

Abstract
Catechol-O-methyltransferase (COMT) is of great importance in pharmacology because it catalyzes the metabolism (methylation) of endogenous and xenobiotic catechols. Moreover, inhibition of COMT is the drug target in the management of central nervous system (CNS) disorders such as Parkinson's disease due to its role in regulation of the dopamine level in the brain. The X-ray crystal structures for COMT have been available since 1994. The active sites for cofactor and substrate/inhibitor binding are well resolved to an atomic level, providing valuable insights into the catalytic mechanisms as well as the role of magnesium ions in catalysis. Determination of how the substrates/inhibitors bind to the protein leads to a structure-based approach that has resulted in potent and selective inhibitors. This review focuses on the design of two types of inhibitors (nitrocatechol-type and bisubstrate inhibitors) for COMT using the protein structures.
Keywords: catechol-O-methyltransferase, COMT, crystal structure, drug discovery, drug metabolism
Introduction
Catechol O-methyltransferase (COMT, EC 2.1.1.6) catalyzes the methylation reaction (generally classified as phase II metabolism) whereby a methyl group from the cofactor S-adenosyl-L-methionine (SAM) is transferred to one of the catecholic hydroxyls [1]. COMT exists in two forms. The soluble form (S-COMT) is located in cytosol and the membrane-bound form (MB-COMT) is anchored to the rough endoplasmic reticulum. The two forms differ only by a 50 residue long extension in the MB-form, which is the signal sequence for membrane anchoring [2]. S-COMT is the predominant form in most tissues (highest levels are found in the liver and kidney) with the exception that MB-COMT predominates in the brain [3,4]. MB-COMT may be more relevant in inactivation of catecholaminergic neurotransmitters, whereas S-COMT plays a more important role in inactivation of endogenous and xenobiotic catechols in other tissues. The methylation reaction by COMT occurs via a sequentially ordered mechanism [2]. The binding sequence of SAM, the Mg2+ ion and the substrate to the enzyme is strictly maintained in a catalytic circle. SAM firstly binds to the enzyme, followed by the Mg2+ ion and the substrate [5,6].
COMT is the primary enzyme that inactivates the catechol neurotransmitter dopamine and the drug L-dopa [7]. L-dopa is used in the clinical treatment of central nervous system (CNS) disorders such as Parkinson's disease [8] and possibly others (e.g. schizophrenia [9,10] and depression [11]. Its efficacy is associated with the level of dopamine converted from the drug. Studies have shown that inhibition of COMT activity results in marked reduction of the body clearance of L-dopa and dopamine [1], leading to a sustained level of dopamine in the brain and improved efficacy (Figure 1A, B) [12,13]. The important role of COMT in treatment of Parkinson's disease has promoted an area wherein the aims are to design potent and selective COMT inhibitors. Several of these inhibitors have been used as adjuncts to L-dopa therapy.
Figure 1.

A/B) COMT inhibitors can modulate the pharmacokinetics of L-dopa. A) COMT-mediated methylation is the major clearance pathway for L-dopa and dopamine in peripheral tissues. B) Schematic representation of the changes in the pharmacokinetic profile of L-dopa, showing increased bioavailability and elimination half-life. MEC: minimal effective concentration. C) The tertiary structure of the COMT enzyme (using human COMT (PDB code: 3BWM) as an example), depicting the overall fold of a COMT structure
A number of crystal structures are available for human and rat COMTs (Table 1). The rat and human COMTs share 81% sequence identity and both belong to the highly structurally conserved SAM-dependent methyltransferase fold family (class I) [14]. The crystal structures have provided rationales why COMT accepts a wide range of structurally variable substrates with the only strict requirement that the substrate must have a catechol structure. More importantly, these structures have been used for searching and designing COMT inhibitors that would enhance the L-dopa treatment of Parkinson's disease.
Table 1.
List of available crystal structures for COMT registered at http://www.pdb.org
| NAT | PDB entries | In complex with | Reference |
|---|---|---|---|
| Human COMT (108V) | 3BWM | SAM, Mg2+, DNC | [16] |
| Human COMT (108 M) | 3BWY | SAM, Mg2+, DNC | [16] |
| Rat COMT | 1VID | SAM, Mg2+, DNC | [15] |
| Rat COMT | 2ZVJ | SAM, Mg2+, 4-phenyl-7,8-dihydroxycoumarin | [17] |
| Rat COMT | 2CL5 | SAM, Mg2+, SAM, Mg2+, BIA 8–176 | [22] |
| Rat COMT | 1JR4 | SAM, Mg2+, ‘bisubstrate’ | [24] |
| Rat COMT | 1H1D | SAM, Mg2+, BIA 3–335 | [23] |
| Rat COMT | 3HVH | Mg2+, N6-methyladenine-containing bisubstrate | [19] |
| Rat COMT | 3HVI | Mg2+, N6-ethyladenine-containing bisubstrate | [19] |
| Rat COMT | 3HVJ | Mg2+, N6-propyladenine-containing bisubstrate | [19] |
| Rat COMT | 3HVK | Mg2+, purine-containing bisubstrate | [19] |
| Rat COMT | 3NW9 | Mg2+, methylpurine-containing bisubstrate | [20] |
| Rat COMT | 3OE4 | Mg2+, purine-containing bisubstrate inhibitor | [20] |
| Rat COMT | 3OE5 | Mg2+, a pyridylsulfanyl-containing inhibitor | [20] |
| Rat COMT | 3OZR | Mg2+, a bisubstrate (no substituent in the adenine site) | [20] |
