

Key Points
Phenotypic and genotypic profiling of MDM2 in DLBCL.
MDM2 as a negative regulator of p53 tumor suppressor function.
Abstract
MDM2 is a key negative regulator of the tumor suppressor p53, however, the prognostic significance of MDM2 overexpression in diffuse large B-cell lymphoma (DLBCL) has not been defined convincingly. In a p53 genetically–defined large cohort of de novo DLBCL patients treated with rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP) chemotherapy, we assessed MDM2 and p53 expression by immunohistochemistry (n = 478), MDM2 gene amplification by fluorescence in situ hybridization (n = 364), and a single nucleotide polymorphism in the MDM2 promoter, SNP309, by SNP genotyping assay (n = 108). Our results show that MDM2 overexpression, unlike p53 overexpression, is not a significant prognostic factor in overall DLBCL. Both MDM2 and p53 overexpression do not predict for an adverse clinical outcome in patients with wild-type p53 but predicts for significantly poorer survival in patients with mutated p53. Variable p53 activities may ultimately determine the survival differences, as suggested by the gene expression profiling analysis. MDM2 amplification was observed in 3 of 364 (0.8%) patients with high MDM2 expression. The presence of SNP309 did not correlate with MDM2 expression and survival. This study indicates that evaluation of MDM2 and p53 expression correlating with TP53 genetic status is essential to assess their prognostic significance and is important for designing therapeutic strategies that target the MDM2-p53 interaction.
Introduction
MDM2/Hdm2, the human homolog of murine double minute 2 (Mdm2) or p53 E3 ubiquitin protein ligase homolog (mouse) (Homo sapiens), is a key negative regulator of the tumor suppressor p531 and has other p53-independent functions.2 The MDM2 gene is transactivated by p53, and thus p53 degradation by MDM2 forms the other direction of a negative-feedback loop. MDM2 is frequently overexpressed in cancer, but its prognostic importance has been elusive in many disease entities.3 MDM2 overexpression has been shown to facilitate B-cell lymphomagenesis in vivo4 and to inactivate the tumor suppressor function of wild-type p53 (WT-p53) in vitro.5 MDM2 overexpression, however, has correlated inconsistently with adverse clinical outcomes in patients with hematologic malignancies.6,7
Several factors could account for the inconsistent results: (1) Small study sizes; (2) different cutoffs for MDM2 expression; (3) unclear expression and function of MDM2 isoforms8; and (4) posttranslational modifications or subcellular localization of MDM2.3 In stress conditions, both p53 and MDM2 are modified (eg, phosphorylation by ATM), resulting in reduced affinity and increased degradation of MDM2.9 In addition, MDM2 nuclear entry is inhibited via induction of p53-responsive PTEN,10 and p53-inducible p21 maintains another positive feedback loop.11 A fifth possible factor is oscillation of the p53-MDM2 autoregulatory feedback loop (Figure 1C), which has not been recognized by the previous prognostic studies. Elegant models and laboratory observations have shown that cellular levels of WT-p53 and MDM2 fluctuate in an oscillatory fashion in response to stress, such as DNA damage, hypoxia, or oncogene activation, and that the numbers of pulses and the fraction of cells with oscillatory pulses increase with the strength of DNA damage.9-23 The oscillatory kinetics and the variable amplitude of p53/MDM2 pulses over cell population may affect the measurement of MDM2 expression using immunohistochemistry (IHC), a commonly used method in clinical diagnostic and prognostic studies. If the study cohort is too small, or the cutoff is inappropriately established, the survival difference between 2 groups may not be truly reflected. To obtain evaluable results beyond the “noise” caused by oscillation, a large cohort of cases is essential.
Figure 1.
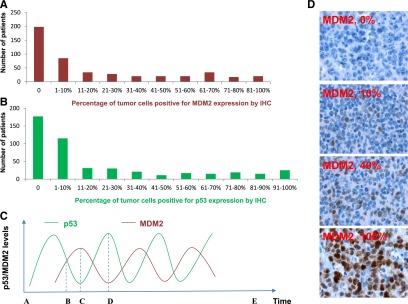
MDM2 and p53 expression in DLBCL patients treated with R-CHOP. (A-B) Histogram showing the distribution of MDM2 and p53 expression levels in the DLBCL cohort. X-axis, percentage of immunopositive cells in tumors; Y-axis, numbers of DLBCL patients. (C) Illustration of p53 and MDM2 kinetic pulses in tumor cells under stress conditions. In a single cell, because of the fluctuating p53/MDM2 levels in oscillatory pulses, the IHC pattern is (1) p53–/MDM2– at zero time point A or E after cellular stress is removed; (2) p53+/MDM2+ at time point B; (3) p53–/MDM2+ at time point C; and (4) p53+/MDM2– at time point D. Same patterns over cell population may be generated by evaluation of the percentage of MDM2+ cells and defining overexpression by certain cutoffs. (D) Representative MDM2 immunohistochemical staining patterns.
