

Abstract
AIMS
Axitinib is a potent and selective second generation inhibitor of vascular endothelial growth factor receptors 1, 2 and 3 approved for second line treatment of advanced renal cell carcinoma. The objectives of this analysis were to assess plasma pharmacokinetics and identify covariates that may explain variability in axitinib disposition following single dose administration in healthy volunteers.
METHODS
Plasma concentration–time data from 337 healthy volunteers in 10 phase I studies were analyzed, using non-linear mixed effects modelling (nonmem) to estimate population pharmacokinetic parameters and evaluate relationships between parameters and food, formulation, demographic factors, measures of renal and hepatic function and metabolic genotypes (UGT1A1*28 and CYP2C19).
RESULTS
A two compartment structural model with first order absorption and lag time best described axitinib pharmacokinetics. Population estimates for systemic clearance (CL), central volume of distribution (Vc), absorption rate constant (ka) and absolute bioavailability (F) were 17.0 l h−1, 45.3 l, 0.523 h−1 and 46.5%, respectively. With axitinib Form IV, ka and F increased in the fasted state by 207% and 33.8%, respectively. For Form XLI (marketed formulation), F was 15% lower compared with Form IV. CL was not significantly influenced by any of the covariates studied. Body weight significantly affected Vc, but the effect was within the estimated interindividual variability for Vc.
CONCLUSIONS
The analysis established a model that adequately characterizes axitinib pharmacokinetics in healthy volunteers. Vc was found to increase with body weight. However, no change in plasma exposures is expected with change in body weight; hence no dose adjustment is warranted.
Keywords: axitinib, CYP2C19 metabolizing status, population pharmacokinetics, UGT1A1*28 genotype, vascular endothelial growth factor receptor inhibitor
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT.
Axitinib is a potent and selective second generation inhibitor of vascular endothelial growth factor receptors 1, 2 and 3.
It has demonstrated superior efficacy over sorafenib in a randomized phase III clinical trial in patients with metastatic renal cell carcinoma (mRCC), whose disease progressed following one prior first line systemic therapy.
WHAT THIS STUDY ADDS.
A population pharmacokinetic model was developed and the effects of various demographics, measures of renal and hepatic function and genetic factors on axitinib disposition were evaluated in healthy volunteers in the absence of confounding factors that are often associated with cancer patients.
Of the covariates tested, body weight was found to affect significantly interindividual variability in axitinib pharmacokinetics.
The model developed here is useful in assessing the impact of other factors on axitinib plasma exposure.
Introduction
Axitinib is a potent and selective second generation inhibitor of vascular endothelial growth factor receptors (VEGFR) 1, 2 and 3 [1–3]. In several phase II clinical studies, this anti-angiogenic agent has shown antitumour activity as a single agent against various solid tumours, including metastatic renal cell carcinoma (mRCC), thyroid cancer and non-small cell lung cancer [4–7]. Axitinib is generally well tolerated, with a safety profile similar to that reported for other anti-angiogenic tyrosine kinase inhibitors, such as sorafenib and sunitinib [8,9]. Recently, in a randomized phase III clinical trial (AXIS), axitinib demonstrated superior efficacy over sorafenib as second line therapy for mRCC [10]. Axitinib is now approved in the United States for the treatment of advanced RCC after failure of a prior systemic therapy. It is also approved in several other countries.
A pharmacokinetic (PK) analysis in the first-in-human phase I dose-finding study, in which patients with advanced solid tumours received axitinib doses ranging from 5 mg twice daily up to 30 mg twice daily, demonstrated that plasma concentrations for axitinib reached a peak within 2 to 6 h after oral dosing and declined with a clinically relevant plasma half-life of 2 to 5 h in the fed state [11]. The rate and extent of absorption were higher in the fasted state. Axitinib PK were dose-dependent up to 20 mg twice daily. The maximum tolerated dose was determined to be 5 mg twice daily and recommended as a starting dose in subsequent phase II clinical trials. Axitinib dose increase (to a maximum of 10 mg twice daily) or reduction is permitted, based on individual tolerability. Studies have been conducted using two different crystal polymorphs for axitinib, Form IV and Form XLI, film-coated immediate-release (FCIR) tablets, in either fed or fasted states. Form XLI, with greater thermodynamic stability, improved photostability and more favourable physical properties, is the commercial formulation.
Axitinib is metabolized in the liver, primarily by cytochrome P450 (CYP) 3A4 and 3A5, and to a lesser extent, by CYP1A2 and CYP2C19 (<10% each), and uridine diphosphate glucuronosyltransferase (UGT) 1A1 [12]. The two major metabolites identified in human plasma following administration of a radiolabelled axitinib were M7 (N-glucuronide) and M12 (sulfoxide product), both of which were considered pharmacologically inactive since these metabolites were ≥400-fold less active against phosphorylation of VEGFR-2 compared with axitinib [13]. While in vitro studies have shown axitinib to be a weak substrate for efflux transporters, P-glycoprotein and breast cancer resistance protein, axitinib is not expected to inhibit these transporters at the therapeutic plasma concentrations [13]. Axitinib elimination occurs mainly via hepatobiliary excretion.
Axitinib PK following a single dose have been studied in more than 500 healthy volunteers [13–18] and approximately 90 patients with advanced solid tumours [11,19–22]. As seen with other orally administered drugs, including tyrosine kinase inhibitors [23–25], axitinib PK were variable in healthy volunteers as well as in cancer patients. Since not only toxicity but also clinical benefit of axitinib may be affected by its plasma exposure, it is critical to identify clinical factors (e.g. demographic factors such as age, body weight, gender and race) that contribute to the variability in axitinib PK. Additionally, genetic polymorphisms that are associated with decreased drug-metabolizing enzyme or transporter activity have been characterised. Thus, any factors that affect the activity and/or expression of these enzymes and transporters could potentially contribute to the variability in axitinib plasma exposure, resulting in altered clinical efficacy and/or toxicity.