| Rat COMT | 3OZS | Mg2+, a trifluoromethyl-imidazolyl-containing inhibitor | [20] |
| Rat COMT | 3OZT | Mg2+, a 4-oxo-pyridinyl-containing inhibitor | [20] |
| Rat COMT | 3NWB | Mg2+, a fluorinated desoxyribose-containing bisubstrate | [21] |
| Rat COMT | 3NWE | Mg2+, a methylated desoxyribose bisubstrate | [21] |
| Rat COMT | 3U81 | SAH | [21] |
| Rat COMT | 3S68 | Mg2+, SAM and Tolcapone | [21] |
| Rat COMT | 3R6T | Mg2+, a bisubstrate inhibitor | [21] |
| Rat COMT | 2ZTH | Mg2+, SAM | [18] |
| Rat COMT | 2ZLB | [18] |
3D structure of COMT
The crystal structures of human and rat COMTs reveal that the enzymes adopt a similar structural fold [15,16]. Specifically, the COMT enzyme is composed of a seven-stranded β-sheet core (arranged in an order of 3214576) sandwiched between two sets of α-helices (helices α1–α5 on one side and helices α6–α8 on the other side) (Figure 1C). In the β-sheet, strand 7 is antiparallel to the others (Figure 1C). The cofactor SAM interacts with conserved residues along the first half of the core β-sheet (β1–β4) (Figure 2A) [16]. E90 (β2) forms hydrogen bonds with the ribose hydroxyl groups of SAM (Figure 2A). The adenine ring of SAM forms hydrogen bonds with S119 and Q120 (α6) and van der Waals interactions with residues I91 (β2), A118 (β3–α6 loop) and W143 (β4–α7 loop) (Figure 2A). The methionine portion of SAM is coordinated through the hydrogen bonds with residues V42 (α2-α3 loop), S72 (α4), D141 (β4) and the hydrophobic interactions with M40, V42, and Y68 (Figure 2A). The methyl group (CH3) attached to the methionine sulfur atom in SAM is oriented toward the substrate binding site and specifically towards the catechol oxygen atom to be methylated (Figure 2A).
Figure 2.
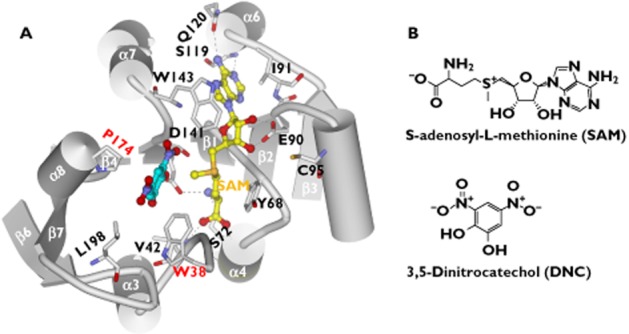
The substrate-binding and SAM-binding sites in the COMT protein (PDB code: 3BWM). A) Molecular interactions of SAM and DNC with the binding site residues. The dashed lines indicate hydrogen bonds. B) Chemical structures of SAM (S-adenosyl-L-methionine) and DNC (3,5-dinitrocatechol)
The substrate-binding site is a shallow pocket defined by M40, L198, W143 and ‘gatekeeper’ residues W38 and P174 (Figure 2A). All these residues are hydrophobic, suggesting that van der Waals contacts are the main forces for ligand binding (Figure 2A). The magnesium ion (Mg2+), a necessary cofactor for the methylation, is present in all the COMT crystals complexed with a ligand [15–24]. The Mg2+ ion is octahedrally coordinated in the active site by the side chains of D141, D169, N170, the two hydroxyl groups of the catechol substrate, and a water molecule [16]. This is direct evidence that Mg2+ participates in the enzymatic reaction by facilitating the substrate binding. It thus explains why the ion is required for COMT mediated catalysis. It is noteworthy that the Mg2+ ion also lowers the pKa of the catechol hydroxyls, making them more easily ionized [5].
The Km values for methylation of catechol substrates are generally smaller in human COMT compared with rat COMT [25]. Comparison of the active sites of rat and human COMT provided a rationale why the two proteins differ in Km value and other kinetic properties [16]. The Mg2+-ligand distances in the crystal of human COMT are shorter than those in that of rat COMT, indicating that substrate binding is stronger (smaller Km value) in human protein [16]. Although most of residues are identical in the SAM binding sites of the two proteins, three residues are found to be different, namely, M89, M91 and Y95 in the rat protein, but I89, I91 and C95 in the human protein. The residues in the rat protein are bulkier and thus interact more closely with SAM compared with the human protein. Moreover, the substrate-binding site contains two charged substitutions (R201 and E202 in humans contrasting with M201 and K202 in rats). Direct interactions of the two residues with the substrate appear to be unlikely. However, they may affect Vmax as well as substrate binding and release due to their location at the pocket entrance [16].
Nitrocatechol-type inhibitors
Nitro-substituted catechols have been found to inhibit COMT activity with varied inhibition potency. They possess the same binding motif as the catechol substrates, but the presence of the strong electron-withdrawing nitro function hinders their reactivity toward O-methylation [26]. 3,5-dinitrocaatechol (DNC, Figure 2B) is a competitive inhibitor of COMT. The crystal structure of the COMT-DNC complex reveals how a nitrocatechol-type inhibitor interacts with the protein. The DNC molecule occupies the substrate-binding site, which is consistent with the competitive inhibition mechanism (Figure 2A). The 3-nitro group of DNC has favourable van der Waals interactions with W143, whereas the benzene ring of DNC forms edge-to-face π-π interactions with W38 (Figure 2A). W38 is very important for high affinity binding of catechols [15]. Substitution of residue 38 from tryptophan to arginine in pig COMT reduces the affinity of catechol compounds by 10∼1000-fold [27]. Tolcapone and entecapone are two nitrocatechol-type inhibitors that have been introduced into the drug market [1,12]. They are used as adjuncts to L-dopa therapy in the management of Parkinson's disease, although each of them has problems either in pharmacokinetics, clinical efficacy or in toxicity [28–30].