In comparison, MDM2 function toward mutant p53 (MUT-p53) and the kinetics of MDM2 and MUT-p53 levels under stress are not well defined. In a mouse model, MDM2 and DNA damage regulate MUT-p53 levels in a manner similar to WT-p53.24 However, most MUT-p53s have lost the ability to transactivate MDM2, so basal MDM2 levels cannot compensate for increased MUT-p53 because of the sustained stress.24 Conversely, in a panel of cell lines, mutual stabilization of MDM2 (increased half-life time from 30 minutes to 2 hours) and MUT-p53 was observed.25
The prognostic significance of MDM2 expression is further complicated by other nononcogenic functions of MDM2. MDM2 monoubiquitinates p53, leading to only nuclear export and not p53 degradation.12 MDM2 promotes p53 translation by binding to p53 mRNA, simultaneously impairing its E3 ligase activity.26 After exposure to the p53-activating drug RITA, MDM2 can enhance the apoptotic response induced by p53 through downregulation of p21 (cell-cycle inhibitor) at both the transcriptional and posttranscriptional levels.27 In normal human or murine cells, MDM2 induces G0/G1 cell-cycle arrest.28 These MDM2 functions suggest that MDM2 expression can be a positive prognostic factor in certain cellular contexts. In mouse models, Mdm2 appeared to be both a tumor suppressor and an oncoprotein depending on its cellular levels.29
Mdm2 exerts p53-independent tumorigenesis in mouse models, but the mechanisms are unclear,29,30 although several proteins (such as p73, p63, RB, Sp1, E2F1, L5) have been identified to have interaction with MDM2.2 However, the principal role of Mdm2 as a negative p53 regulator is supported by the similar survival curves and tumor spectrum of p53−/−Mdm2−/−Mdm4−/− and p53−/− mice,31 or the similar tumor incidence in p53−/−Mdm2−/− and p53−/− mice.29 On the other hand, few p53-independent mechanisms upregulating MDM2 are known.12 In many tumors, MDM2 overexpression is not caused by gene amplification. The single nucleotide polymorphism (SNP) 309 T→G in the MDM2 promoter is associated with an increased affinity with the transcriptional activator Sp1, resulting in elevated MDM2 expression, in a gender-specific (females) and hormonal-dependent manner.32,33 On the other hand, in Burkitt lymphoma cells, MDM2 overexpression was caused by enhanced translation.5
To explore the prognostic significance of MDM2 expression and mechanisms of MDM2 overexpression, and to recognize the possible factors that could complicate the analysis of the results, we assessed for MDM2 expression, MDM2 amplification and polymorphism, p53 expression, and TP53 genetic status in a large cohort of patients with diffuse large B-cell lymphoma (DLBCL).
Materials and methods
Patients
The initial study cohort consisted of 478 de novo DLBCL patients treated with rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP) therapy in 29 medical centers. The diagnostic criteria, review process, treatment regimens, treatment response criteria, and eligibility/exclusion criteria have been described previously.34,35 At last follow-up, 36% (173/478) of the patients had died. The median follow-up interval for the 305 censored patients was 56.5 months (range, 12.0-180.3). The validation set consisted of another 227 DLBCL patients with similar clinical features and treatment regimens. This study was approved as being of minimal to no risk or as exempt by the Institutional Review Boards of all participating centers, and at The University of Texas MD Anderson Cancer Center. This study was conducted in accordance with the Declaration of Helsinki.
p53 and MDM2 immunohistochemical staining
Biopsy samples obtained at diagnosis were fixed in formalin and embedded in paraffin, and tissue microarrays (TMAs) were constructed using tissue array (Beecher Instruments, Silver Spring, MD).36 IHC was performed on TMAs using antibodies DO-7 for p53 (Dako) and IF2 for MDM2 (N-terminal37) (Calbiochem, Billerica, MA), as described previously.34,36 Expression levels of p53 and MDM2 for each sample were determined by counting the percentage of positive cells in TMA cores, in combination with the assessment of intensity. Each TMA core contained approximately 500 tumor cells. Six pathologists in addition to the pathologist at each contributing medical center independently evaluated the tissue cores by light microscopy for p53 and MDM2 expression without knowledge of the clinical outcomes. Discrepancies were resolved by joint review on a multihead microscope.
TP53 mutations and SNP309 genotyping of MDM2
p53 mutation status was determined by resequencing microarray using p53 AmpliChip (Roche Molecular Systems, Pleasanton, CA), as described previously.34
The SNP309 (rs2279744) status of the MDM2 promoter, was determined by using a Taqman SNP genotyping assay, as previously described.32 Primers were purchased from Applied Biosystems (Foster City, CA).
Gene expression profiling
Gene expression profiling (GEP) was performed using Affymetrix GeneChip Human Genome HG-U133 Plus 2.0 chips.36 The GEO accession number is GSE#31312. Normalized microarray data were analyzed for differential expression between MDM2+ and MDM2– patients, or p53+ and p53– patients with WT-p53 or MUT-p53. Univariate analysis was performed to identify differentially expressed genes (DEGs) using the Student t test. The P values obtained by multiple t tests were corrected for false discovery rate (FDR) using the BUM method. DEGs were identified with P value cutoffs of .0012 to .0035 for respective comparisons at an FDR of .30.
Fluorescence in situ hybridization for MDM2 gene amplification analysis
A bacterial artificial chromosome clone (RP11-1064P9, obtained from the BACPAC Resource Center of the Children’s Hospital Oakland Research Institute, http://bacpac.chori.org/home.htm) localized on chromosomal region 12q13-15 was used as a probe for MDM2 fluorescence in situ hybridization (FISH) and labeled with Spectrum Orange. A control region from the centromere of chromosome 12 was used as a ploidy reference and labeled with Spectrum Green. Dual-color FISH was performed in 4 micron sections of the TMAs. Images were captured and archived using Cytovision software (Applied Imaging, Santa Clara, CA). Fluorescence signals were scored by 2 independent investigators by counting the number of MDM2 and reference probe signals in 150 to 250 well-defined nuclei. A sample was scored as amplified when the ratio between the test and control signals was >2.
Statistical analysis
Clinical and laboratory features at the time of presentation between different DLBCL subgroups were compared using the χ2 test. Overall survival (OS) was calculated from the time of diagnosis to the time of death from any cause. Progression-free survival (PFS) was calculated from the time of diagnosis to the time of progression or death from any cause.35 Patients who remained alive or were progression-free were censored at the last follow-up. Univariate and multivariate analyses for survival of the study cohort were performed with IBM SPSS statistics 19.0 (Armonk, NY) using the Cox proportional hazards regression model and the forward-stepwise method. OS and PFS curves of different groups were analyzed by GraphPad Prism 5 software using the Kaplan-Meier method, and differences were compared using the log-rank (Mantel-Cox) test. All differences with P ≤ .05 were considered to be statistically significant.