A population PK approach is often used to develop a model to describe PK of a drug and to investigate potential covariates that may contribute to PK variability. Although the ultimate goal of conducting a population PK analysis is to identify factors responsible for PK variability in a target patient population, an analysis using data from patients is often confounded by the effect of concomitant medications and/or treatment for comorbidity, compromised end-stage organ function or underlying disease. Thus, the aims of this population PK analysis were to develop a model that adequately describes axitinib plasma PK following oral, single dose administration in healthy volunteers only and to evaluate the potential influence of demographics, measures of renal and hepatic function and genetic factors in drug metabolizing enzymes.
Methods
Study design
Details of the 10 phase I studies included in the current analysis are summarized in Table 1. For some studies, only subsets of the data that were clinically relevant were included in the analysis. All clinical studies were approved by the Institutional Review Boards (IRBs) or ethics committees and were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. The IRBs were as follows: study 1, the Research Consultants' Review Committee, Austin, TX, USA, studies 2 and 3, PRACS Institute IRB, Fargo, ND, USA, study 4, RCRC IRB, Austin, TX, USA, study 5, IntegReview, Austin, TX, USA, study 6, Aspire IRB, La Mesa, CA, USA and study 8, Independent Investigational Review Board, Inc, Plantation, FL, USA. The ethics committee for study 9 was Comite d'Ethique de l'Hôpital Erasme, Bruxelles, Belgium and for studies 7 and 10, Comite d'Ethique de l'Hôpital Erasme, Bruxelles, Belgium and Parkway Independent Ethics Committee, Parkway Hospitals, Singapore. Written informed consent was obtained from all subjects prior to study entry.
Table 1.
Summary of studies included in axitinib population pharmacokinetic analysis
| Study | Study design | Enrolled/analyzed (n) | Treatment (for those included in analysis) | Full PK sampling time points post dosing (h) |
|---|---|---|---|---|
| 1 [14] | R, SB, 2-way CO, DDI with ketoconazole | 35/32*† | 5 mg, oral, Form IV, o/n fast | Pre-dose (0), 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48 |
| 2 [15] | OL, food effect on PK | 42/41‡ | Tx 1: 5 mg, oral, Form IV, o/n fast | 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 36, 48 |
| Tx 2: 5 mg, oral, Form IV, fed | ||||
| 3 | OL, 2-way CO, absolute bioavailability study | 16/16 | Tx 1: 5 mg, Form IV, oral, o/n fast | 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 36 |
| Tx 2: 1 mg, Form IV, i.v., o/n fast | ||||
| Tx 3: 5 mg, Form IV, oral, fed | ||||
| 4 | R, OL, CO, relative bioavailability study | 40/20§ | 5 mg, oral, Form IV, o/n fast | 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 36 |
| 5 | R, OL, 2-sequence, 3-period CO, bioequivalence study | 40/40 | 5 mg, oral, Form IV, o/n fast | 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 32 |
| 6 [16] | R, OL, 2-period, 2-Tx CO, | 40/40† | 5 mg, oral, Form IV, o/n fast | 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 32 |
| DDI with rifampin | ||||
| 7 | R, OL, 4-sequence, 4-period, CO, relative bioavailability study | 56/54¶ | Tx 1: 5 mg, oral, Form IV, fed | 0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 32 |
| Tx 2: 5 mg, oral, Form XLI, fed | ||||
| 8 [18] | OL, PG, subjects with normal hepatic function or mild or moderate hepatic impairment | 24/8** | 5 mg, oral, Form IV, fed | 0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 12, 16, 24, 36, 48, 96, 144 |
| 9 | R, OL, 4-sequence, 4-period, CO, relative bioavailability study | 20/20 | 5 mg, oral, Form IV, fed | 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 16, 24, 36, 48 |
| 10 | R, OL, 2-sequence, 4-period, CO, bioequivalence study | 68/66* | 5 mg, oral, Form IV, o/n fast | 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 32 |
Reasons for excluding individual data from analysis:
Did not receive axitinib;
Only data following administration of axitinib alone were included in the analysis;
Not completed the study;
Received axitinib dose <5 mg in a spray-dried dispersion;
Received axitinib in a spray-dried dispersion;
Due to impaired hepatic function. CO, crossover; DDI, drug–drug interaction; i.v., intravenous; OL, open-label; o/n, overnight; PG, parallel-group; PK, pharmacokinetics; R, randomized;SB, single-blind; Tx, treatment.
PK assessments
Serial blood samples for PK analysis were collected before dosing and at various time intervals following administration of a single dose of axitinib (Table 1). Axitinib concentrations were determined in plasma samples, using a validated high performance liquid chromatography with tandem mass spectrometric detection method (Charles River Discovery and Development Services, Shrewsbury, MA, USA) [11]. Linear range, precision (% coefficient of variation) and bias (% relative error) were 0.1–25 ng ml−1, <16% and <13%, respectively, in studies 1, 2, 3, 4 and 6, 0.1–100 ng ml−1, <16% and <13%, respectively, in study 5 and 0.5–100 ng ml−1, <8% and <7%, respectively, in studies 7, 8, 9 and 10.
Pharmacogenomic assessments
DNA was extracted from whole blood using the Qiagen's QIAamp® kit (QIAGEN, Hilden, Germany). Genotyping for UGT1A1*28 variant (seven TA repeats) was conducted using the MassARRAY® assay (Sequenom Inc, San Diego, CA, USA). The MassEXTEND® assay (Sequenom Inc) used a beadless and label-free primer extension chemistry to generate allele-specific diagnostic products. Following extension reaction, MassEXTEND® clean resin was added to the reaction to remove extraneous salts. Aliquots (15 nl) of samples were spotted onto the 384-SpectroCHIP™ bioarray pad, placed into the matrix-assisted laser desorption/ionization–time of flight mass spectrometry (MALDI-TOF-MS) and the mass and correlating genotype were determined in real time with MassARRAY RT® software (SpectroTYPER™ v 3.1; Sequenom Inc).
