β-arrestin1 and β-arrestin2, proteins that are known to regulate β2-adrenergic receptor signalling and trafficking. AGAP2 co-localized with β-arrestin2 on the plasma membrane, and knockdown of AGAP2 expression reduced plasma membrane association of β-arrestin2 upon β2-adrenergic receptor activation. AGAP2 also co-localized with internalized β2-adrenergic receptors on endosomes, and overexpression of AGAP2 slowed accumulation of β2-adrenergic receptor in the perinuclear recycling endosomes. In contrast, knockdown of AGAP2 expression prevented the recycling of the β2-adrenergic receptor back to the plasma membrane. In addition, AGAP2 formed a complex with endogenous ERK (extracellular-signal-regulated kinase) and overexpression of AGAP2 potentiated ERK phosphorylation induced by β2-adrenergic receptors. Taken together, these results support the hypothesis that AGAP2 plays a role in the signalling and recycling of β2-adrenergic receptors.
Keywords: AGAP2, β-arrestin2, Arf GTPase-activating protein, endocytosis, G-protein-coupled receptor, recycling endosome
INTRODUCTION
β2ARs (β2-adrenergic receptors) are members of the GPCR (G-protein-coupled receptor) superfamily and couple to both Gs and Gi proteins [1,2]. Agonist binding to β2ARs induces conformational changes in the receptors which function as guanine-nucleotide-exchange factors for the heterotrimeric G-proteins that, in turn, activate downstream signalling via generation of soluble second messengers such as cAMP. β2ARs, similar to many other GPCRs, are regulated by two groups of proteins: GRKs (GPCR kinases) and β-arrestins. Upon receptor activation, GRKs translocate to the plasma membrane to phosphorylate agonist-occupied activated β2ARs. Phosphorylated β2ARs exhibit a higher affinity for β-arrestins than for G-proteins, therefore the receptors are uncoupled from G-proteins [3].
In addition to physically hindering GPCR binding to G-proteins, β-arrestins play additional roles to dampen signalling from GPCRs. For example, β-arrestins can recruit phosphodiesterase to the vicinity of activated β2ARs to accelerate the degradation of cAMP [4]. Another mechanism of β-arrestin-mediated regulation of β2AR signalling is receptor internalization. In that sense, β-arrestins were found to bind to the critical components of the endocytic machinery that are required for β2AR internalization [5].
β2ARs, together with other GPCRs, have been shown to undergo constitutive and agonist-induced internalization [6]. Agonist-induced β2AR internalization is largely mediated through clathrin-coated vesicles. The clathrin adaptor protein AP2 is a critical component of clathrin-dependent endocytosis in that AP2 binds both cargo and clathrin so that AP2 links cargo selection and clathrin assembly [7]. β-Arrestins were found to bind both AP2 and clathrin [8,9], consistent with a role for β-arrestins in promoting clathrin-dependent endocytosis of GPCRs.
Arfs (ADP-ribosylation factors) are small GTP-binding proteins that regulate protein trafficking and actin remodelling [10]. Hydrolysis of GTP bound to Arf proteins is catalysed by a family of enzymes termed Arf GAPs (GTPase-activating proteins) [11,12]. Emerging evidence has suggested the involvement of Arf and Arf GAPs in GPCR trafficking. For example, knockdown of Arf6 with siRNA (small interfering RNA) reduced β2AR endocytosis [13]. Similarly, overexpression of GIT1, an Arf GAP that promotes GTP hydrolysis on Arf, also inhibited β2AR endocytosis [14,15]. It has been shown that ubiquitination of β-arrestin2 upon β2AR activation is essential for β2AR endocytosis [16]. Similarly, S-nitrosylation of β-arrestin2 is also involved in β2AR endocytosis [17], demonstrating the complexity of the regulatory mechanisms of β2AR signalling and trafficking.
AGAP [Arf GAP with GTP-binding-protein-like, ankyrin repeat and PH (pleckstrin homology) domains] 2 also referred to as PIKE-A (phosphatidylinositol 3-kinase enhancer A), was originally identified as being amplified in glioblastoma [18], and subsequent studies suggest its up-regulation in many other human cancers [19]. AGAP2 was shown to bind activated Akt and to promote invasion of glioblastoma cells [18]. AGAP2 was also shown to transform NIH 3T3 cells and to protect glioblastoma cells from apoptosis [19]. We have shown that AGAP2 binds the clathrin adaptor protein AP1 and regulates recycling of transferrin receptors [20], and AGAP2 interacts with the focal adhesion kinase to regulate focal adhesion remodelling [21]. In the present paper, we report that AGAP2 forms a complex with β-arrestin and regulates recycling of β2ARs. AGAP2 co-localized with overexpressed β2AR, and reduced expression of endogenous AGAP2 trapped β2AR in the perinuclear region. Functionally, forced overexpression of AGAP2 potentiated β2AR-induced activation of ERK (extracellular-signal-regulated kinase). Therefore AGAP2 may play a role in β2AR signalling and trafficking.
MATERIALS AND METHODS
Reagents
Anti-FLAG and anti-HA (haemagglutinin) antibodies were purchased from Sigma; anti-ERK antibody was from Santa Cruz Biotechnology; anti-phospho-ERK and anti-β-arrestin1/2 antibodies were from Cell Signaling Technology; anti-LAMP1 (lysosome-associated membrane protein 1) antibody was from Hybridoma Bank. FITC- and rhodamine-conjugated secondary antibodies were from Jackson ImmunoResearch, and rhodamine-conjugated phalloidin was from Invitrogen. shRNAs (short hairpin RNAs) targeting AGAP2 were from OpenBiosystem and the generation of the HEK (human embryonic kidney)-293 cell line with stable overexpression or knockdown of AGAP2 has been described previously [21]. β-Arrestin1–HA and β-arrestin2–HA expression vectors and anti-β-arrestin antibody were provided by Dr Y. Daaka (Department of Urology, University of Florida, Gainesville, FL, U.S.A.); FLAG–β2AR cDNA was provided by Dr Y. Xiang (Department of Molecular and Integrative Physiology, University of Illinois, Urbana, IL, U.S.A.); GFP (green fluorescent protein)–β2AR plasmid was provided by Dr J.L. Benovic (Biochemistry and Molecular Biology, Thomas Jefferson University Medical Center, Philadelphia, PA, U.S.A.); GST (glutathione transferase)-β-arrestin2 cDNA was provided by Dr M.G. Scott (INSERM, Institut Cochin, Paris, France); His6–AGAP2 protein was provided by Dr P.A. Randazzo (Center for Cancer Research, National Cancer Institute, Bethesda, MD, U.S.A.). Lipofectamine™ 2000 was from Invitrogen. FBS (fetal bovine serum) was from HyClone, and DMEM (Dulbecco’s modified Eagle’s medium) and penicillin/streptomycin were from Mediatech. ISO (isoprenaline, also known as isoproterenol), fibronectin, BSA and phosphatase inhibitor cocktails were from Sigma. Alprenolol was from Tocris and 125I-cyanopindolol was from PerkinElmer.
Cell culture, transfection and immunofluorescence
HEK-293 or U87 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Transfection of cDNA was performed using Lipofectamine™ 2000. For β2AR internalization, cells were starved in 0.2% FBS overnight, trypsinized, resuspended and reseeded on fibronectin (10 μg/ml)-coated coverslips for 6 h. Cells were then stimulated with ISO (1 μM) for 0–45 min as indicated and fixed. For β2AR recycling, cells were treated with ISO (1 μM) for 10 min, washed twice with PBS and once with medium, and then incubated in medium containing 0.2% FBS for 15 or 30 min, and harvested for plasma membrane preparation or fixed. Cells were processed for immunofluorescence staining with appropriate antibodies and the slides were examined using a Leica Confocal Microscope (TCS SP5) equipped with a × 63/1.4 numerical aperture oilimmersion lens. Images were captured and analysed using the application suite Advanced Fluorescence 2.0.2 software (Leica).
Immunoprecipitation and Western blot analysis
Cells were washed with PBS and lysed in lysis buffer [25 mM Tris/HCl, pH 8.0, 100 mM NaCl, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 1 mM EDTA, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 2 μg/ml pepstatin A]. Cell lysates were cleared by centrifugation and incubated with anti-FLAG antibodies overnight for immunoprecipitation, followed by incubation with Protein A/G beads for 1 h at 4°C. Protein A/G beads were washed with lysis buffer and immunoprecipitated proteins were boiled in SDS/PAGE sample buffer. Samples were resolved by SDS/PAGE (10 or 15% gel) followed by Western blot. For detection of phosphorylated ERK, phosphatase inhibitors (Cocktail 2 and 3) and sodium orthovanadate (1 mM) were added to the lysis buffer and BSA was used instead of non-fat dried skimmed milk powder for blocking and primary antibody incubation. Densitometry was performed using ImageJ software (NIH).