Results
MDM2 and p53 expression
Our results showed variable p53/MDM2 levels (0-100%) among 478 patients and all combinations of IHC staining patterns (+/+, +/−, −/−, −/+) (Table 1A-B), which could possibly be explained by the oscillations of p53/MDM2 levels in patients with WT-p53 (Figure 1C) and other regulatory factors that affect p53 and MDM2 levels. The mean percentage of MDM2-positive cells in 478 patients was 22%. The mean percentage of p53-positive cells was 23% in 474 patients with p53 expression data available. Based on occurrence distribution of different MDM2 or p53 expression levels (Figure 1A-B), both MDM2 and p53 overexpression (MDM2+, p53+) were defined as >10% of cells positive for IHC staining, which is similar to cutoffs used by others in earlier lymphoma studies of MDM2 and p53 expression.6,38,39 Using the >10% cutoff, 193 of 478 (40.4%) DLBCLs had MDM2 overexpression and 182 of 474 (38.4%) DLBCLs had p53 overexpression. The frequency of MDM2 overexpression was higher (but not significant) in DLBCLs with WT-p53 (41.9%, 156/372) than in DLBCLs with MUT-p53 (34.9%, 37/106). Correspondingly, there is a higher mean level of MDM2 expression in DLBCL patients with WT-p53 than DLBCLs with MUT-p53 (24% vs 17%, Table 1A). Comparatively, p53 overexpression is significantly more common in DLBCLs with MUT-p53 than with WT-p53 (71.4% vs 29.1%) (P < .0001, see case numbers in Table 1A), and has a higher mean level of p53 expression in DLBCL with MUT-p53 (54% vs 14%). MUT-p53 was overexpressed compared with WT-p53 in both MDM2– and MDM2+ DLBCLs (Table 1B), suggesting that overexpression was not readily explained by reduction in MDM2-mediated degradation alone.
Table 1.
MDM2 and p53 expression in DLBCL patients treated with R-CHOP
A. MDM2 and p53 expression in DLBCL patients with WT-p53 or MUT-p53
| Overall | WT-p53 | MUT-p53 | |
|---|---|---|---|
| Number of MDM2– patients | 285 | 216 | 69 |
| Number of MDM2+ patients | 193 | 156 | 37 |
| Mean MDM2 expression (% cells) | 22% | 24% | 17% |
| Number of p53– patients | 292 | 262 | 30 |
| Number of p53+ patients | 182 | 107 | 75 |
| Mean p53 expression (% cells) | 23% | 14% | 54% |
B. MDM2 and p53 expression in DLBCL patients with or without p53 overexpression
| p53+ | p53– | Mean p53 expression (% cells) | |
|---|---|---|---|
| MDM2– patients | 79 | 204 | 19% |
| WT-p53/MDM2– | 32 | 183 | 9% |
| MUT-p53/MDM2– | 47 | 21 | 39% |
| MDM2+ patients | 103 | 88 | 28% |
| WT-p53/MDM2+ | 75 | 79 | 17% |
| MUT-p53/MDM2+ | 28 | 9 | 50% |
Also notable, all patients (n = 16) with non-sense, splice-site, or frame-shift p53 mutations had a p53-negative immunophenotype; all but 4 patients were also MDM2–.3 In contrast, 84% of patients with missense mutations had a p53+ phenotype, and 72% of patients had a high IHC score (≥50%) of tumor cells (mean level, 64%). MDM2 expression had no good correlation with missense-MUT-p53+ phenotype: p53+/MDM2–: 52.7%; p53+/MDM2+: 30.8%; p53–/MDM2–: 11.0%; p53–/MDM2+: 5.5%.
Clinical features of the cohort and univariate and multivariate analysis
Clinical features of the patients were compared between MDM2– and MDM2+ DLBCL in the entire cohort and in subcohorts of patients with WT- or MUT-p53 (Table 2A).
Table 2.
Clinical characteristics of 478 de novo DLBCL and multivariate survival analysis
A. Clinical characteristics of patients with de novo DLBCL treated with R-CHOP: comparison between patients with or without MDM2 overexpression or with different SNP309 genotypes
| Overall (n = 478) | WT-p53 (n = 372) | MUT-p53 (n = 106) | SNP (n = 108) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDM2– | MDM2+ | P | MDM2– | MDM2+ | P | MDM2– | MDM2+ | P | T/T | T/G or G/G | P | |
| n | 285 | 193 | 216 | 156 | 69 | 37 | 41 | 67 | ||||
| Age, y | ||||||||||||
| <60 | 124 | 68 | 95 | 56 | 29 | 12 | 17 | 22 | ||||
| ≥60 | 161 | 125 | .07 | 121 | 100 | .12 | 40 | 25 | .33 | 24 | 45 | .37 |
| Gender | ||||||||||||
| F | 128 | 69 | 97 | 57 | 31 | 12 | 10 | 23 | ||||
| M | 157 | 124 | .046 | 119 | 99 | .11 | 38 | 25 | .21 | 31 | 44 | .28 |
| Stage | ||||||||||||
| I-II | 136 | 79 | 101 | 67 | 35 | 12 | 17 | 25 | ||||
| III-IV | 137 | 108 | .11 | 103 | 83 | .37 | 34 | 25 | .071 | 24 | 49 | .41 |
| B-symptoms | ||||||||||||
| No | 158 | 123 | 118 | 101 | 40 | 22 | 26 | 45 | ||||
| Yes | 80 | 51 | .35 | 63 | 40 | .22 | 17 | 11 | .73 | 12 | 18 | .75 |
| LDH | ||||||||||||
| Normal | 83 | 64 | 69 | 52 | 14 | 12 | 14 | 19 | ||||
| Elevated | 180 | 103 | .17 | 130 | 80 | .50 | 50 | 23 | .23 | 18 | 30 | .82 |
| No. of extranodal sites | ||||||||||||
| 0-1 | 217 | 138 | 164 | 111 | 53 | 27 | 41 | 52 | ||||
| ≥2 | 55 | 45 | .27 | 41 | 36 | .31 | 14 | 9 | .63 | 0 | 15 | .0011 |
| Performance status | ||||||||||||
| 0-1 | 218 | 137 | 163 | 108 | 55 | 29 | 28 | 47 | ||||
| ≥2 | 30 | 27 | .21 | 23 | 23 | .20 | 7 | 4 | .90 | 5 | 8 | .94 |
| Size of largest tumor | ||||||||||||
| <5 cm | 131 | 87 | 101 | 75 | 30 | 12 | 22 | 33 | ||||
| ≥5 cm | 88 | 62 | .78 | 63 | 45 | .88 | 25 | 17 | .25 | 8 | 18 | .42 |
| IPI risk group | ||||||||||||
| 0-2 | 170 | 99 | 130 | 78 | 40 | 21 | 25 | 36 | ||||
| 3-5 | 83 | 71 | .064 | 61 | 57 | .062 | 22 | 14 | .67 | 10 | 20 | .50 |
| Therapy response | ||||||||||||
| CR | 225 | 139 | .081 | 176 | 123 | .53 | 49 | 16 | .0051 | 28 | 49 | .59 |
| PR | 33 | 30 | 22 | 20 | 11 | 10 | 7 | 9 | ||||
| SD | 13 | 4 | 8 | 3 | 5 | 1 | 1 | 1 | ||||
| PD | 14 | 20 | 10 | 10 | 4 | 10 | 5 | 8 | ||||
Univariate analysis regarding survival indicated that an International Prognostic Index (IPI) score of >2 and the 5 risk factors composing the IPI (age, stage, serum lactate dehydrogenase level, performance status, extranodal sites), as well as B-symptoms, tumor size, TP53 mutation, and p53 overexpression, were significantly associated with poorer survival. MDM2 overexpression was not associated with poorer survival.