Genotyping for the CYP2C19*2 (G681A), *3 (G636A), and *17 (C-806T) alleles was conducted with validated TaqMan® Allelic Discrimination assays (applied biosystems™, Life Technologies Corp, Carlsbad, CA, USA) [26], using oligonucleotide probes labelled with a 5′ reporter dye (VIC or FAM [6-carboxyfluoroscein]) and a 3′-quencher dye (TAMRA [6-carboxytetramethylrhodamine] or a non-fluorescent quencher), following the manufacturer's instructions.
Statistical analysis
Plasma concentration−time data were analyzed using non-linear mixed effects modelling (nonmem 7, level 1.0, ICON Developmental Solutions; Ellicott City, MD, USA) to estimate population PK parameters [mean and interindividual variability (IIV)] for axitinib and to identify potential covariates to explain any IIV in the parameters. Analyses used first bullet conditional estimation with interaction (FOCEI). Model selection was based on assessment of diagnostic plots and comparison of the nonmem change of minimum objective function values (ΔMOF) between nested models, using the log-likelihood ratio test. The selected base model was analyzed for covariate influence on the IIV error terms. The significance of potential covariates was systematically evaluated in a stepwise forward selection (ΔMOF >–3.84 points, P < 0.05) followed by backward elimination process (ΔMOF >10.83 points, P < 0.001). Simulations were performed to determine the predicted effect of these covariates.
The predictive performance was evaluated by simulating data based on the final model and conducting visual predictive checks (VPCs) between observed and simulated data. Evaluation of model robustness was based on relative standard errors (RSE) of the model parameter estimates determined by non-parametric bootstrapping (n = 200) [Perl-speaks nonmen (PsN), version 3.2.4].
Model development
Log-transformed axitinib plasma concentration–time data were evaluated using one, two or three compartment models. The most conservative model was selected at the P < 0.05 level as evaluated by ΔMOF. IIV was modelled using an exponential error term on each PK parameter and was expressed as a percent (%IIV = √ω2 x 100). The residual error model (for intravenous and oral) was modelled additively in a logarithmic scale.
During base model development, alternative models were investigated to improve estimates and to decrease residual variability (e.g. transit compartment model [27] instead of a lag-time model and the M3 approach to investigate below limit of quantitation data 28). Briefly, the residual variability did improve slightly using these alternative models; however, systemic clearance (CL) was not different between models [29]. Additionally, longer run time limited their usefulness when analyzing axitinib plasma concentration data in large pooled data sets.
Following development of the base model, a total of 11 covariates of clinical interest were systematically evaluated in a stepwise forward selection and significant covariates were combined in a full model. This was followed by backward elimination and significant covariates were retained in the final model. The continuous covariates evaluated were age, body weight, serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), bilirubin and creatinine clearance (CLcr, using Cockcroft–Gault equation), whereas the binary covariates evaluated were gender, race, smoking status, UGT1A1*28 genotype and CYP2C19 inferred phenotype. All of these variables were investigated for potential effect on axitinib CL. Only gender and body weight were investigated for potential effect on central volume of distribution (Vc). Continuous covariates were modelled as a power model centred on a median value:
where θ1 is a base parameter value, X is the covariate of interest, and Xreference is the median value of that covariate. Binary covariates were incorporated as a linear proportional change:
where θ1 is a base parameter value and indicator is a coding variable equal to 1 when the covariate is present and 0 when it is absent. θ2is the proportional change, coded as relative to the most common covariate value.
Results
Subject characteristics
Axitinib plasma concentration data from 337 healthy volunteers in 10 studies were pooled for the population PK analysis. Studies varied with respect to formulation of axitinib (crystal polymorph Form IV vs. Form XLI), route of administration (oral administration in most studies vs. intravenous administration in one study) and food status (fed vs. fasted state) (Table 1). Baseline characteristics and demographics of subjects included in the analysis are summarized in Table 2. The majority of the subjects were young (mean age 34.1 years), White (62%), males (93%) and non-smokers (88%), with normal kidney and liver function as evidenced by mean CLcr (117.2 ml min−1) and serum concentrations of AST (23.9 U l−1), ALT (24.1 U l−1) and bilirubin (0.8 mg dl−1) within normal ranges.
Table 2.
Demographics and baseline characteristics of subjects, route of administration, formulation, and fed/fasted state included in axitinib population pharmacokinetic analysis
| Characteristic | n = 337 |
|---|---|
| Gender, n (%) | |
| Male | 315 (93) |
| Female | 22 (7) |
| Race, n (%) | |
| White | 208 (62) |
| Black | 28 (8) |
| Asian (excluding Japanese) | 59 (18) |
| Japanese | 20 (6) |
| Hispanic | 11 (3) |
| Others | 11 (3) |
| Smoking status, n (%) | |
| Non-smoker | 297 (88) |
| Ex-smoker | 40 (12) |
| UGT1A1*28 genotype, n (%)* | |
| Homozygous wild-type (TA6/TA6) | 173 (51) |
| Heterozygous (TA6/TA7) | 107 (32) |
| Homozygous variant (TA7/TA7) | 35 (10) |
| Other† | 21 (6) |
| Unknown‡ | 1 (N/A) |
| CYP2C19 inferred phenotype, n (%)* | |
| Extensive metabolizer | 312 (93) |
| Ultra-rapid metabolizer | 8 (2) |
| Poor metabolizer | 15 (4) |
| Unknown‡ | 2 (N/A) |
| Age (years) median (mean ± SD) | 31.0 (34.1 ± 11.6) |
| Weight (kg) median (mean ± SD) | 75.0 (76.7 ± 11.6) |
| CLcr (ml min–1) median (mean ± SD) | 115.7 (117.2 ± 24.5) |
| AST (U l–1) median (mean ± SD) | 23.0 (23.9 ± 7.9) |
| ALT (U l–1) median (mean ± SD) | 22.0 (24.1 ± 9.9) |
| Bilirubin (mg dl–1) median (mean ± SD) | 0.7 (0.8 ± 0.4) |
| Route of administration, Form, Fed/Fasted, n (%)§ | |
| Oral, Form IV, Fasted | 231 (69) |
| Oral, Form IV, Fed | 138 (41) |
| Oral, Form XLI, Fed | 54 (16) |
| i.v., Form IV, Fasted | 16 (5) |
Percentage was calculated from the total number of subjects with genotype or inferred phenotype data.