In vitro binding between AGAP2 and β-arrestin2
GST–β-arrestin2 was expressed and purified using glutathione–Sepharose 4B gel. His6–AGAP2 (150 nM) and GST–β-arrestin2 (500 nM) were incubated at room temperature (22–25°C) for 1 h in a binding buffer containing 50 mM Tris/HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM MgCl2, 1 mM DTT (dithiothreitol) and 0.1% Triton X-100. PA (phosphatidic acid; 360 μM), PtdIns(4,5)P2 (45 μM) or PtdIns(3,4,5)P3 (10 μM) was included in binding reactions as indicated to examine the effect of phospholipids on protein interactions. The glutathione–Sepharose 4B gel was washed three times with the binding buffer and the proteins that remained bound to the gel were resolved by SDS/PAGE (10% gel) and visualized by Coomassie Blue staining.
Radioligand receptor binding
Membrane preparation and receptor binding were performed as described previously [22,23]. Briefly, HEK-293 cells with or without stable AGAP2 knockdown were starved overnight and stimulated with ISO for 10 min. Cells were washed with PBS three times and harvested in binding buffer containing 50 mM Tris/HCl, pH7.4, 12.5 mM MgCl2, 2 mM EDTA and protease inhibitors (1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 2 μg/ml pepstatin A). Cells were lysed by a Dounce homogenizer with ten strokes. For receptor recycling, cells were incubated in medium for 30 min after removal of ISO. Cell lysates were centrifuged at 1000 g for 5 min to remove debris and organelles. The supernatants were centrifuged at 40000 g for 15 min and the resulting pellets were washed three times and resuspended in lysis buffer. For receptor binding, 25μg of cell membrane preparation were incubated in binding buffer containing 60 pM 125I-cyanopindolol at 37°C for 1 h. Non-specific binding was determined in the presence of 10 μM alprenolol. Membranes were collected and washed using a cell harvester (Brandel) and the bound radioactivity was quantified using a gamma counter (PerkinElmer).
Statistical analysis
Experiments were repeated at least three times and data are presented as the means ± S.E.M. or as representative images. Statistical significance was calculated by one-way ANOVA with Tukey’s post-hoc test. Graphs were generated using Prism software (GraphPad) and axis labels were generated using Adobe Illustrator.
RESULTS
AGAP2 interacts with β-arrestin2
In order to identify interacting proteins for AGAP2, we overexpressed FLAG–AGAP2 in HEK-293 cells, and looked for binding partners for AGAP2 by immunoprecipitation. We found that β-arrestin proteins co-immunoprecipitated with AGAP2 (Figure 1A). AGAP2 belongs to the multidomain family of Arf GAPs with similar domain structures, and we examined the selectivity of the interaction between AGAP2 and β-arrestins. FLAG–AGAP1 co-immunoprecipitated β-arrestins to a lesser extent than FLAG–AGAP2, whereas ACAP1 (Arf GAP with coiled-coil domains, ankyrin repeats and PH domains 1) and ASAP1 [Arf GAP with SH3 (Src homology 3) domain, ankyrin repeats and PH domains 1] failed to co-immunoprecipitate β-arrestins (Figure 1B), suggesting that AGAP2 has a higher selectivity for interaction with β-arrestins.
Figure 1. AGAP2 formed a complex with β-arrestins.
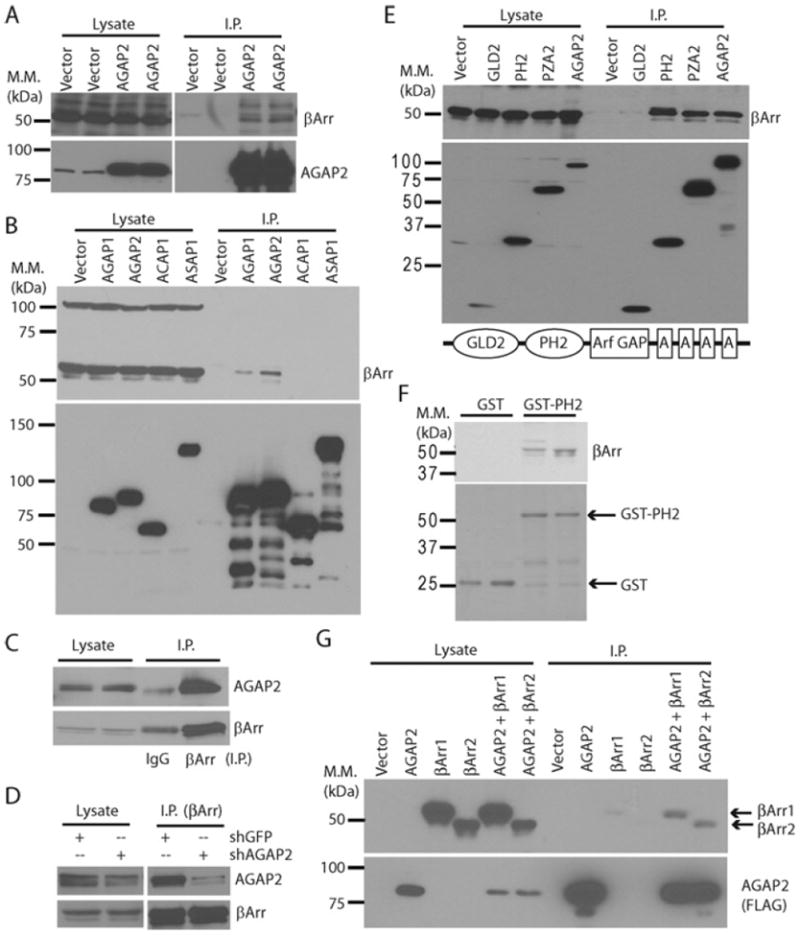
(A) Overexpressed AGAP2 co-immunoprecipitated endogenous β-arrestins. HEK-293 cells were transfected with pcDNA3.1 (Vector) or FLAG–AGAP2 expression vector and the overexpressed FLAG–AGAP2 was immunoprecipitated. The presence of FLAG–AGAP2 or endogenous β-arrestin proteins in cell lysates or immunoprecipitates was examined by Western blotting. (B) AGAP2 bound β-arrestins better than AGAP1. HEK-293 cells were transfected with emtpy vector, or with the expression vectors for FLAG-tagged AGAP1, AGAP2, ACAP1 or ASAP1. Overexpressed proteins were immunoprecipiatated and co-immunoprecipitation of β-arrestins was examined by Western blotting. (C) Co-immunoprecipitation of endogenous AGAP2 with β-arrestin. Immunoprecipitation was performed using anti-β-arrestin antibody or IgG, and precipitated β-arrestins and AGAP2 were examined by Western blotting. (D) Decreased expression of AGAP2 affected its co-immunoprecipitation with β-arrestin. Endogenous β-arrestins were immunoprecipitated from HEK-293 cells with or without stable knockdown of AGAP2. Immunoprecipiated AGAP2 and β-arrestins were examined by Western blotting. (E) AGAP2 interacted with β-arrestins through the PH domain. Expression vectors for FLAG-tagged AGAP2 or its deletion mutants were transfected into HEK-293 cells. Proteins were immunoprecipitated and examined by Western blotting. Schematic diagram of AGAP2 domain structure is shown at the bottom. A, ankyrin repeat. (F) Binding of β-arrestins to the PH2 domain. GST or GST fused to the isolated PH domain of AGAP2 (GST–PH2) were purified and incubated with HEK-293 cell lysates at 4°C overnight. Beads were washed three times and bound proteins were resolved by SDS/PAGE (15% gel) followed by Coomassie Blue staining (bottom panel) or Western blotting (top panel). (G) AGAP2 formed a complex with both β-arrestin1 and β-arrestin2. β-Arrestin1–HA (βArr1) or β-arrestin2–HA (βArr2) were transfected alone or in combination with FLAG–AGAP2. Overexpressed proteins were immunoprecipitated using anti-FLAG antibody, and co-immunoprecipitation of β-arrestins was examined by Western blotting using anti-HA antibody. I.P., immunoprecipitation. Molecular mass (M.M.) is shown on the left-hand side of the Western blots in kDa.
We next examined whether the interaction can be detected with endogenous proteins, and whether the interaction is affected by the level of protein expression. First, we examined the co-immunoprecipitation of AGAP2 with β-arrestins. As shown in Figure 1(C), endogenous AGAP2 co-immunoprecipitated with endogenous β-arrestins, whereas rabbit IgG failed to precipitate either β-arrestins or AGAP2 (note that the IgG heavy chain migrated at a similar rate as did β-arrestins, but the IgG heavy chain exhibited as a single band, whereas β-arrestins presented as double bands). Secondly, we examined whether the expression level of AGAP2 will affect the interaction between AGAP2 and β-arrestins. Endogenous β-arrestins were immunoprecipitated from HEK-293 cells with or without stable knockdown of AGAP2, and the level of β-arrestin immunoprecipitation was similar in control and AGAP2-knockdown cells (Figure 1D). β-Arrestins co-immunoprecipitated endogenous AGAP2 from control HEK-293 cells and the level of bound AGAP2 decreased in cells with stable knockdown of AGAP2 (Figure 1D). Therefore the interaction between AGAP2 and β-arrestins occurs with endogenous proteins.