1-(3,4-dihydroxy-5-nitrophenyl)-2-phenyl-ethanone (BIA 3–202, Figure 3A) is a potent nitrocatechol-type COMT inhibitor that demonstrates selective inhibition of peripheral COMT [31]. The potential benefits of the compound for the treatment of Parkinsonian patients are under clinical evaluation. The X-ray structures predicted that methylation of BIA 3–302 and its analogues (e.g. tolcapone) occurred preferentially at the catechol hydroxyl group in position para (relative to the C1 substituent) over the hydroxyl in position meta [32]. This prediction was confirmed by the in vitro O-methylation experiments [32]. However, the regioselectivity of O-methylation in vivo shifts towards much higher meta : para ratios, with lesser or null amount of the p-O-methylated products being formed [32]. An explanation was proposed for this apparent paradox of regioselectivity [32]. The compounds undergo in vivo O-methylation by COMT at either meta or para catechol hydroxyl groups. In a subsequent step, the p-O-methyl derivatives are selectively demethylated by a microsomal enzyme system. The overall balance is the accumulation of the m-O-methylated metabolites over the para-regioisomers.
Figure 3.
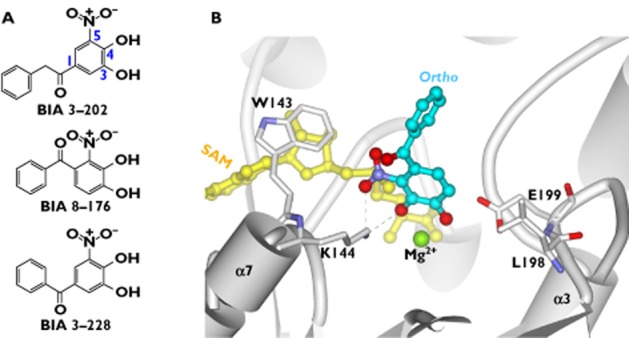
Binding mode of BIA 8–176 in the COMT protein (PDB code: 2CL5), showing that the inhibitor is bound in the ortho (in relation to the nitro group) pose. A) Chemical structures of BIA 3–202, BIA 8–176 and BIA 3–228. B) Molecular interactions of BIA 8–176 with the binding site residues. The dashed lines indicate hydrogen bonds
BIA 8–176 and BIA 3–228 (Figure 3A) are regioisomeric COMT inhibitors that contain the nitrocatechol core substituted with a benzoyl side chain. They differ structurally in that BIA 3–228 contains the benzoyl fragment placed in the meta position (relative to the nitro group), whereas in BIA 8–176, the side chain is at the ‘non-classic’ ortho position. The regioselectivity for O-methylation of the two inhibitors were determined [22]. The atomic interactions of COMT with the inhibitors were compared to explain the observed regioselectivity differences [22]. Structural analyses and docking revealed that interactions involving both the nitrocatechol moiety and the benzoyl side chain affect the energetic balance between ortho and meta poses, although the nitrocatechol moiety appears to play a more important role [22]. In the case of BIA 8–176, the effects of the benzoyl and nitro substituents are additive in determining the binding geometry. Thus the ortho configuration is greatly favoured (Figure 3B). By contrast, in the case of BIA 3–228, the effects of the same substituents on the binding geometry are partially self-compensatory and the energy difference between ortho and meta poses is relatively attenuated. Therefore, the regioselectivity of BIA 3–228 is less pronounced compared with BIA 8–176.
BIA 3–335 (Figure 4A) is a potent, reversible and tight-binding inhibitor of COMT (Ki = 6.0 nm) [33]. It displays a competitive inhibition towards the substrate binding site and non-competitive inhibition towards the SAM binding site [33]. The compound was identified as a promising candidate for clinical evaluation based on the evaluation of a series of nitrocatechol inhibitors to which heteroatom-containing substituents were introduced at C1 [33]. Structural variations of the substituents at C1 are predicted to modulate greatly the inhibitory activity towards COMT [31]. The crystal complex of COMT with BIA 3–335 reveals that the inhibitor binds into the catalytic site [33] (Figure 4B). The C1 substituent has sufficient space to accommodate within the protein structure. In fact, the substituent (side chain) lies within a long groove, where interactions with the hydrophobic residues (e.g. W38 and M201) are prevalent (Figure 4B). This structural knowledge is expected to provide useful guidelines for the design of better inhibitors.
Figure 4.
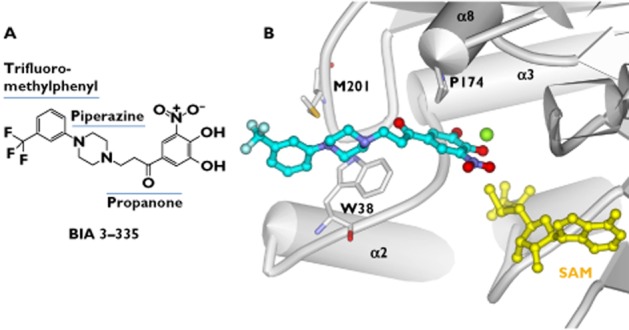
Molecular interactions of COMT with BIA 3–335 (PDB code: 1H1D). A) Chemical structures of BIA 3–335, showing its three components of the C1 substituent. B) Molecular interactions of BIA 3–335 with the binding site residues
Bisubstrate inhibitors
A bisubstrate inhibitor of enzymes is formed by covalently linking a substrate analogue with a cofactor analogue. The first series of bisubstrate inhibitors for COMT were designed and synthesized by Masjost et al. on the basis of the enzyme's crystal structure and molecular docking [34]. Three binding pockets were defined: catechol pocket (for binding catechol), adenosine pocket (for binding the adenosine portion of the SAM cofactor) and methionine pocket (for binding the methionine portion of the SAM cofactor) (Figure 5A, B). The methionine pocket with a polar nature was not considered in the design of the bisubstrate inhibitor because it would be favourably filled with water [34]. Bisubstrate inhibitors should incorporate the adenosine and catechol portions that bind to the corresponding pockets (Figure 5A, B). Connection of the two parts is through an appropriate spacer (linker) [24]. Docking analysis suggested that compound 1 (Figure 5A) provided best fitting compared with other analogues by preserving all original interactions of the catechol and adenosine with the protein. This was confirmed by the activity determination showing that compound 1 has the lowest IC50 value of 2 μm [34]. The bisubstrate inhibitors display much stronger affinity compared with the catechol inhibitors, confirming that targeting of COMT inhibitors to both the catechol and SAM binding site is a viable and more advantageous approach [34].