The prognostic factors according to univariate analysis, and MDM2 >10% and gender that are significantly different between MDM2+ and MDM2– DLBCLs (Table 2A), were then entered into multivariate survival analysis. The IPI >2, TP53 mutations, and B-symptoms remained as the independent prognostic factors, whereas p53 >10% and MDM2 >10%, tumor size >5 cm, and gender were not associated with significantly poorer survival (Table 2B).
B. Multivariate survival analysis
| OS | PFS | |||||
|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | P value | Hazard ratio | 95% CI | P value | |
| Overall cohort | ||||||
| IPI >2 | 3.04 | 1.96-3.96 | <.0001 | 2.59 | 1.8-3.62 | <.0001 |
| B-symptoms | 1.43 | 1.00-2.04 | .048 | 1.49 | 1.06-2.10 | .024 |
| TP53 mutation | 1.59 | 1.06-2.26 | .013 | 1.52 | 1.03-2.17 | .023 |
| MUT-p53 subcohort | ||||||
| IPI >2 | 2.03 | 1.05-3.93 | .035 | 1.95 | 1.02-3.74 | .042 |
| p53 >10% | 2.70 | 1.04-7.04 | .042 | 2.87 | 1.10-7.45 | .031 |
| MDM2 >10% | 2.24 | 1.17-4.27 | .015 | 1.96 | 1.03-3.70 | .041 |
LDH, lactate dehydrogenase; CR, complete remission; PR, partial response; SD, stable disease; PD, progressive disease.
For therapy response, we calculated P values as CR vs other responses. Some clinical features of certain cases were not available.
However, in the MUT-p53 subcohort, the IPI >2, p53 >10%, and MDM2 >10% remained prognostically significant in the multivariate analysis, whereas B-symptoms and tumor size >5 cm did not independently predict a poorer prognosis (Table 2B).
Prognostic significance of MDM2 and p53 overexpression
MDM2 overexpression was not significantly associated with poorer survival for the entire cohort (P values: .18 for OS and .41 for PFS; Figures 2A and 3A), or within patients with WT-p53 (P values: .71 for OS and .99 for PFS; Figures 2C and 3C). However, MDM2 overexpression conferred an inferior OS for patients with MUT-p53 (median OS: 30.0 vs 87.3 months; hazard ratio [HR] = 2.92; 95% confidence interval [CI], 2.33-3.50; P = .0072) (Figure 2E). A similar trend was observed for PFS curves (Figure 3E).
Figure 2.

Impact of MDM2 and p53 expression on OS in de novo DLBCL patients with WT- or MUT-p53.
Figure 3.

Impact of MDM2 and p53 expression on PFS in de novo DLBCL patients with WT- or MUT-p53.
In contrast, p53 overexpression correlated with poorer survival for the entire cohort (P = .017 for OS, and P = .019 for PFS). However, the prognostic significance of p53 overexpression was restricted to DLBCL patients with MUT-p53 (median OS: 33.3 vs 82.4 months; HR = 2.48; 95% CI, 2.0-2.96; P = .016; median PFS: 25.8 vs 82.4 months; HR = 2.16; 95% CI, 1.23-3.79; P = .0073) (Figures 2 and 3B,D,F).
The prognostic significance of MDM2 or p53 overexpression is confirmed in the validation set (supplemental Figure 1).
Concurrent evaluation of p53 and MDM2 expression
Not only the survival curves for p53 and MDM2 overexpression showed similar patterns in the WT-p53 and MUT-p53 subgroups (Figures 2 and 3C-F): patients with p53+/MDM2−, p53–/MDM2+, or p53+/MDM2+ had comparable survival rates (Figure 4A-D), reinforcing the idea that MDM2+ and p53+ patients are prognostic equivalents in the WT- or MUT-p53 subgroups. If stratifying patients with either MDM2 or p53 overexpression as a single group, the impact of their overexpression on survival was similar to that of p53 overexpression (Figure 4E-F for the MUT-p53 subgroup).
Figure 4.

Concurrent evaluation of p53 and MDM2 overexpression in DLBCL.
Differentially expressed genes
DEGs were identified by comparing GEPs of different groups: MDM2+ vs MDM2– DLBCLs with WT-p53 (DEGs1: 157) or MUT-p53 (DEGs2: 302), as well as p53+ vs p53– DLBCLs with WT-p53 (DEGs3: 547) or MUT-p53 (DEGs4: 0) (Figure 5A-C). Although DEGs1 and DEGs2 shared 11 genes (supplemental Table 1), they appeared to be p53- and MDM2-independent, suggesting the different and heterogeneous transcription programs in the MUT-p53 subgroup. Common 49 genes (supplemental Table 2) between DEGs1 and DEGs3 include CDKN1A/p21, MDM2, MDM4, and ATM. Albeit the stress transmitter and p53 activator, ATM was downregulated both in MDM2+ and p53+ DLBCLs with WT-p53, probably by p53-inducible Wip1.12 Only one gene (probable ATP-dependent RNA helicase DDX59) overlaps DEGs2 and DEGs3.
Figure 5.

Comparison and characterization of differentially expressed genes in subgroups of DLBCL patients treated with R-CHOP. (A) DEGs in patients with MDM2+ and MDM2– DLBCL with WT-p53. (B) DEGs in patients with MDM2+ and MDM2– DLBCL with MUT-p53. (C) DEGs in patients with p53+ and p53– DLBCL with WT-p53. (D) MDM2 amplification shown by FISH. In every single cell, the orange signals (MDM2), by at least two times, outnumber the green signals (centromere 12). (E-F) Impact of MDM2 SNP309 on OS and PFS in DLBCL patients.