Distinct domains of the AGAP subfamily of Arf GAPs have been shown to interact with other proteins. For example, the GLD (G-protein-like domain) of AGAP1 was recently shown to interact with Rho GTPases [24], and the PH domain of AGAP2 was shown to interact with AP1 [20]. We next tried to identify the domain of AGAP2 that mediates the interaction with β-arrestins. FLAG-tagged AGAP2 or its deletion mutants were overexpressed and immunoprecipitated. β-Arrestins were found to co-immunoprecipitate with AGAP2 proteins containing the PH2 domain, including the isolated PH2 domain, PZA2 (containing PH2, Arf GAP and ankyrin repeat domain, the suffix number 2 denoting domains from AGAP2) and AGAP2 (Figure 1E), suggesting that the PH2 domain is the primary site for interaction with β-arrestins. To provide further support for these results, we performed in vitro pull-down assays. As shown in Figure 1(F), GST by itself failed to bind β-arrestins, whereas the PH2 domain fused to GST (GST–PH2) successfully precipitated endogenous β-arrestins, supporting the notion that the PH2 domain of AGAP2 is the major binding site for β-arrestins.
As the antibody that we used to detect β-arrestin proteins can recognize both β-arrestin1 and β-arrestin2 (Figures 1B and 1E), we next examined interaction of AGAP2 with epitope-tagged β-arrestin1 or β-arrestin2. β-Arrestin1–HA or β-arrestin2–HA was overexpressed alone or together with FLAG–AGAP2, and the overexpressed proteins were immunoprecipitated with anti-FLAG antibody. As shown in Figure 1(G), either β-arrestin1 or β-arrestin2 co-immunoprecipitated with AGAP2. Therefore AGAP2 forms a complex with both β-arrestin1 and β-arrestin2.
PtdIns(4,5)P2 potentiates the binding between AGAP2 and β-arrestin2
The PH2 domain of AGAP2 binds phospholipids, and the function of AGAP2 was shown to be regulated by various phospholipids, including PtdIns(4,5)P2 and PtdIns(3,4,5)P3 [20]. We next examined whether the interaction between AGAP2 and β-arrestin2 is regulated by phospholipids. GST or GST–β-arrestin2 was incubated with His6–AGAP2 in the presence or absence of phospholipids and binding of His6–AGAP2 was examined by SDS/PAGE (10% gel) followed by gel staining. AGAP2 was shown to bind GST–β-arrestin2 but not GST, and the specific interaction between AGAP2 and β-arrestin2 required the presence of Triton X-100 micelles (Figure 2A), suggesting that the interaction of these two proteins is dependent on membrane. PtdIns(4,5)P2 increased the binding of AGAP2 to β-arrestin2 (Figure 2), whereas PtdIns(3,4,5)P3 was much less effective (Figure 2). PA did not enhance binding between AGAP2 and β-arrestin2 (Figure 2), and when presented in combination with PtdIns(4,5)P2, PA inhibited binding between AGAP2 and β-arrestin2 that was potentiated by PtdIns(4,5)P2 (Figure 2). Given that PA potentiated the activation of the GAP activity of AGAP2 by either PtdIns(4,5)P2 or PtdIns(3,4,5)P3 [20], it can be envisaged that distinct phospholipids and upstream signals elicit selective activation of AGAP2 function, i.e. GTP hydrolysis on Arf or association with β-arrestins.
Figure 2. PtdIns(4,5)P2 enhanced the interaction between AGAP2 and β-arrestin2.
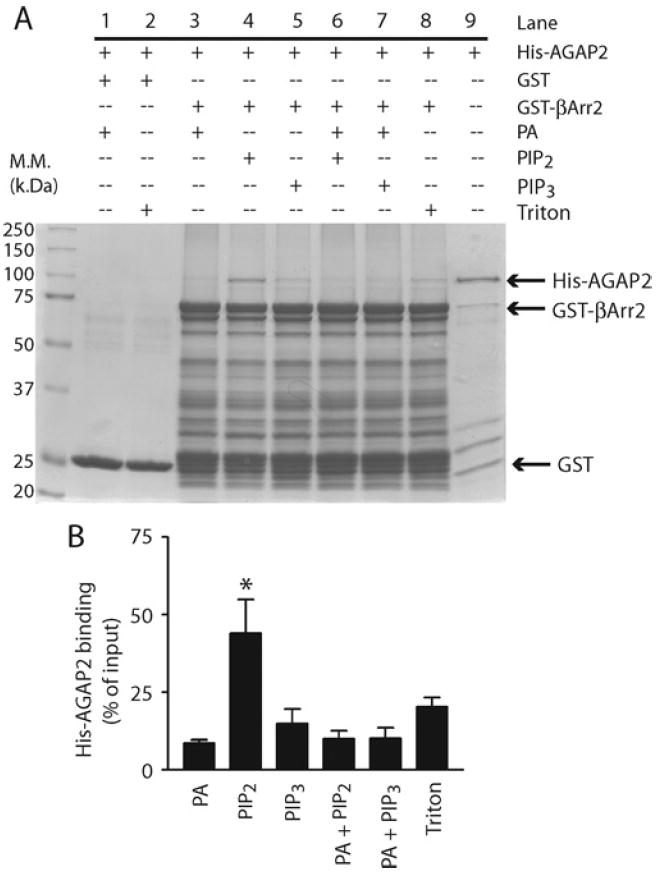
(A) Effect of phospholipids on AGAP2 binding to β-arrestin2 (βArr2). Purified His6–AGAP2 (150 nM) was incubated with GST–β-arrestin2 (500 nM) at room temperature for 1 h in the presence or absence of phospholipids at the following concentrations: PA, 360 μM; PtdIns(4,5)P2 (PIP2), 45 μM; PtdIns(3,4,5)P3 (PIP3), 10 μM; presented in micelles with 0.1% Trition X-100. Glutathione beads were washed three times and bound proteins were resolved by SDS/PAGE (10% gel) and visualized by Coomassie Blue staining. Molecular mass (M.M.) is shown on the left-hand side of the Western blot in kDa. (B) Quantification of His6–AGAP2 binding to GST–β-arrestin2. The binding assay was repeated three times and analysed by densitometry. *P < 0.05 compared with Triton X-100.
AGAP2 is involved in the regulation of β-arrestin2 membrane association
β-Arrestin proteins function to regulate signalling and trafficking of β-adrenergic receptors. Interaction with β-arrestin1 or β-arrestin2 suggests that AGAP2 has a potential role in β-adrenergic receptor signalling and/or trafficking. First, we examined the subcellular distribution of FLAG–AGAP2, since no antibody is available to detect endogenous AGAP2 by immunofluorescence. FLAG–AGAP2 is diffusely distributed in the cytosol (Figure 3A), and a fraction of FLAG–AGAP2 translocated to the plasma membrane (Figure 3D, arrows) upon β2AR activation by stimulation with the agonist ISO in HEK-293 cells that endogenously express only β2AR [25]. To examine the interaction of AGAP2 with β-arrestins in this process, we overexpressed β-arrestin2–HA because β-arrestin2 has a higher affinity for β2ARs than β-arrestin1 in cells. As can be expected, overexpressed β-arrestin2 is diffusely distributed in the cytosol (Figure 3B) and translocated to the plasma membrane upon β2AR activation (Figure 3E, arrows). Interestingly, AGAP2 and β-arrestin2 co-localized on the plasma membrane following activation of β2AR (Figure 3F, arrows).
Figure 3. AGAP2 affects plasma membrane association of β-arrestin2.