Figure 5.
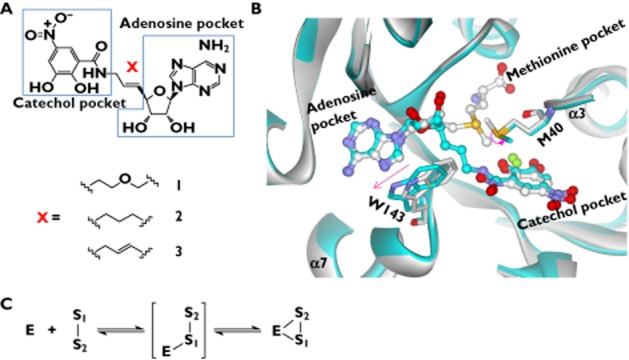
Binding mode of the bisubstrate inhibitor in the COMT protein. A) Chemical structures of the bisubstrate inhibitors 1–3. B) comparison of the binding sites of the bisubstrate inhibitor 3 (PDB code: 1JR4) and the catechol inhibitor DNC (PDB code: 1VID). The bisubstrate-binding protein and DNC-binding protein are indicated in cyan and white, respectively. The bisubstrate and DNC are also indicated in cyan and white (for carbons only), respectively. Arrows denote the changes in residue conformation.C): Bisubstrate inhibitor binds to the SAM pocket prior to binding to the catechol pocket in COMT. S1 is the adenosine portion of a bisubstrate inhibitor, whereas S2 is the catechol portion
The mechanism of enzyme inhibition by compound 1 was determined [34,35]. The compound exhibits competitive kinetics for the SAM and non-competitive kinetics for the catechol binding site. Dialysis assay indicated that the inhibitor binds reversibly but its dissociation from the enzyme is rather slow, a feature of a tight binding inhibitor [34]. The non-competitive inhibition pattern (with respect to the catechol binding site) appears to be contradictory to the fact that the inhibitor binds to the catechol site the same as the substrates. Two possible reasons have been proposed to explain the observed kinetics [34]. First, binding of the bisubstrate inhibitor to the SAM binding pocket induces alterations in the binding characteristics of the catechol site compared with when SAM is bound. This leads to a non-competitive mechanism for the binding of the bisubstrate inhibitor 1 to the catechol binding site. Second, the bisubstrate nature of the inhibitor influences the affinity of its catechol residue for the catechol binding site. This could result in a situation where the catechol residue of inhibitor 1 is ‘locked’ into the binding site. In both cases, the bisubstrate inhibitor must dock into the SAM binding site before it binds to the catechol binding pocket (Figure 5C).
Lerner et al. determined the effects of the linker structure on the affinity of the bisubstrate inhibitors [24]. The authors found that compound 3 with a propene as the linker is the most potent inhibitor (IC50 = 9 nm), followed by compounds 2 and 1 (Figure 5A). Kinetic analysis shows a competitive inhibition mechanism for compound 3 with regard to the SAM binding site and a more complex inhibition mechanism with regard to the catechol binding site. The crystal complex of COMT with compound 3 reveals that the inhibitor occupies, as predicted, both the SAM and catechol binding sites (Figure 5B). Binding of the bisubstrate inhibitor requires only small structural changes of the protein at residues W143 and M40 compared with the protein with the substrate and SAM bound (Figure 5B) [24]. The side chain of W143 is shifted by only 1.2 Å and M40 is rotated by 79°. These small changes are sufficient to make space for the fitting of the linker of the bisubstrate inhibitor in the protein (Figure 5B).
Paulini et al. synthesized a series of bisubstrate inhibitors that lack the nitro group on the catechol moiety [36]. In order to enhance the binding affinity, large hydrophobic groups (e.g. 4-fluorophenyl ring) were used to replace the nitro group. This was based on the structural analyses of the COMT-inhibitor complex [36], which suggested that the replacements are able to form favourable apolar interactions with the hydrophobic cleft formed by W38, V173, P174 and L198. The structure-based predictions were confirmed by the experimental data [36]. The inhibitors demonstrated strong inhibitory potency with an IC50 value of 21–29 nm, highlighting that the 5-nitrocatechol anchor is not required for high affinity inhibition [36]. Those inhibitors that lack a nitrocatechol core are of considerable interest due to less toxicity concerns [37].
The bisubstrate-COMT crystal (pdb code: 1JR4) shows that the adenine moiety forms two hydrogen bonds to a water molecule [19]. It was reasoned that replacement of this water by a hydrophobic residue of the ligand would result in a free energy gain and a higher binding affinity [19,38]. To test the hypothesis, novel bisubstrate inhibitors were synthesized that contain N6-alkyl substituents. The N6-alkyl substituents were predicted to displace the water molecule. Crystal structures of COMT with the inhibitors revealed that the water indeed was replaced by the substituents (methyl, ethyl, propyl and hydroxyethyl) (Figure 6A) [19]. Also a gain, albeit not significant, in the binding affinity was achieved [19]. The IC50 and Ki values for N6-alkylated ligands are slightly lower than those measured for the unsubstituted ones. The N6-alkylated inhibitors bind with similar strength as the non-alkylated ligand, although energetic gain was attained by displacement of the water molecule [19]. This is because the N6-alkylated ligands are bound in an s-trans conformation that is energy unfavourable. The energy costs (∼1.8 kcal mol−1) of the unfavourable conformation must be compensated by the energetic gains resulting from the replacement of water. As a result, the net energy is not altered and there is no significant improvement in the binding affinity of the N6-alkylated ligands [19].