Three main genotypes were derived for the UGT1A1*28 polymorphism based on the number (i.e. 6 or 7) of promoter TA repeats: (1) TA6/TA6 (homozygous wild-type), (2) TA6/TA7 (heterozygous) or (3) TA7/TA7 (homozygous variant). All other polymorphisms (i.e. number of TA repeats different from 6 or 7) were classified as ‘other.’
No genotyping samples were collected. These subjects were grouped with the reference group during the covariate testing.
Studies varied with respect to route of administration, formulation and fed/fasted; hence, subjects who crossed over to different treatments were counted more than once. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CLcr, creatinine clearance; i.v., intravenous; N/A, not applicable.
Subjects were genotyped for three single nucleotide polymorphisms in the CYP2C19 gene (*2, *3, *17) and the number of TA repeats in the promoter region of UGT1A1 gene (UGT1A1*28). The three main genotypes were derived for the UGT1A1*28 polymorphism based on the number (6 or 7) of TA repeats: (1) TA6/TA6 (homozygous wild-type), (2) TA6/TA7 (heterozygous) or (3) TA7/TA7 (homozygous variant). Of subjects genotyped for UGT1A1*28, 51% were categorized as ‘homozygous wild-type’, 10% as ‘homozygous variant’ and 32% as ‘heterozygous’ (Table 2). Subjects who had a number of TA repeats different from 6 or 7 were classified as ‘other’ (6%). Of subjects analyzed for CYP2C19 inferred phenotype, 93% were extensive metabolizers (heterozygous CYP2C19 variants), 2% were ultra-rapid metabolizers (homozygous for CYP2C19*17) and 4% were poor metabolizers (two null alleles for CYP2C19*2 or *3 variants) (Table 2). Subjects with missing genotypes were coded as ‘Unknown’ in the data set and were grouped with the reference group for covariate testing.
PK base model
The base model used to describe axitinib PK was a linear two compartment structural model defined in terms of CL, Vc, inter-compartmental clearance (Q), volume of distribution in the peripheral compartment (Vp), first-bullet absorption rate constant (ka) and absorption lag time (tlag). Absolute bioavailability (F) after oral administration was estimated using the intravenous data (study 3). The model included IIV on CL, Vc, Q, Vp and ka and covariance between CL and Vc, as well as between Q and Vp. The effects of food and formulation were included in the base model, as they improved and stabilized the model prior to covariate testing. PK parameter estimates and 95% confidence interval (CI) (based on standard errors generated from the nonmem covariance step) for the base model with food and formulation effects are provided in Table 3. Large variability in axitinib PK was evident from the estimated residual error standard deviation of 50.9% for oral administration and 34.2% for intravenous administration of axitinib, which could not be reduced by introduction of inter-occasion variability in the model.
Table 3.
Axitinib pharmacokinetic parameter estimates for base and final models
| Parameter | Base | Final | ||||
|---|---|---|---|---|---|---|
| Estimate | %RSE* | 95% CI† | Estimate | %RSE* | 95% CI† | |
| Structural model parameters | ||||||
| CL (l h−1) | 17.1 | 8.2 | 14.6, 20.1 | 17.0 | 6.6 (8.2) | 14.9, 19.4 |
| Vc (l) | 46.6 | 7.6 | 40.2, 54.0 | 45.3 | 6.4 (8.3) | 40.0, 51.3 |
| Weight effect on Vc | – | – | – | 0.758 | 14 (14) | 0.556, 0.960 |
| Q (l h−1) | 1.73 | 8.2 | 1.47, 2.03 | 1.74 | 9.2 (16) | 1.45, 2.08 |
| Vp (l) | 44.7 | 20 | 30.5, 65.6 | 45.9 | 25 (57) | 28.0, 75.3 |
| ka (h−1) Form IV, Fed | 0.530 | 6.8 | 0.464, 0.605 | 0.523 | 7.6 (7.0) | 0.450, 0.607 |
| Fasting | 2.10 | 13 | 1.55, 2.65 | 2.07 | 14 (14) | 1.49, 2.65 |
| F, Form IV, Fed | 0.469 | 7.8 | 0.403, 0.546 | 0.465 | 7.2 (8.4) | 0.403, 0.536 |
| Fasting | 0.323 | 15 | 0.228, 0.418 | 0.338 | 15 (14) | 0.240, 0.436 |
| Form XLI | −0.147 | 24 | −0.216, −0.0780 | −0.150 | 23 (23) | −0.219, −0.0814 |
| tlag (h)‡ | 0.457 | 0.32 | 0.454, 0.460 | 0.457 | – | – |
| Interindividual variability model parameters§ | ||||||
| ω2 CL | 0.270 | 11 | 0.216, 0.337 | 0.272 | 14 (11) | 0.208, 0.355 |
| ω2Vc | 0.140 | 15 | 0.104, 0.188 | 0.0949 | 25 (18) | 0.0579, 0.155 |
| ω2 Q | 0.380 | 23 | 0.244, 0.591 | 0.406 | 22 (27) | 0.266, 0.619 |
| ω2Vp | 1.06 | 34 | 0.549, 2.05 | 1.07 | 33 (44) | 0.566, 2.02 |
| ω2 ka | 0.476 | 14 | 0.360, 0.629 | 0.506 | 13 (13) | 0.392, 0.654 |
| ω CL ω Vc | 0.158 | 14 | 0.114, 0.202 | 0.141 | 22 (15) | 0.0812, 0.201 |
| ω Q ω Vp | 0.593 | 28 | 0.272, 0.914 | 0.619 | 26 (35) | 0.300, 0.938 |
| Residual error model parameters | ||||||
| Oral, % | 50.9 | 2.7 | 48.3, 53.7 | 50.9 | 2.7 (2.9) | 48.3, 53.7 |
| Intravenous, % | 34.2 | 15 | 25.5, 45.9 | 34.8 | 16 (14) | 25.4, 47.7 |
%RSE of model parameter estimates obtained from the nonmem covariance step. %RSE shown in parenthesis for the final model were obtained from non-parametric bootstrap of 200 samples (91% successful minimization).