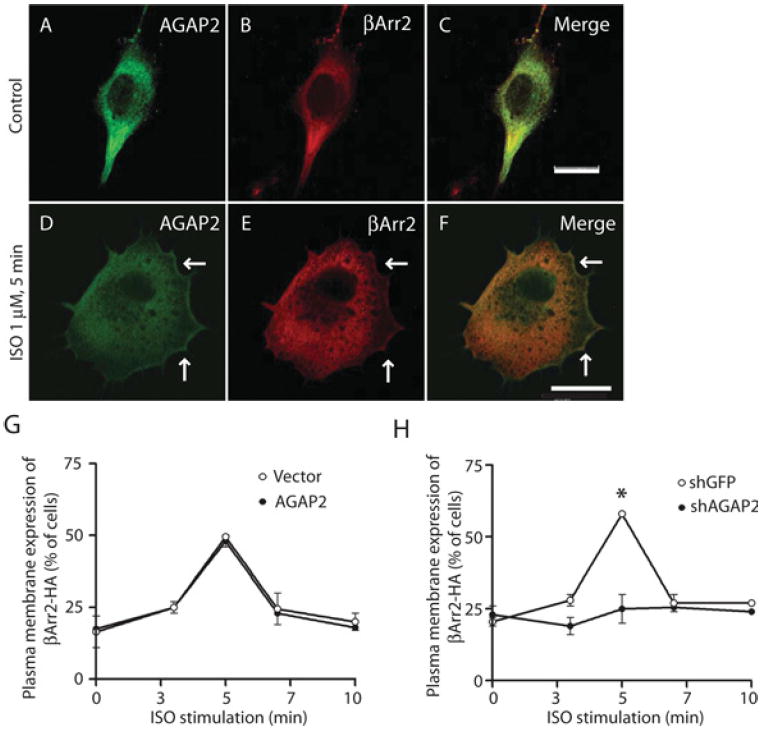
(A–C) Distribution of AGAP2 and β-arrestin2 (βArr2) in resting cells. FLAG–AGAP2 and β-arrestin2–HA expression vectors were transfected into HEK-293 cells for 24 h and immunofluorescence staining was performed to examine the intracellular distribution of FLAG–AGAP2 (A) and β-arrestin2–HA (B). (D–F) Co-localization of AGAP2 and β-arrestin2 upon β2AR activation. Cells were transfected as described above and stimulated with ISO (1 μM, 5 min) before being processed for immunofluorescence. Plasma membrane translocation of AGAP2 (D) or β-arrestin2 (E) is indicated by arrows, and co-localization of both proteins on the plasma membrane was shown in the merged picture (F, arrows). (G) AGAP2 overexpression did not affect plasma membrane association of β-arrestin2. The β-arrestin2–HA expression vector was transfected into HEK-293 cells that stably express pcDNA3.1 or FLAG–AGAP2. Cells were stimulated with ISO (1 μM) for the indicated times before being fixed and processed for immunofluorescence. Plasma membrane association of β-Arrestin2–HA was examined under the microscope. (H) Knockdown of AGAP2 inhibited plasma membrane association of β-arrestin2. β-Arrestin2–HA expression vector was transfected into HEK-293 cells that stably express GFP- or AGAP2-targeting shRNA with verified knockdown of protein expression. Plasma membrane association of β-arrestin2–HA was examined as described in (G). *P < 0.05. Scale bars, 20 μm.
We next examined whether AGAP2 plays a role in the plasma membrane translocation of β-arrestin2 upon β2AR activation. First, we overexpressed β-arrestin2–HA in HEK-293 cells that stably overexpress empty vector or FLAG–AGAP2. In cells stably transfected with empty vector, activation of β2AR resulted in a time-dependent membrane association of β-arrestin2–HA, which peaked at 5 min and returned to the basal level at 10 min following β2AR activation. In cells that stably overexpress FLAG–AGAP2, plasma membrane association of β-arrestin2–HA is virtually the same as that observed in empty-vector-expressing cells (Figure 3G). In cells stably transfected with GFP-targeting shRNA, β2AR activation induced time-dependent translocation of β-arrestin2 to the plasma membrane (Figure 3H). However, in cells with AGAP2 knockdown by shRNA, activation of β2AR failed to induce plasma membrane expression of β-arrestin2 beyond the basal level (Figure 3H). Similar results were obtained using U87 cells with stable overexpression or knockdown of AGAP2 (results not shown), suggesting that this effect of AGAP2 knockdown on β-arrestin2 is not cell-type specific. Therefore, AGAP2 may play a role in the plasma membrane association of β-arrestin2 as induced by β2AR activation.
AGAP2 plays a role in β2AR trafficking
β-Arrestin2 is known to regulate signalling and trafficking of β2ARs. Interaction of AGAP2 with β-arrestin2 suggests that AGAP2 may play a role in β2AR signalling and trafficking. We then examined whether AGAP2 co-localized with β2ARs. Transiently overexpressed GFP–β2AR was distributed to the plasma membrane (Figures 4A and 4D, arrows), with some receptors observed on the punctate structures near the nucleus. Stably overexpressed FLAG–AGAP2 was diffusely distributed in the cytosol, with formation of dorsal ruffle-like ring structures in approximately 20–30% of cells (Figures 4B and 4E, arrowhead). Upon stimulation with ISO for 10 min, GFP–β2ARs were internalized and were observed on punctate structures (Figures 4G and 4J), presumably endosomes. FLAG–AGAP2 also appeared on these punctate structures (Figures 4H and 4K) and co-localized with β2ARs (Figures 4I and 4L, yellowdots), suggesting that AGAP2 may affect β2AR trafficking.
Figure 4. AGAP2 co-localized with internalized β2AR.
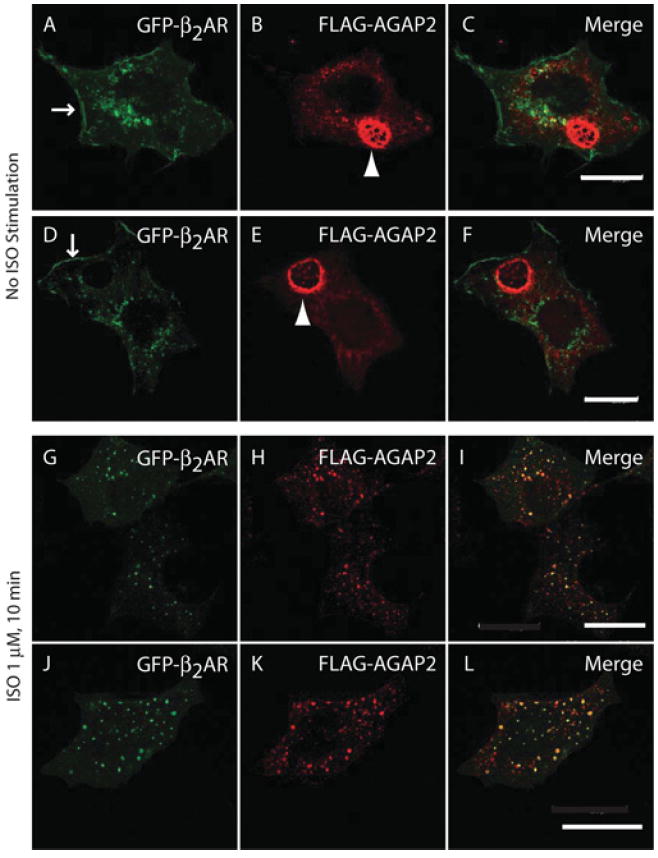
(A–F) Distribution of GFP–β2AR (A and D) and FLAG–AGAP2 (B and E) in HEK-293 cells. HEK-293 cells that stably express FLAG–AGAP2 were transfected with an expression vector for GFP–β2AR for 24 h. Cells were trypsinized and replated on fibronectin (10 μg/ml)-coated coverslips in DMEM with 0.2% FBS for 6 h. FLAG–AGAP2 was detected by staining with an anti-FLAG antibody. GFP–β2AR was distributed on the plasma membrane (A and D; arrows) and intracellular punctate structures. FLAG–AGAP2 was diffusely distributed in the cytosol and formed ring-like structures (B and E; arrowhead). (G–L) AGAP2 co-localized with internalized β2AR. Cells were treated with ISO (1 μM, 10 min) and stained using an anti-FLAG antibody. Both GFP–β2AR (G and J) and FLAG–AGAP2 (H and K) were distributed and co-localized on the punctate structures (I and L). Scale bars, 20 μm.
To explore the role for AGAP2 in β2AR trafficking, we examined the internalization pattern of β2AR in cells with AGAP2 overexpression or knockdown. First, we tested whether overexpression of AGAP2 affected β2AR internalization. In HEK-293 cells stably transfected with the empty vector pcDNA3.1, transiently overexpressed GFP–β2AR was distributed on the plasma membrane under resting conditions (Figures 5A and 5B). Stimulation with ISO for 5 min resulted in internalization of GFP–β2ARs, which were distributed on punctate structures throughout the cytosol (Figures 5C and 5D). At 15 min of ISO stimulation, most of the GFP–β2ARs were concentrated around the nucleus, reflecting the perinuclear recycling endosomes (Figures 5E and 5F, arrows). Longer stimulation (30 and 45 min) led to recycling of the receptors. Although GFP–β2AR puncta were still found in the perinuclear region (Figures 5G–5J, arrows), some receptors could be detected near the cell periphery (Figures 5G–5J, arrowhead). In cells overexpressing AGAP2, the pattern of GFP–β2AR internalization was different. GFP–β2ARs were present on the plasma membrane without receptor activation (Figures 5K and 5L). Stimulation with ISO from 5 to 30 min failed to induce apparent accumulation of GFP–β2ARs at the perinuclear region (Figures 5M–5R), which was only obvious at 45 min after receptor activation (Figures 5S and 5T, arrow). One possible explanation for the delayed arrival of β2ARs at the perinuclear recycling endosomes is that AGAP2 facilitated fast recycling of GFP–β2ARs from early endosomes to the plasma membrane, consistent with the reported role for AGAP2 on the fast recycling of transferrin receptors [20].
Figure 5. AGAP2 regulated intracellular distribution of β2AR.