Figure 6.

Structure-activity relationships for the bisubstrate inhibitors. A) Comparison of the binding sites of N6-ethylated bisubstrate inhibitor (PDB code: 3HVI) and bisubstrate inhibitor 3 (PDB code: 1JR4), showing that the water is replaced by the ethyl substituent. The bisubstrate-binding site and the DNC-binding site are indicated in cyan and white, respectively. The dashed lines indicate hydrogen bonds. B) Summary of the structure-activity relationships for the bisubstrate inhibitors, highlighting that structural variations of the catechol, ribose and adenine moieties modulate the affinities of bisubstrate inhibitors toward COMT. C) Comparison of the binding sites of the bisubstrate inhibitor 3 (PDB code: 1JR4) and its 3′-fluoro derivative (PDB code: 3NWB). The binding sites of the bisubstrate inhibitor 3 and of the 3′-fluoro derivative are indicated in white and purple, respectively. The dashed lines denote hydrogen bonds. The solid lines denote distances
The effects of ribose modification on ligand affinity were investigated [21,39]. Surprisingly, the minor change from the ribose ether oxygen atom to the CH2 unit of a carbocyclic cyclopentane core, in most cases, leads to complete loss of the binding affinity. Although the exact reason is unknown, it was speculated that steric congestion in the ribose binding site (a narrow channel) that connects the adenine and catechol sites, as well as conformational changes upon replacement of the ribose moiety may play an important role [39].
The IC50 value of the 2′-deoxyribose derivative is increased by almost three orders of magnitude to 28 μm. In contrast, the 3′-deoxyribose derivative has only a ∼4-fold elevated IC50 value of 40 nm [21]. This clearly shows that the main energetic contribution for ribose binding originates from interactions of the 2′-hydroxyl group with the protein (Figure 6B). On the basis of structural analyses, Ellermann et al. illustrated the subtle differences of 2′-hydroxyl and 3′-hydroxyl groups in interacting with the protein [21], providing explanations why 2′-hydroxyl plays a more important role in determining ligand affinity. First, both H atoms of the ribose hydroxyl groups interact with the syn lone pairs of E90. However, the angular deviation of the hydrogen bond from the acceptor plane π-system is different. For the 2′-hydroxyl group, this angle is ∼5° and it is 25° for the 3′-hydroxyl group, indicating a much weaker interaction for the 3′-hydrolxyl [40]. Second, the acidity of the 2′-hydroxyl group is higher compared with the 3′-hydroxyl group owing to the inductive effects from the glycosidic bond and the ring O atom [21]. Thus, the 2′-hydroxyl group is more prone to share an H atom with an acceptor and should form a stronger hydrogen bond to E90. Third, the COMT protein itself better solvates the E90 O atom contacting the 3′-hydroxyl than the O atom contacting the 2′-hydroxyl, thereby increasing the basicity and hydrogen-bond acceptor strength of the latter.
Since 3′-hydroxyl is not essential for high affinity binding, replacements of this group have been explored to accomplish a high potency of inhibition [21]. Molecule modelling suggested the active site of COMT could accommodate small 3′-substituents trans to the 2′-hydroxyl group of the ribose moiety. The 3′-fluoro derivative and others with inverted chirality at C3′ were synthesized [21]. The rationale for the fluorine substituent was to enhance affinity of the compound by its strong σ-inductive effect, lowering the pKa value of the 2′-hydroxyl group and strengthening the hydrogen bond to the side chain of E90 [41]. As expected, the 3′-fluoro derivative is a very potent COMT inhibitor with an IC50 value of 11 nm. The binding mode of the inhibitor in the protein was confirmed by the X-ray crystal structure. The introduction of a 3′-fluoro substituent leads to subtle conformational changes around the ribose moiety (Figure 5C) [21]. The ribose ring retains the ‘south’ conformation, indicating that inversion of configuration does not change the pucker in the enzyme complex (Figure 5C). The 3′-fluorine substituent is located in a rather hydrophobic environment and favourably contacts the edges of the Y95 and W143 planes and the terminal methyl group of M40 (Figure 5C).
Ellermann et al. explored the effects of structural alterations of the adenine moiety on the affinity of bisubstrate inhibitors [20]. Novel bisubstrate inhibitors with adenine replacements (including thiopyridine, purine, N-methyladenine and 6-methylpurine) were developed by structure-based design [20]. Comparison of the activities of the purine inhibitor and the benzimidazole analogue shows that the heteroatoms in the six-membered ring of the purine moiety are crucial for strong binding to COMT. Further comparisons of the activities among the adenine-modified inhibitors identified the N(1) nitrogen heteroatom as one of the key determinants of affinity [20]. The N(1) of adenine is hydrogen bonded to the backbone NH of S119. By contrast, the nitrogen heteroatoms N(3) and N(7) are unnecessary for activity and thus can be replaced without a large energetic penalty (Figure 6B). The 6-methylpurine inhibitor (IC50 = 6 nm) is the most potent ligand, displaying an ∼5-fold higher binding affinity than the reference compound, demonstrating that the exocyclic NH2 of adenine is not necessary for strong binding (Figure 6B). The methyl group contributes to an increase in inhibitory activity of a factor of ∼26 probably through hydrophobic interactions with the pocket.