Confidence interval was calculated as estimate × exp (± 1.96 %RSE/100), or for quantities that could be positive or negative (covariate parameters and covariances), it was calculated as estimate ± 1.96 %RSE/100 to allow the interval to span 0.
The final model was run again with tlag fixed to 0.457 in bullet to obtain a successful covariance step and calculate %RSE. Estimates with fixed tlag matched the final run or differed by no more than 2 in the last significant digit.
Interindividual variability was calculated as √ω2 × 100. CI, confidence interval; CL, systemic clearance; F, absolute bioavailability; ka, first-bullet rate of absorption; Q, peripheral clearance; RSE, relative standard error; tlag, absorption lag time; Vc, central volume of distribution; Vp, peripheral volume of distribution.
Diagnostic plots of the base model demonstrated a good balance between the observed concentrations and the population and individual predictions (Figure 1A, B, respectively). There was no systematic bias in conditional weighted residuals over the predicted population concentrations as well as over time (Figure 1C, D, respectively). Since the predicted etas (i.e. IIV random effects) for CL and Vc did not differ substantially across studies (Figure 2A, B, respectively), pooling of the 10 studies was retained in the analysis.
Figure 1.

Diagnostic plots for the base model: (A) observed vs. population predicted concentrations; (B) observed vs. individual predicted concentrations; (C) conditional weighted residuals vs. population predicted concentrations; (D) conditional weighted residuals vs. time. Dashed lines represent lines of identity (A,B) or null value (C,D) and solid lines depict smooth (LOESS) trends. LOESS, locally weighted scatter plot smoothing
Figure 2.
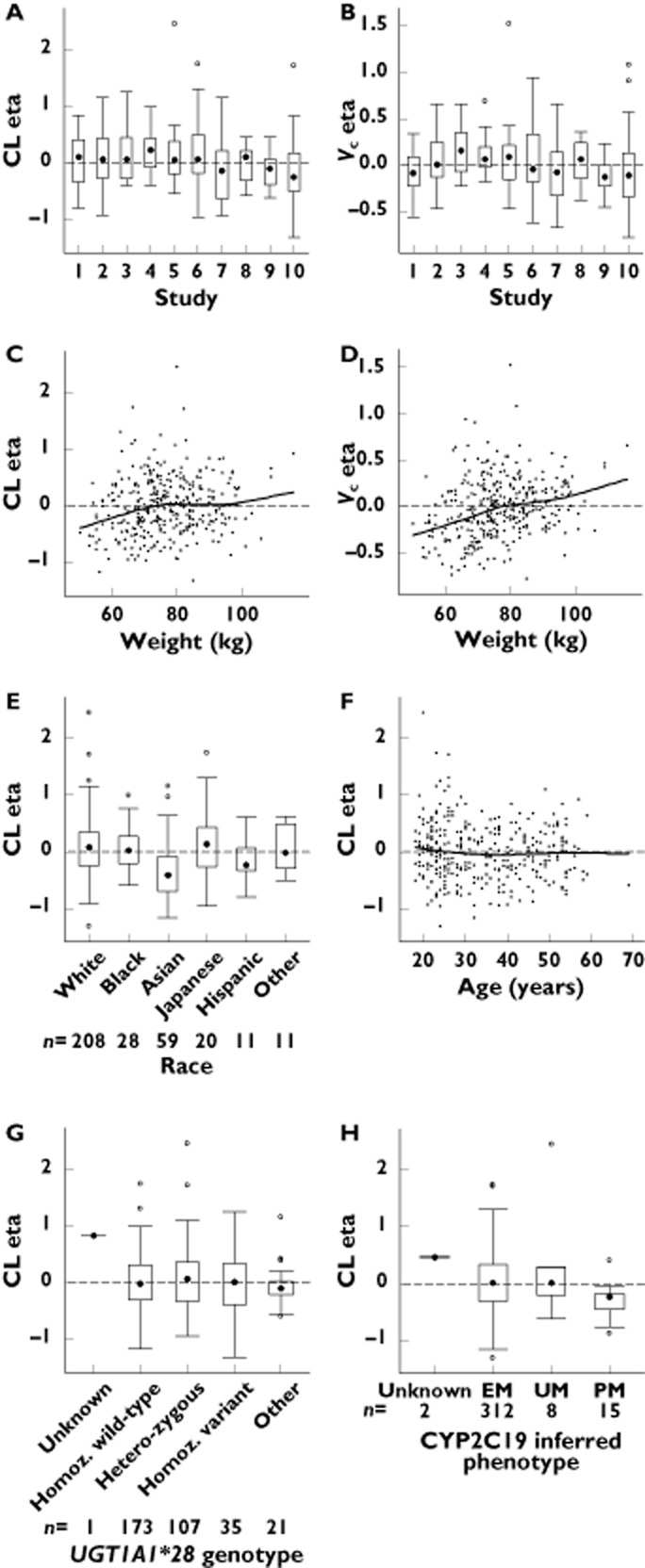
Parameter−covariate relationships for the base model: (A) CL eta vs. individual study; (B) Vc eta vs. individual study; (C) CL eta vs. body weight; (D) Vc eta vs. body weight; (E) CL eta vs. race; (F) CL eta vs. age; (G) CL eta vs. UGT1A1*28 genotype; (H) CL eta vs. CYP2C19 inferred phenotype. CL, systemic clearance; etas, individual random effects; Vc, volume of distribution in the central compartment; Homoz. wild-type, homozygous wild-type (TA6/TA6); heterozygous, (TA6/TA7); Homoz. variant, homozygous variant (TA7/TA7); EM, extensive metabolizer; UM, ultra-rapid metabolizer; PM, poor metabolizer
The eta plots suggested potential increases in CL and Vc with body weight and possible decreases in CL for Asian ethnicity and age (Figure 2C–F). There were no notable effects of UGT1A1*28 variant or CYP2C19 inferred phenotype (i.e. extensive, ultra-rapid or poor metabolizer) on CL based on eta plots (Figure 2G, H, respectively). The eta shrinkage estimates were as follows: 3% for CL, 15% for Vc, 36% for Q, 40% for Vp and 14% for ka. The eta shrinkage estimate for CL, Vc and ka were considered adequate and enabled the use of empirical Bayes estimates (EBEs) as a diagnostic tool to evaluate the predictive performance of the model [30].
Full model and final model
The potential impacts of body weight, gender, age, race, smoking status (i.e. evaluation of CYP1A2 induction), CLcr, ALT, AST, bilirubin, UGT1A1*28 polymorphism and CYP2C19 metabolizing status on axitinib CL and of body weight and gender on Vc were then systematically evaluated in a stepwise forward selection process. The full model included body weight on CL [point estimate (%RSE): 0.558 (34%)] and Vc [1.11 (12%)] and Asian ethnicity [–0.106 (41%)] and age [–0.168 (34%)] on CL. However, they accounted only for a small portion of the %IIV in CL and Vc, which decreased (for base to full model) from 52.0% to 50.2% and from 37.4% to 30.5% for CL and Vc, respectively. Inclusion of gender, age, smoking status, CLcr, ALT, AST, bilirubin, UGT1A1*28 polymorphism and CYP2C19 metabolizing status did not alter MOF substantially and, hence, they were not considered as significant covariates. The PK parameter estimates for the full model were similar to those for the base model (data not shown).