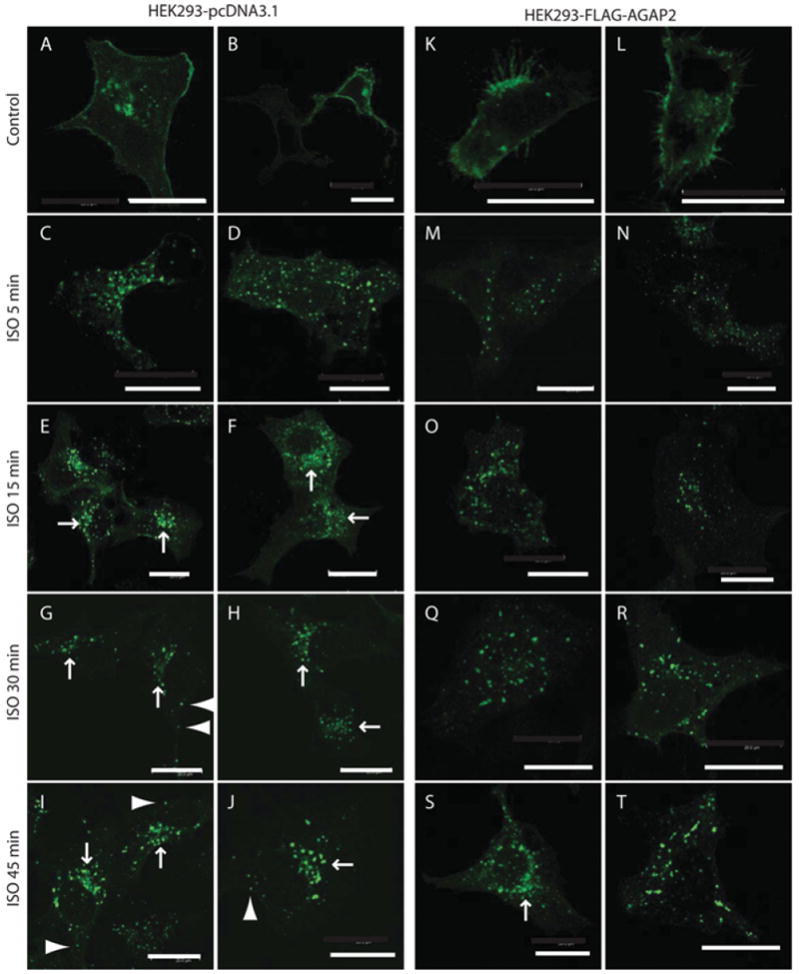
(A–J) Distribution of GFP–β2AR in HEK-293 cells that are stably transfected with the pcDNA3.1 expression vector. GFP–β2ARs were detected on the plasma membrane in resting cells (A and B). ISO stimulation for 5 min resulted in internalization of GFP–β2ARs (C and D), and the receptors reached the perinuclear recycling endosomes by 15 min (E and F; arrows). At 30 min (G and H) and 45 min (I and J), GFP–β2ARs were still observed in the perinuclear region (as indicated by the arrows), but a fraction of GFP–β2ARs already recycled back to the periphery of the cells (arrowhead). (K–T) Distribution of GFP–β2AR in HEK-293 cells that stably overexpress FLAG–AGAP2. GFP–β2ARs distributed on the plasma membrane in resting cells (K and L). Receptor activation by ISO from 5 min to 30 min resulted in distribution of GFP–β2AR on the endosomes throughout the cytosol (M–R). At 45 min, perinuclear concentration of GFP–β2AR could be observed (S, arrow). Scale bars, 20 μm.
Next, we examined the effect of AGAP2 knockdown on the internalization of GFP–β2ARs. In HEK-293 cells stably overexpressing shGFP, FLAG–β2AR was distributed on the plasma membrane (Figures 6A and 6B), and ISO stimulation resulted in FLAG–β2AR internalization (Figures 6C and 6D, and results not shown) that was similar to the internalization pattern observed in HEK-293-pcDNA3.1 cells. In cells with stable knockdown of AGAP2, FLAG–β2AR was found at the plasma membranes in resting cells (Figures 6E and 6F) and were enriched in the perinuclear region at 15 min following receptor activation (Figures 6G and 6H), consistent with a potential role for AGAP2 in β2AR recycling.
Figure 6. AGAP2 regulated β2AR recycling.
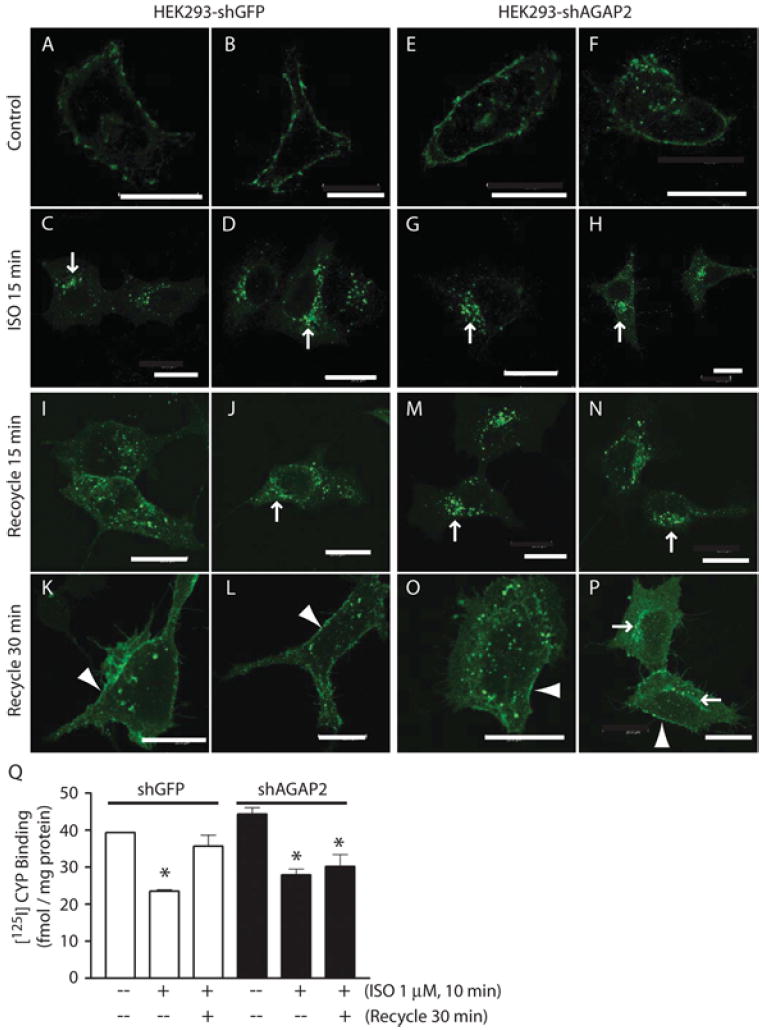
(A–D) Distribution of FLAG–β2AR in control HEK-293 cells. Cells were transfected with FLAG–β2AR cDNA for 24 h and replated on fibronectin-coated coverslips in DMEM containing 0.2% FBS for 6 h. FLAG–β2ARs, as stained using anti-FLAG antibody, were detected on the plasma membrane of resting cells (A and B) and were concentrated on the perinuclear recycling endosomes at 15 min of ISO stimulation (C and D, arrows). (E–H) Distribution of FLAG–β2AR in HEK-293 cells with stable knockdown of AGAP2. Cells were transfected with FLAG–β2AR cDNA and processed as described above. FLAG-tagged β2ARs were distributed on the plasma membrane of resting cells (E and F), and were enriched on the perinuclear recycling endosomes at 15 min of ISO stimulation (G and H, arrows). (I–L) Recycling of FLAG-β2AR in control HEK-293 cells. Cells were stimulated with ISO for 10 min, washed and incubated with DMEM containing 0.2% FBS for 15 min (I and J) or 30 min (K and L). At 15 min, FLAG-tagged β2ARs were recycling from the perinuclear region to the periphery and by 30 min they reached the plasma membrane (K and L, arrowhead). (M–P) Recycling of FLAG–β2ARs in HEK-293 cells with stable knockdown of AGAP2. At 15 min of recycling, most of the FLAG–β2AR signals were still present in the perinuclear region (M and N, arrows). By 30 min, FLAG-tagged β2ARs were present in perinuclear regions (P, arrows) or on endosomes evenly distributed throughout the cytosol (O). A small fraction of FLAG-tagged β2ARs reached plasma membrane (O and P, arrowhead). Scale bars, 20 μm. (Q) AGAP2 affected recycling of endogenous β2ARs. HEK-293 cells with or without stable AGAP2 knockdown were treated, or not, with ISO (1 μM, 10 min). Cells were washed and lysed to prepare plasma membrane, or washed and incubated with medium for an additional 30 min before plasma membrane preparation. The amount of β2ARs was determined by 125I-cyanopindolol binding and non-specific binding was determined using alprenolol. *P <0.05 compared with control.