To identify genes and pathways responsible for the different clinical outcomes of MDM2+ vs MDM2– DLBCL patients with MUT-p53, DEGs2 with fold change >2 were examined (supplemental Table 3). In MDM2+ DLBCL patients, 8 genes with known function (http://www.uniprot.org) were expressed at significantly higher levels (1.66- to 2.64-fold change, P < .0023), whereas 21 genes were expressed at significantly lower levels (1.66- to 2.46-fold change, P < .0023). None of these genes has been reported to interact with MDM2, but CXCL5,40 MBD4,41 PAK2, ATG7,42 and DCUN1D143 have been reported to interact with the TP53 pathway, and many genes downregulated in MDM2+ patients are related to DNA repair or cell death. Upregulation of CXCL5 suggests gain-of-function of MUT-p53s in MDM2+ DLBCL,40 whereas downregulated MBD4, PAK2, ATG7, and DCUN1D1 in MDM2+ DLBCL suggest loss-of-function of MUT-p53s. Interestingly, not all upregulated genes in patients with MDM2+ DLBCL are oncogenic (such as MAD/MXD1 antagonizing MYC function), and there are several oncogenes downregulated in MDM2+ DLBCLs (such as ATAD2 and DCUN1D1). When we further compared the GEPs of MDM2+ DLBCLs with WT-p53 vs MDM2+ DLBCLs with MUT-p53, only 14 transcripts were significantly differentially expressed (P cutoff: .000113; FDR: .30), which appears to indicate the simultaneous presence of WT-p53 transcription activities in some MDM2+ patients with MUT-p53.
MDM2 amplification
MDM2 amplification was identified by FISH (Figure 5D) in 3 of 364 patients (0.8%). All 3 patients had strongly MDM2+ tumors (100%, 95%, and 60%). The 2 patients with 100% and 95% tumor cells expressing MDM2 had WT-p53, and the patient with 60% MDM2+ cells had MUT-p53. These tumors had 90%, 70%, and 0% p53-positive cells, respectively. All 3 patients were alive and censored at last follow-up.
MDM2 SNP309 polymorphism and survival
SNP309 genotyping was analyzed in 108 patients treated with R-CHOP. Clinical features of patients with homogenous T/T genotype vs with T→G polymorphism were compared (Table 2A). The only significantly different variant is the number of extranodal sites. Most female patients were elderly. In this cohort, no significant difference in the mean MDM2 expression levels was detected in patients with different SNP309 genotypes (Table 1C).
C. Numbers of DLBCL patients with different SNP309 genotypes and the mean MDM2 expression level
| Genotype | Number of patients | Mean MDM2 expression |
|---|---|---|
| G/G | 10 | 33% |
| G/T | 57 | 42% |
| T/T | 41 | 42% |
OS and PFS were compared among patients with G/G (n = 10), G/T (n = 57), and T/T (n = 41) genotypes. The SNP309 T→G change did not correlate with poorer survival in DLBCL patients (Figure 5E-F).
Discussion
In contrast with several reports in the literature for other types of human cancer, we show in DLBCL patients that MDM2 overexpression has no significant (P > .05) adverse impact on survival in the entire cohort (Figures 2 and 3A) and in the WT-p53 subcohort (Figures 2C and 3C) with a >10% cutoff or with other cutoffs of 20% to 70%, suggesting that suppression of p53 by MDM2 in tumor cells did not significantly affect WT-p53 function under stress conditions. Possible explanations include: (1) Posttranslational modifications of p53 and MDM2 resulted in reduced repression by MDM2, and more degradation of tumorigenic MDM29,28 rather than degradation of p53; (2) interaction of p53/MDM2 can be regulated by many other factors (eg, ARF, L11, MDM4)1; (3) during the time delay of MDM2 repression, the tumor suppressor function of WT-p53 is already exerted; and (4) other MDM2 functions positive for the p53 function may also exist.26-28 Correspondingly, MDM2 overexpression did not significantly affect the clinical outcomes of DLBCL patients with WT-p53 in our study, which may explain why clinical trials of MDM2 inhibitors have not shown impressive efficacy.1
Similarly, p53 overexpression by IHC did not predict poorer survival in patients with WT-p53 (Figures 2 and 3D). This is true for cutoffs of 10% to 70%. Overexpression of WT-p53 in tumor cells could be caused by high endogenous cellular stress (DNA damage or oncogene activation), which increases the fraction of cells with oscillatory p53/MDM2 pulses17 (only part of which were detected by IHC). The correlation between levels of p53 reflecting pretreatment cellular stress level and clinical outcomes was not significant, probably because after chemotherapy treatment, p53 would be activated to high levels as a result of the widespread DNA damage rendered by chemotherapy or radiation. What determined the tumor cell fate and the therapeutic response depended on whether p53 functions properly after the treatment, not the p53 expression level before treatment.
In contrast, in DLBCL patients with MUT-p53, p53 overexpression correlated with significantly worse survival (Figures 2 and 3F), which stayed significant with cutoffs of 10% to 60% (P values are marginal for 40-60%). Because all non-sense, splice-site, and frame-shift p53 mutants had a p53-negative phenotype (accounting for 53% of MUT-p53– cases) and MUT-p53+ patients carried exclusively missense mutants with probable dominant oncogenic gain-of-function, the different prognosis of MUT-p53+ and MUT-p53– arms may be attributed to the variable loss-of-function and gain-of-function of MUT-p53s.44 However, the survival difference between patients with missense p53 mutations and other patients is not significant. Moreover, after excluding other types of p53 mutants, patients with missense p53 mutants were still stratified into 2 groups, with significant different survival according to p53 overexpression or not before chemotherapy. In a mouse model, MUT-p53 was stabilized by oncogene activation.45 Higher MUT-p53 expression levels at diagnosis may indicate higher DNA damage, sustained oncogene activation, loss-of-function (including MDM2 activation), and gain-of-function of MUT-p53 at presentation; chemotherapy probably further stabilized and increased the levels of MUT-p5345 and added more DNA damage to tumor cells because of the absence of WT-p53 function. Therefore, pretreatment MUT-p53 expression level correlated with patient survival even after chemotherapy.