The final model was reached in three backward elimination steps. After accounting for the food effect on ka and F for Form IV and formulation effect (Form IV vs. Form XLI) on F, body weight on Vc was the only significant covariate retained in the final model. The PK parameter estimates for the final model were similar to those for the base model (Table 3). The eta shrinkage estimates were also low and considered adequate for using EBEs for diagnostic plots: shrinkage estimate was 3% for CL, 13% for Vc, 35% for Q, 40% for Vp and 12% for ka. As with the base model, diagnostic plots for the final model did not reveal any major concerns with the relationship between observed and predicted data (Figure 3A–D). In the final model, population estimates (%IIV) for axitinib CL and Vc were 17.0 l h−1 (52.2%) and 45.3 l (30.8%), respectively. The estimated tlag from axitinib administration to the start of the first bullet absorption was 0.457 h. Population mean estimates (%IIV) for Q and Vp were 1.74 l h−1 (63.7%) and 45.9 l (103%), respectively. Although IIV estimates were high for Q and Vp, their influence on the tail portion of the concentration−time curve was modest, as demonstrated in simulation of variability in Q and Vp (data not shown). The RSE for the model parameter estimates were also obtained from non-parametric bootstrap of 200 samples and showed that the estimates were not statistically different from those of the nonmem covariance step (Table 3).
Figure 3.

Diagnostic plots for the final model: (A) observed vs. population predicted concentrations; (B) observed vs. individual predicted concentrations; (C) conditional weighted residuals vs. population predicted concentrations; (D) conditional weighted residuals vs. time. Dashed lines represent lines of identity (A,B) or null value (C,D) and solid lines depict smooth (LOESS) trends. LOESS, locally weighted scatter plot smoothing
When axitinib was administered as crystal polymorph Form IV, ka and F increased by 207% and 33.8%, respectively, in the fasted state compared with the fed state. When administered as Form XLI in the fed state, F was reduced by 15% (as shown by the negative value −0.147 in Table 3) compared with Form IV. A food effect for Form XLI could not be tested since data from a prospective food effect study with Form XLI was not available at the time of this analysis.
The model predicted increases in Vc of axitinib with body weight. The typical Vc can be defined by:
where 75.0 kg is the population median body weight. Based on the 95% CI, in an individual with a low body weight of 62 kg (10th percentile), Vc is predicted to decrease by 10% to 17% relative to a subject with median 75 kg body weight. Conversely, for an individual with a high body weight of 93 kg (90th percentile), Vc is expected to be 13% to 23% higher compared with an individual with a median 75 kg body weight. However, these changes are less than the estimated IIV of 30.8% for Vc. Furthermore, the effect on Vc alone does not affect overall axitinib exposure (area under plasma concentration–time curve).
To assess further the effect of body weight on Vc, simulations were performed to predict steady-state exposures of axitinib at 5 mg oral dosing in Form XLI administered in the fed state. The results were obtained for subjects with body weight at the 10th or 90th percentile and compared with subjects with median body weight. The typical peak plasma concentration (Cmax) for a lighter weight subject was found to increase by approximately 7% (from 19.3 to 20.6 ng ml−1) relative to a subject with median body weight, whereas Cmax for a heavier subject is predicted to decrease by approximately 8% (from 19.3 to 17.9 ng ml−1). These changes in Cmax were considered of limited clinical significance.