To provide direct evidence for the involvement of AGAP2 in β2AR recycling, we treated the cells with ISO for 10 min, washed away ISO, and let the cells recover. In control cells, FLAG-tagged β2ARs were distributed on punctate structures in the cytosol, with only slight enrichment in the perinuclear region after 15 min of recycling (Figures 6I and 6J, arrow). However, in cells with AGAP2 knockdown, β2AR were mainly detected in the perinuclear region after 15 min of recovery (Figures 6M and 6N, arrows). At 30 min, most of the β2AR recycled back to the plasma membrane in control cells (Figures 6K and 6L, arrowhead), and knockdown of AGAP2 retarded the recycling of β2AR. At 30 min, a large fraction of β2ARs were still located on the punctate structures throughout the cytosol or in the perinuclear region (Figures 6O and 6P, arrows). Plasma membrane association of β2AR was only detected in limited regions (Figures 6O and 6P, arrowhead).
The effect of AGAP2 on β2AR recycling was further examined by radioligand receptor binding. Under resting conditions, plasma membrane β2AR levels were similar in HEK-293 cells with or without AGAP2 knockdown (Figure 6Q). ISO treatment resulted in a decrease in plasma membrane β2AR levels in both cells (Figure 6Q), consistent with receptor internalization. At 30 min following ISO removal, plasma membrane β2ARs returned to control levels in cells without AGAP2 knockdown (Figure 6Q). In cells with AGAP2 knockdown, however, plasma membrane β2AR levels remained decreased at 30 min of recovery, supporting a critical role for AGAP2 in β2AR recycling.
AGAP2 does not affect lysosomal targeting of β2ARs
Internalized β2ARs can be recycled to the plasma membrane for continual signalling, or can be targeted to lysosomes for degradation, leading to receptor down-regulation. We examined whether AGAP2 affected lysosomal targeting of β2ARs. In resting cells, the staining of the lysosomal marker LAMP1 was distributed in a compact area close to the nucleus (Figures 7A–7C) or on punctate structures throughout the cytosol (Figures 7G–7I). ISO treatment for 6 h resulted in extensive co-localization of β2AR with the lysosomal marker LAMP1 (Figures 7D–7F), suggesting that β2ARs were trafficked to lysosomes. In cells overexpressing FLAG–AGAP2, the pattern of β2AR co-localization with LAMP1 (Figures 7J–7L) was similar to that in control cells. Therefore AGAP2 is less likely to affect lysosomal targeting of β2ARs.
Figure 7. Overexpression of AGAP2 did not affect β2AR trafficking to lysosomes.
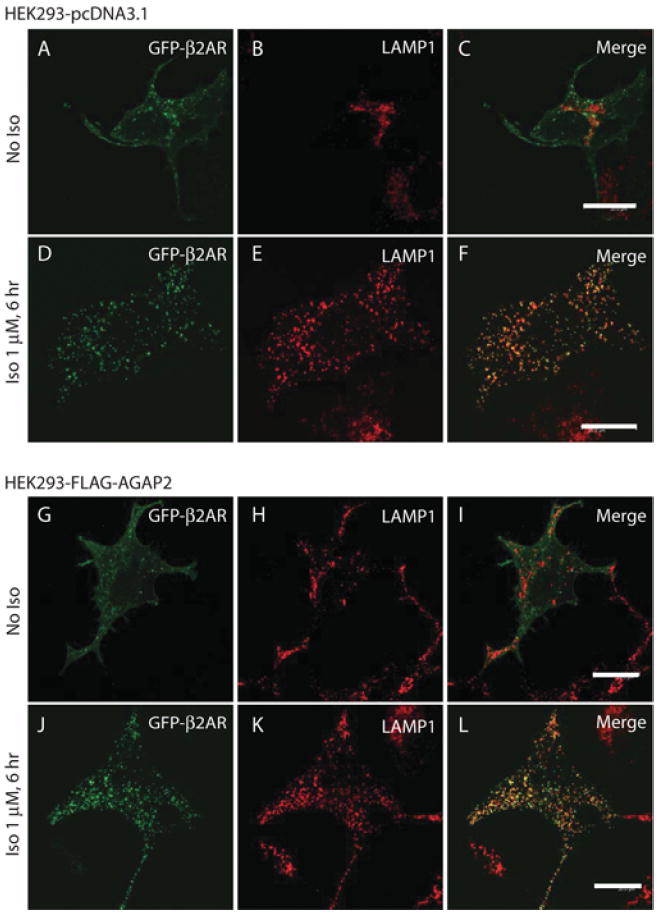
(A–F) Distribution of GFP–β2AR in control HEK-293 cells. HEK-293 cells stably expressing pcDNA3.1 vector were transfected with GFP–β2AR cDNA for 24 h, replated on fibronectin (10 μg/ml)-coated coverslips in DMEM containing 0.2% FBS for 6 h and treated with vehicle (No ISO) or ISO (1 μM) for 6 h. Cells were stained with anti-LAMP1 antibody. LAMP1 was observed in compact regions near the nucleus in resting cells (B). ISO treatment for 6 h resulted in internalization of GFP-tagged β2ARs which co-localized with LAMP1 (D–F), suggesting trafficking of GFP–β2AR to lysosomes. (G–L) Distribution of GFP–β2AR in HEK-293 cells with stable overexpression of FLAG–AGAP2. Cells were transfected and processed as described above. LAMP1 staining was observed in punctate structures near the nucleus and throughout the cytosol of the resting cells (H). In ISO treated cells, GFP-tagged β2ARs were internalized and co-localized with LAMP1 (J–L). Scale bars, 20 μm.
AGAP2 potentiates β2AR-induced activation of ERK
β2ARs have been shown to induce ERK activation (as determined by phosphorylation of ERK) that exhibits two peaks, the first one being G-protein-dependent, and the second one being β-arrestin-dependent [26,27]. Interaction with β-arrestins suggests that AGAP2 may affect β2AR-induced ERK activation. To test this possibility, β2AR was activated in HEK-293 cells that were stably transfected with empty vector or FLAG–AGAP2. Activation of β2ARs resulted in an increased level of phosphorylated ERK, suggesting its activation, and the time course of ERK phosphorylation showed two peaks (Figures 8A and 8B). In cells that overexpress AGAP2 (Figure 8A, bottom panel), phospho-ERK signals were higher than those in control cells (Figure 8A, top panel), whereas the expression level of ERK remained unchanged (Figure 8A, middle panel), suggesting that AGAP2 potentiated β2AR-induced ERK activation. Quantification of phospho-ERK signals by densitometry indicated that AGAP2 did not affect the first peak, but enhanced the second peak of ERK activation (Figure 8B), reinforcing the idea that AGAP2 is involved in β-arrestin-dependent signalling of β2AR.
Figure 8. AGAP2 enhanced β2AR-induced ERK activation.
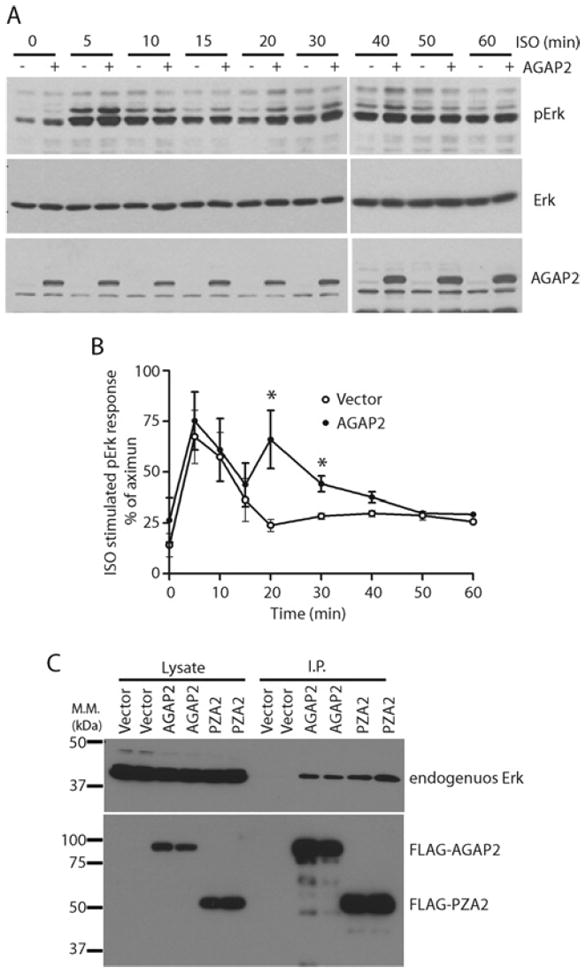
(A) AGAP2 overexpression enhanced β2AR-induced ERK activation. HEK-293 cells stably overexpressing empty vector or FLAG–AGAP2 were starved in Opti-MEM overnight and treated with ISO (10 μM) for the indicated times. Cell lysates were centrifuged and used for detection of phospho-ERK by Western blotting. Total ERK and FLAG–AGAP2 were also detected by Western blotting. (B) Quantification of phospho-ERK signals at different times by densitometry. *P < 0.05 compared with vector. (C) AGAP2 formed a complex with ERK. FLAG–AGAP2 or FLAG–PZA2 was transiently expressed in HEK-293 cells and the overexpressed proteins were immunoprecipitated using an anti-FLAG antibody. Co-immunoprecipitation of endogenous ERK was examined by Western blotting. I.P., immunoprecipitation. Molecular mass (M.M.) is shown on the left-hand side of the Western blots in kDa.