Other inhibitors
4-phenyl-7, 8-dihydroxycoumarin (4PCM, Figure 7A), a coumarin derivative, is a non-nitrocatechol inhibitor of COMT. The interest in developing this type of inhibitors is because nitrocatechol structures (e.g. tolcapone) have concerns about their hepatotoxicity [37,42]. The crystal complex of COMT with 4PCM reveals how the inhibitor is recognized by the protein [17]. The carbonyl oxygen of 4PCM faces to the side chain of W143 and the phenyl ring substitute at the 4-position is exposed to the solvent region (Figure 7B) [17]. K144 has an electrostatic interaction with 1-position of oxygen atom of 4PCM. It also makes a hydrogen bond to the carbonyl oxygen of 4PCM via a water molecule (Figure 7B) [17]. Further, the carbonyl oxygen of 4PCM may form the CH-O hydrogen bond with CE3 of W143 [17]. It is predicted that substitution of hydrophobic groups to the phenyl ring will be a promising approach to enhance the inhibitory activity because W38 is available for favourable hydrophobic interactions with the inhibitor molecule at the area around the phenyl ring [17].
Figure 7.
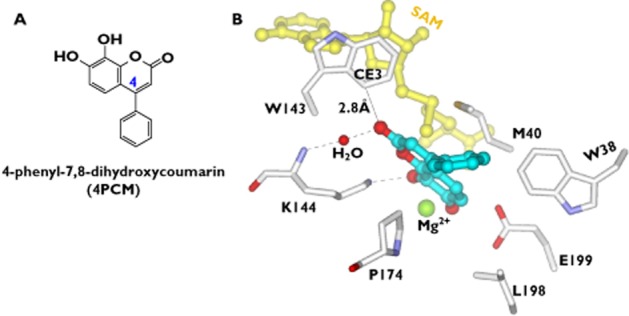
Molecular interactions of COMT with 4PCM (PDB code: 2ZVJ). A) Chemical structure of 4PCM (4-phenyl-7,8-dihydroxycoumarin). B) Molecular interactions of 4PCM with the binding site residues. The dashed lines indicate hydrogen bonds
More recently, a number of novel heterocyclic COMT inhibitors were derived from in vitro screening [43]. 1,2,4-oxadiazoles substituted with a pyridine N-oxide motif were found to have reduced toxicity risk and were endowed with longer duration of inhibition. Of note, opicapone [2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide, also known as BIA 9–1067] was selected for further pharmacological studies and was found to be a purely peripheral inhibitor of COMT with a unprecedented duration of action [43]. In addition, it presents favourable pharmacodynamics with L-dopa, resulting in stable and sustained plasma L-dopa concentrations over prolonged periods [43]. In fact, opicapone is currently under phase III clinical trials for the therapy of Parkinson's disease [44,45]. Studies involving human subjects have confirmed that opicapone is a very long acting inhibitor and a once daily regimen may be effective [44,45]. Computational analyses indicate that the long acting inhibition primarily depends on the catalytic rate constant (Kcat) of the inhibitor's O-methylation rather than the rate constant of dissociation (Koff) of the enzyme-inhibitor complex [46].
Conclusion
COMT catalyzes methylation (classified as phase II metabolism) which is a major clearance pathway for catecholic compounds. In recent years, COMT has become intensively studied largely due to its role in regulation of the dopamine level in the brain. Inhibition of COMT is an important approach for developing new therapeutic treatments for CNS disorders such as Parkinson's disease. The crystal structures for COMT have facilitated a deeper understanding of the atomic interactions of the substrates/inhibitors with the protein. It is not surprising that these structures have been frequently used in the design of novel inhibitors with a high success rate. The nitrocatechol-type inhibitors were designed based on their interactions with the catechol pocket. By contrast, the bisubstrate inhibitors targeted both the catechol pocket and the SAM pocket. Novel inhibitors without a nitrocatechol core have been available to alleviate possible inhibitor toxicity. Although many inhibitors are proven to be potent and selective in in vitro experiments, whether they can become drugs requires rigorous in vivo efficacy and toxicity testing.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare BW had support from Jinan University for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Acknowledgments
The authors are grateful to Dr Wen-cai Ye for his help in preparing this work.
References
- 1.Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 2.Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melén K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–4210. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- 3.Tenhunen J, Salminen M, Lundström K, Kiviluoto T, Savolainen R, Ulmanen I. Genomic organization of the human catechol O-methyltransferase gene and its expression from two distinct promoters. Eur J Biochem. 1994;223:1049–1059. doi: 10.1111/j.1432-1033.1994.tb19083.x. [DOI] [PubMed] [Google Scholar]
- 4.Karhunen T, Tilgmann C, Ulmanen I, Julkunen I, Panula P. Distribution of catechol-O-methyltransferase enzyme in rat tissues. J Histochem Cytochem. 1994;42:1079–1090. doi: 10.1177/42.8.8027527. [DOI] [PubMed] [Google Scholar]
- 5.Zheng YJ, Bruice TC. A theoretical examination of the factors controlling the catalytic efficiency of a transmethylation enzyme: catechol O-methyltransferase. J Am Chem Soc. 1997;119:8137–8145. [Google Scholar]