Predictive performance of final model
The final model was evaluated by VPCs from 1000 nonmem simulations, with IIV and residual variability included. VPCs were stratified by study and shown in Figure 4 (representatives from four studies). The observed data fell mostly within the 95% CI of the simulated data. Based on these plots, the population model was considered to fit the data reasonably well.
Figure 4.

Visual predictive check showing observed and simulated axitinib concentrations plotted against time following oral 5-mg dose of axitinib in (A) study 1: Form IV in subjects after overnight fasting; (B) study 2: Form IV in subjects either fed or after overnight fasting; (C) study 6: Form IV in subjects after overnight fasting; (D) study 7: Form IV and Form XLI in subjects fed moderate-fat diet. Open circles represent observed individual points. The grey shaded areas indicate the 95% CI of the simulated data, with the solid lines representing the simulated median. The grey horizontal dashed lines represent lower limit of quantitation (LLOQ).  , simulated 95% CI;
, simulated 95% CI;  , simulated median;
, simulated median;  , observed;
, observed;  , LLOQ
, LLOQ
Discussion
This analysis utilized a population-based approach to assess the plasma PK of axitinib using pooled data from 337 healthy volunteers in 10 phase I clinical studies. The second objective was to evaluate the effect of clinically relevant covariates that may explain variability in plasma exposure following administration of a single 5 mg oral dose of axitinib in healthy subjects. Since other tyrosine kinase inhibitors have previously reported correlations between drug exposure and increased toxicity and/or efficacy [23,24], it is critical to identify demographic, biologic and/or genetic factors that contribute to the variability in axitinib PK.
The disposition of axitinib was best characterized by a linear, two compartment model with first bullet absorption and lag time. The lag time from dose administration to the beginning of the first bullet absorption was estimated to be 0.457 h. CL (%IIV) and Vc (%IIV) were estimated to be 17 l h−1 (52.2%) and 45.3 l (30.8%), respectively. Various covariates were tested throughout the modelling process for potential effects on axitinib CL and Vc. The effects of food and different crystal polymorph formulations were evaluated on ka and F. In earlier clinical trials, axitinib was administered as crystal polymorph Form IV FCIR tablets. Subsequently, a new crystal polymorph Form XLI with more favourable properties was discovered, evaluated as a clinical candidate and implemented for commercial use. The population PK results showed that for Form IV, ka and F of axitinib were increased by 207% and 33.8%, respectively, in the fasted state compared with the fed state. In the fed state, F of Form XLI was 15% lower than that of Form IV. The food effect on Form XLI could not be tested in this analysis since data from the study measuring plasma PK for Form XLI administered with food vs. after fasting were not available at the time of this analysis. Recently, however, results of the food effect study directly comparing the two crystal polymorph formulations showed that the food effect was not clinically meaningful for Form XLI [15]. Hence, the recommendation is that axitinib can be taken with or without food.
After inclusion of the aforementioned effects in the model, body weight was found to affect significantly axitinib Vc. However, the predicted changes based on body weight were less than the estimated IIV of 30.8% for Vc. In addition, the effect of body weight on Vc translates marginally to altered Cmax, but not to overall plasma exposures. Taken together, these data indicate that no dosing adjustment of axitinib is needed based on body weight. None of the other covariates studied, including demographic factors such as age, gender, race, measures of renal (CLcr) and hepatic function (AST, ALT or bilirubin) and metabolic genotypes and inferred phenotypes (UGT1A1*28 and CYP2C19, respectively), was found to affect significantly the disposition of axitinib. It is of note that when axitinib PK data from young healthy volunteers were expanded to include additional data from patients with advanced renal cell carcinoma, age over 60 years and Japanese ethnicity were found to be associated with decreased CL by ∼20% [31]. However, together, they accounted for <5% of the CL. Furthermore, simulations showed substantial overlap between the concentration profiles of >60-year-old Japanese with light body weight and those of <60-year-old heavy non-Japanese. Hence no dose adjustments are recommended on the basis of age or race.
The absence of significant association between axitinib CL and CLcr, AST, ALT or bilirubin in healthy volunteers was not unexpected, since the levels of these laboratory measurements were within normal ranges, indicative of normal kidney and liver function. In this population PK analysis, only laboratory values of AST, ALT and bilirubin were used as markers of hepatic function. However, a formal hepatic impairment study was previously conducted in subjects using the Child-Pugh classification (a more comprehensive assessment of hepatic function). That study showed that mild hepatic impairment did not alter axitinib plasma exposures compared with normal hepatic function. However, subjects with moderate hepatic impairment demonstrated a two-fold higher axitinib plasma exposure, necessitating axitinib dose adjustments in these patients [18]. Smoking status was included in this analysis since smoking is known to induce CYP1A2, which is partly responsible for axitinib metabolism. The lack of effect of smoking on axitinib CL observed in healthy volunteers in this analysis is similar to the finding reported for another tyrosine kinase inhibitor, imatinib, which is also metabolized partly by CYP1A2, in cancer patients [32]. Interestingly, smoking was found to be associated with increased risks of adverse effects and reduced overall survival in those cancer patients treated with imatinib.