We next examined whether knockdown of AGAP2 affected β2AR-induced ERK activation. Efforts in this regard were unsuccessful in that the basal ERK activity was consistently, although unexpectedly, elevated following knockdown of AGAP2. β-Arrestins regulate ERK activation on signalling endosomes or signalosomes. Given that AGAP2 forms a complex with β-arrestins (Figure 1), and AGAP2 was known to regulate endosomal functions, it is possible that AGAP2 functions as scaffold to facilitate ERK activation. We examined whether AGAP2 complexes with ERK. Immunoprecipitates of overexpressed AGAP2, or its truncated mutant PZA2, contained endogenous ERK (Figure 8C), suggesting that AGAP2 forms a complex with ERK to co-ordinate its activation. Therefore depletion of AGAP2 may alter the subcellular distribution of ERK and thereby the interaction of ERK with other kinases or phosphatases, leading to high ERK activity in cells with AGAP2 knockdown.
DISCUSSION
β2AR activation has been shown to regulate Arf6 activation via the β-arrestin-associated guanine-nucleotide-exchange factor ARNO (Arf nucleotide-binding-site opener), and Arf6 activity is involved in both endocytosis and recycling of activated β2ARs [28]. Knockdown of Arf6 by siRNA reduced β2AR endocytosis [13], whereas stimulation of β2AR with agonists resulted in activation of Arf6 that inhibited Rab4-dependent fast recycling of β2AR [29]. This is consistent with an earlier study showing that internalized β2ARs undergo agonist-dependent Rab4-mediated fast recycling and agonist-independent slower recycling [30]. We report in the present paper that AGAP2 is involved in the recycling of β2ARs, consistent with our previous findings that AGAP2 promoted Rab4-dependent fast recycling [20]. The involvement of Arf proteins in AGAP2-promoted fast recycling of β2AR is not clear at present. In vitro studies suggest AGAP2 preferred Arf1 over Arf6 as a substrate [20], but the possibility exists that AGAP2 regulates Arf6 in cells. In addition, AGAP2 may regulate fast recycling of β2AR in an Arf-independent manner.
Regulation of β2AR trafficking by the GIT family Arf GAPs has been well documented. Overexpression of the Arf GAP GIT1 or GIT2, which hydrolyse GTP on Arf6 [31], inhibited β2AR endocytosis [14,15]. Several members of the AZAP (Arf GAP with ankyrin repeats and PH domains) subfamily Arf GAPs have been shown to affect EGFR (epidermal growth factor receptor) trafficking [32]. ASAP1 has an N-terminal BAR (Bin/amphiphysin/Rvs) domain that may facilitate formation of membrane curvature and promote formation of transport intermediates, and ASAP1 was found to affect the recycling of EGFRs [33]. Additional mechanisms are involved for ASAP1 to regulate EGFR trafficking. For example, interaction between ASAP1 and the adaptor protein CIN85 (Cbl-interacting protein of 85 kDa) contributed to ASAP1-potentiated EGFR recycling [34]. In addition, the association with CIN85 may be required for different Arf GAPs to regulate EGFR trafficking at different stages. In that case, ARAP1 (Arf GAP with Rho GAP, ankyrin repeats and PH domains) binds CIN85 to drive the exit of EGFRs from the endosomal pathway prior to EGFR ubiquitination [35]. AGAP2 was shown to promote the fast recycling of transferrin receptors [20]. However, regulation of GPCR trafficking by the AZAP family of Arf GAPs has not been reported prior to the present study.
The results of the present study suggest that the PH2 domain of AGAP2 is the major binding site for β-arrestins. The AZAP family of Arf GAPs all contain a PH domain that is essential for the GAP activity of these proteins and is specifically regulated by phospholipids [36,37]. AGAP2-mediated GTP hydrolysis on Arf was activated by PtdIns(4,5)P2 or PtdIns(3,4,5)P3 and potentiated by PA [20]. In the present study we showed that AGAP2 binding to β-arrestin2 was promoted by PtdIns(4,5)P2 with PA playing an inhibitory role, suggesting distinct outputs of AGAP2 function as determined by upstream signals that elevate intracellular levels of distinct phospholipids. Similarly, β-arrestins were shown to interact with and regulate the function of PTEN (phosphatase and tensin homologue deleted on chromosome 10). Depending on the upstream signals that activate RhoA, β-arrestins either activate the lipid phosphatase activity of PTEN or inhibit the phosphatase-independent anti-migratory effect of PTEN [38].
We have proposed that the AZAP subfamily of multidomain Arf GAPs may serve as coat components during vesicle trafficking [32]. Several pieces of evidence from the present study support this idea. First, AGAP2 formed a complex with β-arrestins, which are known to bind clathrin heavy chain and AP2, critical components of clathrin-dependent endocytosis. Secondly, AGAP2 co-localized with β2ARs during receptor endocytosis, presumably on endosomes. Thirdly, knockdown of AGAP2 prevented β2AR recycling to the plasma membrane. As β2ARs are class A receptors that form a loose association with β-arrestins that dissociate from β2ARs shortly after receptor internalization [39], AGAP2 may remain for a longer period of time on β2AR-containing endosomes, consistent with its potential role in β2AR recycling.
Activation of several GPCRs has been shown to elicit two peaks of ERK activation as determined by the phosphorylation of ERK, which can be G-protein-mediated or β-arrestin-mediated. The first clue about β-arrestin in ERK activation came from a study where inhibition of endocytosis abolished β2AR-induced ERK activation [40]. For class B receptors, β-arrestins associate tightly with the receptor-containing endosomes or signalosomes, which may contribute to the late onset and longer-lasting phase of ERK activation. β2ARs belong to class A receptors, but still elicited the second phase, longer-lasting ERK activation [41]. The G-protein-dependent ERK activation may involve both Rap and Ras, and the β-arrestin-dependent ERK activation is more complex and may involve scaffolding of Src [26,27]. Since β-arrestins only form loose complexes with β2ARs, the distribution of phospho-ERK following β2AR activation remain unclear. One possibility is that β2AR elicited phospho-ERK will be cytosolic. Another possibility is that AGAP2 or other adaptor proteins may serve as a scaffold for endosomal retention of phospho-ERK. Our observation from the present study that knockdown of AGAP2 resulted in high basal phospho-ERK suggests a more complex role for AGAP2 in ERK regulation. One possibility is that AGAP2 scaffolds ERK to regulate its interaction with kinase(s) that activate ERK or with phosphatase(s) that inactivate ERK.
Evidence is accumulating that GPCRs, including β2AR, play important roles in human cancer initiation and progression [42,43]. Activation of β2AR increased the growth, invasion and angiogenesis of ovarian cancers [44], and promoted β-arrestin1-dependent activation of MDM2 (murine double minute 2), which binds to and degrades p53, leading to DNA damage [25]. AGAP2 was shown to be up-regulated in various human cancers [19] and to enhance focal adhesion remodelling and cancer cell migration [21]. In the present study, we provide evidence that AGAP2 forms a complex with β-arrestin to promote recycling of internalized β2ARs and β2AR-induced ERK activation. An increased ERK activity may contribute to the tumorigenic effects of β2AR and AGAP2.
Acknowledgments
We thank Dr Y. Daaka for advice and suggestions, and for providing the β-arrestin1–HA and β-arrestin2–HA cDNAs, and anti-β-arrestin antibody, and for a critical reading of the paper prior to submission. We thank Dr J. Benovic for GFP–β2AR cDNA, Dr Y. Xiang for FLAG–β2AR cDNA and Dr M. Scott for GST–β-arrestin cDNA. We thank Dr P. Randazzo for providing His6–AGAP2 proteins and for suggestions. We thank Dr Y. Daaka and Dr V. Ramkumar for helping with the receptor binding assays.
FUNDING
This work was supported by the National Institutes of Health [grant number K22CA124578 (to Z.N.)].