- 6.Tsao D, Diatchenko L, Dokholyan NV. Structural mechanism of S-adenosyl methionine binding to catechol O-methyltransferase. PLoS ONE. 2011;6:e24287. doi: 10.1371/journal.pone.0024287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guldberg HC, Marsden CA. Catechol-O-methyl transferase: pharmacological aspects and physiological role. Pharmacol Rev. 1975;27:135–206. [PubMed] [Google Scholar]
- 8.Kostić VS. COMT inhibition in the treatment of Parkinson's disease: neuroprotection and future perspectives. Adv Exp Med Biol. 2004;541:75–90. doi: 10.1007/978-1-4419-8969-7_5. [DOI] [PubMed] [Google Scholar]
- 9.Sazci A, Ergul E, Kucukali I, Kilic G, Kaya G, Kara I. Catechol-O-methyltransferase gene Val108/158Met polymorphism, and susceptibility to schizophrenia: association is more significant in women. Brain Res Mol Brain Res. 2004;132:51–56. doi: 10.1016/j.molbrainres.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Handoko HY, Nyholt DR, Hayward NK, Nertney DA, Hannah DE, Windus LC, McCormack CM, Smith HJ, Filippich C, James MR, Mowry BJ. Separate and interacting effects within the catechol-O-methyltransferase (COMT) are associated with schizophrenia. Mol Psychiatry. 2005;10:589–597. doi: 10.1038/sj.mp.4001606. [DOI] [PubMed] [Google Scholar]
- 11.Fava M, Rosenbaum JF, Kolsky AR, Alpert JE, Nierenberg AA, Spillmann M, Moore C, Renshaw P, Bottiglieri T, Moroz G, Magni G. Open study of the catechol-O-methyltransferase inhibitor tolcapone in major depressive disorder. J Clin Psychopharmacol. 1999;19:329–335. doi: 10.1097/00004714-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Männistö PT, Ulmanen I, Lundström K, Taskinen J, Tenhunen J, Tilgmann C, Kaakkola S. Characteristics of catechol O-methyl-transferase (COMT) and properties of selective COMT inhibitors. Prog Drug Res. 1992;39:291–350. doi: 10.1007/978-3-0348-7144-0_9. [DOI] [PubMed] [Google Scholar]
- 13.Bonifati V, Meco G. New, selective catechol-O-methyltransferase inhibitors as therapeutic agents in Parkinson's disease. Pharmacol Ther. 1999;81:1–36. doi: 10.1016/s0163-7258(98)00032-1. [DOI] [PubMed] [Google Scholar]
- 14.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 15.Vidgren J, Svensson LA, Liljas A. Crystal structure of catechol O-methyltransferase. Nature. 1994;368:354–358. doi: 10.1038/368354a0. [DOI] [PubMed] [Google Scholar]
- 16.Rutherford K, Le Trong I, Stenkamp RE, Parson WW. Crystal structures of human 108V and 108M catechol O-methyltransferase. J Mol Biol. 2008;380:120–130. doi: 10.1016/j.jmb.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 17.Tsuji E, Okazaki K, Takeda K. Crystal structures of rat catechol-O-methyltransferase complexed with coumarine-based inhibitor. Biochem Biophys Res Commun. 2009;378:494–497. doi: 10.1016/j.bbrc.2008.11.085. [DOI] [PubMed] [Google Scholar]
- 18.Tsuji E, Okazaki K, Isaji M, Takeda K. Crystal structures of the apo and holo form of rat catechol-O-methyltransferase. J Struct Biol. 2009;165:133–139. doi: 10.1016/j.jsb.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Ellermann M, Jakob-Roetne R, Lerner C, Borroni E, Schlatter D, Roth D, Ehler A, Rudolph MG, Diederich F. Molecular recognition at the active site of catechol-O-methyltransferase: energetically favorable replacement of a water molecule imported by a bisubstrate inhibitor. Angew Chem Int Ed Engl. 2009;48:9092–9096. doi: 10.1002/anie.200904410. [DOI] [PubMed] [Google Scholar]
- 20.Ellermann M, Paulini R, Jakob-Roetne R, Lerner C, Borroni E, Roth D, Ehler A, Schweizer WB, Schlatter D, Rudolph MG, Diederich F. Molecular recognition at the active site of catechol-O-methyltransferase (COMT): adenine replacements in bisubstrate inhibitors. Chemistry. 2011;17:6369–6381. doi: 10.1002/chem.201003648. [DOI] [PubMed] [Google Scholar]
- 21.Ellermann M, Lerner C, Burgy G, Ehler A, Bissantz C, Jakob-Roetne R, Paulini R, Allemann O, Tissot H, Grünstein D, Stihle M, Diederich F, Rudolph MG. Catechol-O-methyltransferase in complex with substituted 3′-deoxyribose bisubstrate inhibitors. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 3):253–260. doi: 10.1107/S0907444912001138. [DOI] [PubMed] [Google Scholar]
- 22.Palma PN, Rodrigues ML, Archer M, Bonifácio MJ, Loureiro AI, Learmonth DA, Carrondo MA, Soares-da-Silva P. Comparative study of ortho-and meta-nitrated inhibitors of catechol-O-methyltransferase: interactions with the active site and regioselectivity of O-methylation. Mol Pharmacol. 2006;70:143–153. doi: 10.1124/mol.106.023119. [DOI] [PubMed] [Google Scholar]
- 23.Bonifácio MJ, Archer M, Rodrigues ML, Matias PM, Learmonth DA, Carrondo MA, Soares-Da-Silva P. Kinetics and crystal structure of catechol-O-methyltransferase complex with co-substrate and a novel inhibitor with potential therapeutic application. Mol Pharmacol. 2002;62:795–805. doi: 10.1124/mol.62.4.795. [DOI] [PubMed] [Google Scholar]
- 24.Lerner C, Ruf A, Gramlich V, Masjost B, Zürcher G, Jakob-Roetne R, Borroni E, Diederich F. X-ray crystal structure of a bisubstrate inhibitor bound to the Enzyme catechol-O-methyltransferase: A dramatic effect of inhibitor preorganization on binding affinity. Angew Chem Int Ed Engl. 2001;40:4040–4042. doi: 10.1002/1521-3773(20011105)40:21<4040::AID-ANIE4040>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 25.Lautala P, Ulmanen I, Taskinen J. Molecular mechanisms controlling the rate and specificity of catechol O-methylation by human soluble catechol O-methyltransferase. Mol Pharmacol. 2001;59:393–402. doi: 10.1124/mol.59.2.393. [DOI] [PubMed] [Google Scholar]