Because MUT-p53s have oncogenic gain-of-function, the MDM2-mediated degradation of MUT-p53 should alleviate oncogenesis. Unexpectedly, overexpression of MDM2, which was thought to regulate oncoprotein MUT-p53 in the same way as WT-p53 (MDM2 ┤p53),24 was also correlated with poor survival in our MUT-p53 subcohort, mirroring p53 overexpression (Figures 2 and 3E). To explain this observation, it may be that the impact of MDM2 ┤MUT-p53 on p53 function was simply not as significant as MDM2 ┤WT-p53, or MDM2 ┤MUT-p53 was lost in MUT-p53 DLBCL.46 However, none of the MUT-p53s in our study had mutations in the MDM2-binding domain (N-terminus)46; most MUT-p53s had mutations in the DNA-binding domain (DBD),34 which may have altered conformation and interaction with MDM2 by other sites alternative to the N-terminus, yet could have still been efficiently degraded by MDM2 through a pathway different from WT-p53.47 Conversely, it has been reported in cell lines that MDM2 and MUT-p53 stabilize each other (p53→MDM2 and MDM2→p53).25 Supporting this theory, coexistence of MDM2 and MUT-p53 was observed in lung carcinomas, and the authors speculated that this resulted in a gain-of-function phenotype, stabilizing MDM2, which otherwise has a short half-life.37 Moreover, MDM2 mRNA levels were not significantly different between MDM2+ and MDM2– DLBCLs with MUT-p53, according to our GEP analysis (Figure 5B; supplemental Table 3), suggesting that MDM2 overexpression is caused by an increased stability at the protein level.
Another explanation is the p53-independent oncogenic function of MDM2.2,12 However, this idea is weakened by the fact that the same effect was not shown in the WT-p53 subgroup. In addition, none of the DEGs between MDM2+ and MDM2– patients with MUT-p53 have been found to relate to MDM2 (supplemental Table 3). Instead, 5 genes are related to WT- or MUT-p53. Therefore, it seems likely that the survival difference between MDM2+ and MDM2– patients can ultimately be attributed to the variable function and activities of different MUT-p53s.44 In this setting, MDM2 expression should be evaluated concurrently with MUT-p53 expression for prognostic studies.
Stratification by concomitant evaluation of p53 and MDM2 expression showed that MDM2+ and p53+ patients are prognostic equivalents in WT- or MUT-p53 subgroups (Figure 4), supporting the fact that MDM2+ and p53+ evaluation can be affected by the oscillatory characteristics of p53/MDM2 levels, and that MDM2 overexpression may largely reflect p53 activity, an indication of cellular stress in tumor cells, or the oncogenic activity of MUT-p53.
MDM2 overexpression potentially could be caused by MDM2 gene amplification, enhanced transcription and translation, or reduced degradation. MDM2 SNP309 T→G causes enhanced MDM2 transcription, resulting in attenuated p53 function.32,33 In our study, MDM2 was rarely amplified. Similarly, increased MDM2 expression was not shown in the homozygous (G/G) or heterozygous (G/T) for SNP309 tumor cells among 108 patients in this study, and SNP309 T→G did not correlate with significantly poorer survival. Although similar results have been shown in other reports,48,49 the lack of significance may be attributable to a small number of patients, similar estrogen levels in the SNP309 groups, oscillation of MDM2 pulses, and the possibility that an endogenous MDM2 level could not be distinguished by IHC (which scores a fraction of positive cells but not the intensity).
In our large DLBCL patient cohort, high MDM2 was not necessarily associated with low p53 (64 of 157 MDM2+ patients with WT-p53 had p53+ phenotype), p53 level could be high in patients with WT-p53 (only ∼44% of p53+ patients had MUT-p53), and expression of MDM2 and p53 mutations were not mutually exclusive abnormalities of the TP53 pathway (Figure 2E; Table 1). These observations may reflect the existence of oscillatory p53/MDM2 pulses in tumor cells or other regulatory factors that affect p53 and MDM2 levels (eg, ARF, L11, MDM4, ATM, Akt, p300/CBP, p73).1,2 Nonetheless, several trends were observed by evaluating the mean p53/MDM2 expression levels (Table 1). These trends suggest that MDM2 was induced by WT-p53, whereas the WT-p53 level was low because of the reduced cellular stress by WT-p53 function and increased degradation by MDM2. In contrast, in patients with MUT-p53, MDM2 level was not increased because of the loss-of-function of MUT-p53 (according to the yeast p53-functional assay,50 only 4 MUT-p53s in MDM2– cases and 5 MUT-p53s in MDM2+ cases of this study preserved function to transactivate MDM2); MDM2+ phenotype in 35% of MUT-p53 cases is likely caused by enhanced stabilization (otherwise short half-life of MDM2) by oncogenic activities of MUT-p53. MUT-p53 levels remained high because of the sustained cellular stress, which activated and stabilized MUT-p53 similarly to WT-p5345 and reduced degradation by lower levels of MDM2 (Table 1B).
In conclusion, our results in R-CHOP–treated DLBCL patients show that p53 or MDM2 overexpression predicts a significantly poor survival in patients with MUT-p53 but not in patients with WT-p53. The prognostic value of p53 overexpression is attributable to the poor prognosis of MUT-p53+ but not WT-p53+, and that MUT-p53– patients were not necessary to have a poor survival. Stratification by p53 overexpression can be further improved by concomitant evaluation of MDM2 expression. These findings may explain the inconsistent prognostic value of p53 and MDM2 by several previous reports in the literature and provide direction to the future therapeutic drug designs targeting the p53-MDM2 pathway.