Abbreviations used
- ACAP1
Arf (ADP-ribosylation factor) GAP (GTPase-activating protein) with coiled-coil domains, ankyrin repeats and PH (pleckstrin homology) domains 1
- AGAP
Arf (ADP-ribosylation factor) GAP (GTPase-activating protein) with GTP-binding-protein-like, ankyrin repeats and PH (pleckstrin homology) domains
- AP
adaptor protein
- Arf
ADP-ribosylation factor
- β2AR
β2-adrenergic receptor
- ASAP1
Arf GAP with SH3 (Src homology 3) domain, ankyrin repeats and PH (pleckstrin homology) domains 1
- AZAP
Arf GAP with ankyrin repeats and PH (pleckstrin homology) domains
- CIN85
Cbl-interacting protein of 85 kDa
- DMEM
Dulbecco’s modified Eagle’s medium
- EGFR
epidermal growth factor receptor
- ERK
extracellular-signal-regulated kinase
- FBS
fetal bovine serum
- GAP
GTPase-activating protein
- GFP
green fluorescent protein
- GPCR
G-protein-coupled receptor
- GRK
GPCR kinase
- GST
glutathione transferase
- HA
haemagglutinin
- HEK
human embryonic kidney
- ISO
isoprenaline (also known as isoproterenol)
- LAMP1
lysosome-associated membrane protein 1
- PA
phosphatidic acid
- PH
pleckstrin homology
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
Footnotes
AUTHOR CONTRIBUTION
Yuanjun Wu, Yu Zhao, Xiaojie Ma, Yunjuan Zhu, Jaimin Patel and Zhongzhen Nie performed the experiments. Zhongzhen Nie wrote the paper.
References
- 1.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 2.Koch WJ, Lefkowitz RJ, Rockman HA. Functional consequences of altering myocardial adrenergic receptor signaling. Annu Rev Physiol. 2000;62:237–260. doi: 10.1146/annurev.physiol.62.1.237. [DOI] [PubMed] [Google Scholar]
- 3.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 5.Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 6.Scarselli M, Donaldson JG. Constitutive internalization of G protein-coupled receptors and G proteins via clathrin-independent endocytosis. J Biol Chem. 2009;284:3577–3585. doi: 10.1074/jbc.M806819200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol. 2003;163:203–208. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SSG, Caron MG, Barak LS. The β2-adrenergic receptor/β arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci U S A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodman OB, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 10.D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 11.East MP, Kahn RA. Models for the functions of Arf GAPs. Semin Cell Dev Biol. 2011;22:3–9. doi: 10.1016/j.semcdb.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Randazzo PA, Hirsch DS. Arf GAPs: multifunctional proteins that regulate membrane traffic and actin remodelling. Cell Signaling. 2004;16:401–413. doi: 10.1016/j.cellsig.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Houndolo T, Boulay PL, Claing A. G protein-coupled receptor endocytosis in ADP-ribosylation factor 6-depleted cells. J Biol Chem. 2005;280:5598–5604. doi: 10.1074/jbc.M411456200. [DOI] [PubMed] [Google Scholar]
- 14.Premont RT, Claing A, Vitale N, Freeman JLR, Pitcher JA, Patton WA, Moss J, Vaughan M, Lefkowitz RJ. β2-adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc Natl Acad Sci U S A. 1998;95:14082–14087. doi: 10.1073/pnas.95.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ. The GIT family of ADP-ribosylation factor GTPase-activating proteins – functional diversity of GIT2 through alternative splicing. J Biol Chem. 2000;275:22373–22380. doi: 10.1074/jbc.275.29.22373. [DOI] [PubMed] [Google Scholar]
- 16.Shenoy SK, Xiao KH, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J Biol Chem. 2008;283:22166–22176. doi: 10.1074/jbc.M709668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daaka Y. S-nitrosylation-regulated GPCR signaling. Biochim Biophys Acta. 2012;1820:743–751. doi: 10.1016/j.bbagen.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye KQ. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem. 2004;279:16441–16451. doi: 10.1074/jbc.M312175200. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Hu Y, Hao C, Rempel SA, Ye K. PIKE-A is a proto-oncogene promoting cell growth, transformation and invasion. Oncogene. 2007;26:4918–4927. doi: 10.1038/sj.onc.1210290. [DOI] [PubMed] [Google Scholar]
- 20.Nie ZZ, Fei JJ, Premont RT, Randazzo PA. The Arf GAPs AGAP1 and AGAP2 distinguish between the adaptor protein complexes AP-1 and AP-3. J Cell Sci. 2005;118:3555–3566. doi: 10.1242/jcs.02486. [DOI] [PubMed] [Google Scholar]
- 21.Zhu YJ, Wu YJ, Kim JI, Wang ZM, Daaka Y, Nie ZZ. Arf GTPase-activating protein AGAP2 regulates focal adhesion kinase activity and focal adhesion remodeling. J Biol Chem. 2009;284:13489–13496. doi: 10.1074/jbc.M900469200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nie ZZ, Mei Y, Ramkumar V. Short term desensitization of the A1 adenosine receptors in DDT1MF-2 cells. Mol Pharmacol. 1997;52:456–464. doi: 10.1124/mol.52.3.456. [DOI] [PubMed] [Google Scholar]
- 23.Moniri NH, Daaka Y. Agonist-stimulated reactive oxygen species formation regulates β2-adrenergic receptor signal transduction. Biochem Pharmacol. 2007;74:64–73. doi: 10.1016/j.bcp.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Luo R, Akpan I, Hayashi R, Sramko M, Barr V, Shiba Y, Randazzo PA. GTP-binding protein-like domain of AGAP1 is a protein binding site that allosterically regulates ArfGAP protein catalytic activity. J Biol Chem. 2012;287:17176–17185. doi: 10.1074/jbc.M111.334458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao KH, et al. A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature. 2011;477:349–353. doi: 10.1038/nature10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Defea K. β-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br J Pharmacol. 2008;153:S298–S309. doi: 10.1038/sj.bjp.0707508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claing A, Chen W, Miller WE, Vitale N, Moss J, Premont RT, Lefkowitz RJ. β-Arrestin-mediated ADP-ribosylation factor 6 activation and β2-adrenergic receptor endocytosis. J Biol Chem. 2001;276:42509–42513. doi: 10.1074/jbc.M108399200. [DOI] [PubMed] [Google Scholar]
- 29.Macia E, Partisani M, Paleotti O, Luton F, Franco M. Arf6 negatively controls the rapid recycling of the β2AR. J Cell Sci. 2012;125:4026–4035. doi: 10.1242/jcs.102343. [DOI] [PubMed] [Google Scholar]
- 30.Yudowski GA, Puthenveedu MA, Henry AG, von Zastrow M. Cargo-mediated regulation of a rapid Rab4-dependent recycling pathway. Mol Biol Cell. 2009;20:2774–2784. doi: 10.1091/mbc.E08-08-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vitale N, Patton WA, Moss J, Vaughan M, Lefkowitz RJ, Premont RT. GIT proteins, a novel family of phosphatidylinositol 3,4,5-trisphosphate-stimulated GTPase-activating proteins for ARF6. J Biol Chem. 2000;275:13901–13906. doi: 10.1074/jbc.275.18.13901. [DOI] [PubMed] [Google Scholar]
- 32.Nie ZZ, Randazzo PA. Arf GAPs and membrane traffic. J Cell Sci. 2006;119:1203–1211. doi: 10.1242/jcs.02924. [DOI] [PubMed] [Google Scholar]
- 33.Nie ZZ, Hirsch DS, Luo RB, Jian XY, Stauffer S, Cremesti A, Andrade J, Lebowitz J, Marino M, Ahvazi B, et al. A BAR domain in the N terminus of the Arf GAP ASAP1 affects membrane structure and trafficking of epidermal growth factor receptor. Curr Biol. 2006;16:130–139. doi: 10.1016/j.cub.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 34.Kowanetz K, Husnjak K, Holler D, Kowanetz M, Soubeyran P, Hirsch D, Schmidt MHH, Pavelic K, De Camilli P, Randazzo PA, Dikic I. CIN85 associates with multiple effectors controlling intracellular trafficking of epidermal growth factor receptors. Mol Biol Cell. 2004;15:3155–3166. doi: 10.1091/mbc.E03-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon HY, Kales SC, Luo RB, Lipkowitz S, Randazzo PA. ARAP1 association with CIN85 affects epidermal growth factor receptor endocytic trafficking. Biol Cell. 2011;103:171–184. doi: 10.1042/BC20100154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campa F, Yoon HY, Ha VL, Szentpetery Z, Balla T, Randazzo PA. A PH domain in the Arf GTPase-activating protein (GAP) ARAP1 binds phosphatidylinositiol 3,4,5-trisphosphate and regulates Arf GAP activity independently of recruitment to the plasma membranes. J Biol Chem. 2009;284:28069–28083. doi: 10.1074/jbc.M109.028266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo R, Jenkins LMM, Randazzo PA, Gruschus J. Dynamic interaction between Arf GAP and PH domains of ASAP1 in the regulation of GAP activity. Cell Signaling. 2008;20:1968–1977. doi: 10.1016/j.cellsig.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lima-Fernandes E, Enslen H, Camand E, Kotelevets L, Boularan C, Achour L, Benmerah A, Gibson LCD, Baillie GS, Pitcher JA, et al. Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-Arrestins. EMBO J. 2011;30:2557–2568. doi: 10.1038/emboj.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oakley RH, Laporte SK, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 40.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SSG, Caron MG, Lefkowitz RJ. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 41.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao KH, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β 2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 42.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 43.Daaka Y. G proteins in cancer: the prostate cancer paradigm. Sci STKE. 2004;2004:re2. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 44.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu CH, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]