- 26.Bäckström R, Honkanen E, Pippuri A, Kairisalo P, Pystynen J, Heinola K, Nissinen E, Linden IB, Männistö PT, Kaakkola S. Synthesis of some novel potent and selective catechol O-methyltransferase inhibitors. J Med Chem. 1989;32:841–846. doi: 10.1021/jm00124a017. [DOI] [PubMed] [Google Scholar]
- 27.Piedrafita FJ, Elorriaga C, Fernández-Alvarez E, Nieto O. Inhibition of catechol-O-methyltransferase by N-(3,4-dihydroxyphenyl) maleimide. J Enzyme Inhib. 1990;4:43–50. doi: 10.3109/14756369009030387. [DOI] [PubMed] [Google Scholar]
- 28.Marsala SZ, Gioulis M, Ceravolo R, Tinazzi M. A systematic review of catechol-O-methyltransferase inhibitors: efficacy and safety in clinical practice. Clin Neuropharmacol. 2012;35:185–190. doi: 10.1097/WNF.0b013e31825c034a. [DOI] [PubMed] [Google Scholar]
- 29.Barrow JC. Inhibitors of catechol-O-methyltransferase. CNS Neurol Disord Drug Targets. 2012;11:324–332. doi: 10.2174/187152712800672517. [DOI] [PubMed] [Google Scholar]
- 30.Kaakkola S. Problems with the present inhibitors and a relevance of new and improved COMT inhibitors in Parkinson's disease. Int Rev Neurobiol. 2010;95:207–225. doi: 10.1016/B978-0-12-381326-8.00009-0. [DOI] [PubMed] [Google Scholar]
- 31.Learmonth DA, Vieira-Coelho MA, Benes J, Alves PC, Borges N, Freitas AP, da-Silva PS. Synthesis of 1-(3,4-dihydroxy-5-nitrophenyl)-2-phenyl-ethanone and derivatives as potent and long-acting peripheral inhibitors of catechol-O-methyltransferase. J Med Chem. 2002;45:685–695. doi: 10.1021/jm0109964. [DOI] [PubMed] [Google Scholar]
- 32.Palma PN, Bonifácio MJ, Loureiro AI, Wright LC, Learmonth DA, Soares-da-Silva P. Molecular modeling and metabolic studies of the interaction of catechol-O-methyltransferase and a new nitrocatechol inhibitor. Drug Metab Dispos. 2003;31:250–258. doi: 10.1124/dmd.31.3.250. [DOI] [PubMed] [Google Scholar]
- 33.Learmonth DA, Palma PN, Vieira-Coelho MA, Soares-da-Silva P. Synthesis, biological evaluation, and molecular modeling studies of a novel, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2004;47:6207–6217. doi: 10.1021/jm040848o. [DOI] [PubMed] [Google Scholar]
- 34.Masjost B, Ballmer P, Borroni E, Zürcher G, Winkler FK, Jakob-Roetne R, Diederich F. Structure-based design, synthesis, and in vitro evaluation of bisubstrate inhibitors for catechol O-methyltransferase (COMT) Chemistry. 2000;6:971–982. doi: 10.1002/(sici)1521-3765(20000317)6:6<971::aid-chem971>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 35.Lerner C, Masjost B, Ruf A, Gramlich V, Jakob-Roetne R, Zürcher G, Borroni E, Diederich F. Bisubstrate inhibitors for the enzyme catechol-O-methyltransferase (COMT): influence of inhibitor preorganisation and linker length between the two substrate moieties on binding affinity. Org Biomol Chem. 2003;1:42–49. doi: 10.1039/b208690p. [DOI] [PubMed] [Google Scholar]
- 36.Paulini R, Lerner C, Jakob-Roetne R, Zürcher G, Borroni E, Diederich F. Bisubstrate inhibitors of the enzyme catechol O-methyltransferase (COMT): efficient inhibition despite the lack of a nitro group. Chembiochem. 2004;5:1270–1274. doi: 10.1002/cbic.200400084. [DOI] [PubMed] [Google Scholar]
- 37.Smith KS, Smith PL, Heady TN, Trugman JM, Harman WD, Macdonald TL. In vitro metabolism of tolcapone to reactive intermediates: relevance to tolcapone liver toxicity. Chem Res Toxicol. 2003;16:123–128. doi: 10.1021/tx025569n. [DOI] [PubMed] [Google Scholar]
- 38.Barillari C, Taylor J, Viner R, Essex JW. Classification of water molecules in protein binding sites. J Am Chem Soc. 2007;129:2577–2587. doi: 10.1021/ja066980q. [DOI] [PubMed] [Google Scholar]
- 39.Paulini R, Trindler C, Lerner C, Brändli L, Schweizer WB, Jakob-Roetne R, Zürcher G, Borroni E, Diederich F. Bisubstrate inhibitors of catechol O-methyltransferase (COMT): the crucial role of the ribose structural unit for inhibitor binding affinity. ChemMedChem. 2006;1:340–357. doi: 10.1002/cmdc.200500065. [DOI] [PubMed] [Google Scholar]
- 40.Bissantz C, Kuhn B, Stahl M. A medicinal chemist's guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 42.Fisher A, Croft-Baker J, Davis M, Purcell P, McLean AJ. Entacapone-induced hepatotoxicity and hepatic dysfunction. Mov Disord. 2002;17:1362–1365. doi: 10.1002/mds.10342. [DOI] [PubMed] [Google Scholar]
- 43.Kiss LE, Ferreira HS, Torrão L, Bonifácio MJ, Palma PN, Soares-da-Silva P, Learmonth DA. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53:3396–3411. doi: 10.1021/jm1001524. [DOI] [PubMed] [Google Scholar]
- 44.Almeida L, Rocha JF, Falcao A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52:139–151. doi: 10.1007/s40262-012-0024-7. [DOI] [PubMed] [Google Scholar]
- 45.Rocha JF, Almeida L, Falcao A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013;76:763–775. doi: 10.1111/bcp.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012;33:970–986. doi: 10.1002/jcc.22926. [DOI] [PubMed] [Google Scholar]