Supplementary Material
Acknowledgments
This work was supported by Harold C. and Mary L. Daily Endowment Fellowships at The University of Texas MD Anderson Cancer Center (Z.Y.X.M.); the Stiftung zur Krebsbekaempfung Zurich Grant 269 award (A.T.); the Ministerio de Ciencia e Innovación, Spain (RETICC, SAF2008-03871) and the Spanish Association against Cancer (AECC) (M.A.P.); National Cancer Institute and National Institutes of Health grant 1R01CA160558 (W.H); a University of Texas MD Anderson Cancer Center Institutional Research Grant Award, an Anderson Lymphoma Specialized Programs of Research Excellence (SPORE) Research Development Program Award, an Anderson Myeloma SPORE Research Development Program Award, and Anderson Collaborative Research Funds with High-Throughput Molecular Diagnostics and Roche Molecular Systems (K.H.Y.). This work was also partially supported by the National Cancer Institute and National Institutes of Health grants (R01CA138688, 1RC1CA146299, P50CA136411, and P50CA142509) and by the MD Anderson Cancer Center Support Grant CA016672.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Z.Y.X.-M. and K.H.Y. designed research; Z.Y.X.-M. and K.H.Y. performed research; Z.Y.X.-M., M.B.M., A.T., S.M.-M., W.H., G.C.M., L.K., L.F., C.V., K.D., A.C., W.T., Y.Z., G.B., K.L.R., E.D.H., W.W.L.C., J.H.v.K., Q.H., J.H., W.A., M.P., A.J.M.F., J.N.W., L.W., X.Z., R.S.G., Y.L., M.A.P., L.J.M., and K.H.Y. contributed vital new reagents, resources, and analytical tools under approved IRB and MTA; Z.Y.X.-M., M.B.M., A.T., S.M.-M., C.V., K.D., A.C., Y.Z., G.B., K.L.R., E.D.H., W.W.L.C., X.Z., J.H.v.K., Q.H., J.H., M.P., A.J.M.F., J.N.W., X.Z., R.S.G., Y.L., M.A.P., and K.H.Y. collected clinical and follow-up data under approved IRB and MTA; Z.Y.X.-M., M.B.M., A.T., S.M.-M., W.H., G.C.M., L.F., C.V., K.D., A.C., W.T., Y.Z., G.B., K.L.R., E.D.H., W.W.L.C., J.H.v.K., Q.H., J.H., W.A., M.P., A.J.M.F., J.N.W., L.W., X.Z., R.S.G., C.E.B.-R., S.A.W., Y.L., M.A.P., L.J.M., and K.H.Y. contributed vital strategies, participated in discussions, and provided scientific input; Z.Y.X.-M. and K.H.Y. analyzed data; Z.Y.X.-M. and K.H.Y. performed and supported statistical analysis; and Z.Y.X.-M., L.J.M., and K.H.Y. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ken H. Young, The University of Texas MD Anderson Cancer Center, Department of Hematopathology, 1515 Holcombe Blvd, Houston, TX 77030-4009; e-mail: khyoung@mdanderson.org; and Miguel A. Piris, Servicio de Anatomia Patologica, Hospital Universitario Marques de Valdecilla Fundacion IFIMAV, 39008 Santander, Spain; e-mail: mapiris@humv.es.
References
- 1.Xu-Monette ZY, Medeiros LJ, Li Y, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119(16):3668–3683. doi: 10.1182/blood-2011-11-366062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1(14):993–1000. [PubMed] [Google Scholar]
- 3.Onel K, Cordon-Cardo C. MDM2 and prognosis. Mol Cancer Res. 2004;2(1):1–8. [PubMed] [Google Scholar]
- 4.Wang P, Lushnikova T, Odvody J, Greiner TC, Jones SN, Eischen CM. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27(11):1590–1598. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- 5.Capoulade C, Bressac-de Paillerets B, Lefrère I, et al. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene. 1998;16(12):1603–1610. doi: 10.1038/sj.onc.1201702. [DOI] [PubMed] [Google Scholar]
- 6.Møller MB, Nielsen O, Pedersen NT. Oncoprotein MDM2 overexpression is associated with poor prognosis in distinct non-Hodgkin’s lymphoma entities. Mod Pathol. 1999;12(11):1010–1016. [PubMed] [Google Scholar]
- 7.Solenthaler M, Matutes E, Brito-Babapulle V, Morilla R, Catovsky D. p53 and mdm2 in mantle cell lymphoma in leukemic phase. Haematologica. 2002;87(11):1141–1150. [PubMed] [Google Scholar]
- 8.Dang J, Kuo ML, Eischen CM, Stepanova L, Sherr CJ, Roussel MF. The RING domain of Mdm2 can inhibit cell proliferation. Cancer Res. 2002;62(4):1222–1230. [PubMed] [Google Scholar]
- 9.Wagner J, Ma L, Rice JJ, Hu W, Levine AJ, Stolovitzky GA. p53-Mdm2 loop controlled by a balance of its feedback strength and effective dampening using ATM and delayed feedback. Syst Biol (Stevenage) 2005;152(3):109–118. doi: 10.1049/ip-syb:20050025. [DOI] [PubMed] [Google Scholar]
- 10.Ciliberto A, Novak B, Tyson JJ. Steady states and oscillations in the p53/Mdm2 network. Cell Cycle. 2005;4(3):488–493. doi: 10.4161/cc.4.3.1548. [DOI] [PubMed] [Google Scholar]
- 11.Pang LY, Scott M, Hayward RL, et al. p21(WAF1) is component of a positive feedback loop that maintains the p53 transcriptional program. Cell Cycle. 2011;10(6):932–950. doi: 10.4161/cc.10.6.15012. [DOI] [PubMed] [Google Scholar]
- 12.Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010;17(1):93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 13.Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci USA. 2000;97(21):11250–11255. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monk NA. Oscillatory expression of Hes1, p53, and NF-kappaB driven by transcriptional time delays. Curr Biol. 2003;13(16):1409–1413. doi: 10.1016/s0960-9822(03)00494-9. [DOI] [PubMed] [Google Scholar]
- 15.Hamstra DA, Bhojani MS, Griffin LB, Laxman B, Ross BD, Rehemtulla A. Real-time evaluation of p53 oscillatory behavior in vivo using bioluminescent imaging. Cancer Res. 2006;66(15):7482–7489. doi: 10.1158/0008-5472.CAN-06-1405. [DOI] [PubMed] [Google Scholar]
- 16.Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 17.Ma L, Wagner J, Rice JJ, Hu W, Levine AJ, Stolovitzky GA. A plausible model for the digital response of p53 to DNA damage. Proc Natl Acad Sci USA. 2005;102(40):14266–14271. doi: 10.1073/pnas.0501352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proctor CJ, Gray DA. Explaining oscillations and variability in the p53-Mdm2 system. BMC Syst Biol. 2008;2:75. doi: 10.1186/1752-0509-2-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, et al. Oscillations and variability in the p53 system. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100068. 2006.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puszyński K, Hat B, Lipniacki T. Oscillations and bistability in the stochastic model of p53 regulation. J Theor Biol. 2008;254(2):452–465. doi: 10.1016/j.jtbi.2008.05.039. [DOI] [PubMed] [Google Scholar]
- 21.Hunziker A, Jensen MH, Krishna S. Stress-specific response of the p53-Mdm2 feedback loop. BMC Syst Biol. 2010;4:94. doi: 10.1186/1752-0509-4-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ouattara DA, Abou-Jaoudé W, Kaufman M. From structure to dynamics: frequency tuning in the p53-Mdm2 network. II Differential and stochastic approaches. J Theor Biol. 2010;264(4):1177–1189. doi: 10.1016/j.jtbi.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 23.Kim DH, Rho K, Kim S. A theoretical model for p53 dynamics: identifying optimal therapeutic strategy for its activation and stabilization. Cell Cycle. 2009;8(22):3707–3716. doi: 10.4161/cc.8.22.10023. [DOI] [PubMed] [Google Scholar]
- 24.Terzian T, Suh YA, Iwakuma T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22(10):1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng Y, Chen L, Li C, Lu W, Agrawal S, Chen J. Stabilization of the MDM2 oncoprotein by mutant p53. J Biol Chem. 2001;276(9):6874–6878. doi: 10.1074/jbc.C000781200. [DOI] [PubMed] [Google Scholar]
- 26.Candeias MM, Malbert-Colas L, Powell DJ, et al. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10(9):1098–1105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 27.Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009;15(3):171–183. doi: 10.1016/j.ccr.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 28.Deb SP. Cell cycle regulatory functions of the human oncoprotein MDM2. Mol Cancer Res. 2003;1(14):1009–1016. [PubMed] [Google Scholar]
- 29.McDonnell TJ, Montes de Oca Luna R, Cho S, Amelse LL, Chavez-Reyes A, Lozano G. Loss of one but not two mdm2 null alleles alters the tumour spectrum in p53 null mice. J Pathol. 1999;188(3):322–328. doi: 10.1002/(SICI)1096-9896(199907)188:3<322::AID-PATH372>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 30.Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci USA. 1998;95(26):15608–15612. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barboza JA, Iwakuma T, Terzian T, El-Naggar AK, Lozano G. Mdm2 and Mdm4 loss regulates distinct p53 activities. Mol Cancer Res. 2008;6(6):947–954. doi: 10.1158/1541-7786.MCR-07-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 33.Hu W, Feng Z, Ma L, et al. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007;67(6):2757–2765. doi: 10.1158/0008-5472.CAN-06-2656. [DOI] [PubMed] [Google Scholar]
- 34.Xu-Monette ZY, Wu L, Visco C, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120(19):3986–3996. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheson BD. The International Harmonization Project for response criteria in lymphoma clinical trials. Hematol Oncol Clin North Am. 2007;21(5):841–854. doi: 10.1016/j.hoc.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 36.Visco C, Li Y, Xu-Monette ZY, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia. 2012;26(9):2103–2113. doi: 10.1038/leu.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorgoulis VG, Zoumpourlis V, Rassidakis GZ, et al. A molecular and immunohistochemical study of the MDM2 protein isoforms and p53 gene product in bronchogenic carcinoma. J Pathol. 1996;180(2):129–137. doi: 10.1002/(SICI)1096-9896(199610)180:2<129::AID-PATH646>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Møller MB, Nielsen O, Pedersen NT. Frequent alteration of MDM2 and p53 in the molecular progression of recurring non-Hodgkin’s lymphoma. Histopathology. 2002;41(4):322–330. doi: 10.1046/j.1365-2559.2002.01506.x. [DOI] [PubMed] [Google Scholar]
- 39.Rassidakis GZ, Thomaides A, Wang S, et al. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia. 2005;19(9):1663–1669. doi: 10.1038/sj.leu.2403840. [DOI] [PubMed] [Google Scholar]
- 40.Yeudall WA, Vaughan CA, Miyazaki H, et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis. 2012;33(2):442–451. doi: 10.1093/carcin/bgr270. [DOI] [PubMed] [Google Scholar]
- 41.Ruzov A, Shorning B, Mortusewicz O, Dunican DS, Leonhardt H, Meehan RR. MBD4 and MLH1 are required for apoptotic induction in xDNMT1-depleted embryos. Development. 2009;136(13):2277–2286. doi: 10.1242/dev.032227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee IH, Kawai Y, Fergusson MM, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336(6078):225–228. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O-charoenrat P, Sarkaria I, Talbot SG, et al. SCCRO (DCUN1D1) induces extracellular matrix invasion by activating matrix metalloproteinase 2. Clin Cancer Res. 2008;14(21):6780–6789. doi: 10.1158/1078-0432.CCR-08-0719. [DOI] [PubMed] [Google Scholar]
- 44.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26(12):1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suh YA, Post SM, Elizondo-Fraire AC, et al. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011;71(23):7168–7175. doi: 10.1158/0008-5472.CAN-11-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue T, Geyer RK, Howard D, Yu ZK, Maki CG. MDM2 can promote the ubiquitination, nuclear export, and degradation of p53 in the absence of direct binding. J Biol Chem. 2001;276(48):45255–45260. doi: 10.1074/jbc.M107477200. [DOI] [PubMed] [Google Scholar]
- 47.Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27(23):8284–8295. doi: 10.1128/MCB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bittenbring J, Parisot F, Wabo A, et al. MDM2 gene SNP309 T/G and p53 gene SNP72 G/C do not influence diffuse large B-cell non-Hodgkin lymphoma onset or survival in central European Caucasians. BMC Cancer. 2008;8:116. doi: 10.1186/1471-2407-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartmann E, Fernàndez V, Stoecklein H, Hernández L, Campo E, Rosenwald A. Increased MDM2 expression is associated with inferior survival in mantle-cell lymphoma, but not related to the MDM2 SNP309. Haematologica. 2007;92(4):574–575. doi: 10.3324/haematol.10891. [DOI] [PubMed] [Google Scholar]
- 50.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28(6):622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.