Among the key enzymes known to be involved in the metabolism of axitinib, variants reported to date in the coding region of the CYP3A4 gene are relatively uncommon and do not lead to major changes in catalytic function and/or expression of this enzyme [33–35]. On the other hand, the CYP3A5, UGT1A1 and CYP2C19 genes have known genetic polymorphisms that significantly alter the expression and/or activity of the protein (e.g. the CYP3A5*3 allele with alternative splicing resulting in loss of activity, the UGT1A1*28 allele leading to a reduced expression, the CYP2C19*2 and/or *3 alleles leading to loss of function), which in turn, affect the PK and/or efficacy of some drugs [33,36–40]. Thus, potential effects of genetic variants in the CYP3A5 gene (CYP3A5*3 A6986G) and UGT1A1*28 genes and the CYP2C19 inferred phenotype on axitinib CL were evaluated. Although the allelic frequency of CYP3A5*3 A6986G variant in this population was 83%, a preliminary covariate analysis did not indicate any significant effect of this genetic polymorphism and therefore, it was not included in the formal covariate analyses. The incidence of UGT1A1*28 homozygous variant (10%) or heterozygous (32%) genotype present in this pooled analysis is consistent with the distribution of Caucasians (62%) and Asians (23%), as seen in other study populations [41,42]. Individuals carrying the UGT1A1*28 variant allele have reduced protein expression, leading to a decreased hepatic conjugation and subsequent biliary excretion of bilirubin, its endogenous substrate [43]. Similarly, in those subjects, there is a potential for reduced glucuronidation (and thus excretion) of axitinib. Considering that the glucuronide metabolite accounts for roughly 50% of the circulating axitinib metabolites, it is surprising that UGT1A1*28 polymorphism was not identified as a significant covariate for axitinib CL or Vc. Given that this analysis was conducted in healthy volunteers, additional analyses to investigate the effect of UGT1A1*28 polymorphism on axitinib PK and safety in cancer patients are warranted. With regard to CYP2C19 metabolizing status, the majority of the subjects (93%) were categorized as extensive metabolizers, with 2% and 4% of subjects categorized as ultra-rapid and poor metabolizers, respectively. Since the number of ultra-rapid or poor metabolizers in the pooled data set was relatively small, a lack of the effect of CYP2C19 inferred phenotype on axitinib PK observed in the current analysis may need a confirmatory study using a larger data set. An independently conducted fixed effects meta-analysis of the data set in healthy volunteers pooled from 11 clinical trials (10 of which were included in the analysis described here) showed that the genetic polymorphisms observed for these drug metabolizing enzymes tested, including the CYP3A5, UGT1A1*28 and CYP2C19 genes, or transporters such as P-glycoprotein and breast cancer resistance protein, were not significant predictors of the variability in axitinib PK [44]. The results of the covariate analysis for axitinib PK (i.e. a lack of clinically significant covariates identified in the current study) are similar to those reported for other tyrosine kinase inhibitors 24,[25]. In bullet to optimize clinical benefits of these tyrosine kinase inhibitors, additional studies are warranted to evaluate other factors that may affect their disposition.
A notable characteristic of the population model for axitinib was an IIV of 52.5% for CL and 30.8% for Vc and intra-subject variability (residual standard deviation) of 50.9% and 34.2% for oral and intravenous administration, respectively. The exact cause(s) for this IIV in axitinib PK are yet to be elucidated. However, because axitinib is primarily metabolized by CYP3A4/5, it is speculated that the major source of variability may still be differences in CYP3A4/5 expression and/or activity in the liver and intestine (10-to 40-fold variability in expression of CYP3A4/5 has been reported in healthy subjects [33]). Since axitinib is a low-extraction drug, the metabolic clearance of axitinib may be particularly sensitive to variable levels of hepatic and intestinal metabolizing enzymes. Another possible explanation is variability in plasma binding of axitinib between subjects. With regard to high residual (intra-subject) variability, it is plausible that differences in dissolution and subsequent gastrointestinal absorption of axitinib may be a contributing factor. Since axitinib solubility is pH-dependent, with solubility declining with increasing pH [11], changes in pH values in stomach and duodenum may lead to variable dissolution of axitinib. The intra-subject observed with axitinib PK is comparable with that reported for other approved orally administered agents in this class, such as sunitinib, pazopanib and sorafenib [25,45,46].
This population PK analysis in healthy volunteers was the first step in an overall strategy for axitinib dose–response evaluation in cancer patients. The analysis was conducted using pooled data from healthy volunteers only because they provide the cleanest assessment of drug disposition in bullet to avoid confounding factors often seen in cancer patients, such as those with compromised end-stage organ function, underlying disease or taking concomitant medications for comorbidity that may affect the metabolism of axitinib. This analysis developed a structural PK model that adequately characterized the disposition of axitinib in a controlled population and would be useful in evaluating population PK analyses in cancer patients.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that this study was funded by Pfizer Inc. Brian Hee, Yazdi K. Pithavala and Michael A. Amantea are employed by and own stock in Pfizer Inc and Meghan Brennan and May Garrett were employed by Pfizer Inc at the time of the study. Bill Poland is employed by Pharsight, which received funding from Pfizer Inc to provide modelling and simulation services. Medical writing support was funded by Pfizer Inc and was provided by Mariko Nagashima, PhD, of UBC Scientific Solutions (Southport, CT, USA).
References
- 1.Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, McTigue MA, Murray BW, Kania RS, O'Connor P, Shalinsky DR, Bender SL. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–7283. doi: 10.1158/1078-0432.CCR-08-0652. [DOI] [PubMed] [Google Scholar]
- 2.Choueiri TK. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr Opin Investig Drugs. 2008;9:658–671. [PubMed] [Google Scholar]
- 3.Kelly RJ, Rixe O. Axitinib – a selective inhibitor of the vascular endothelial growth factor (VEGF) receptor. Target Oncol. 2009;4:297–305. doi: 10.1007/s11523-009-0126-9. [DOI] [PubMed] [Google Scholar]
- 4.Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, Trask PC, Kim S, Rini BI. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975–984. doi: 10.1016/S1470-2045(07)70285-1. [DOI] [PubMed] [Google Scholar]
- 5.Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, Kane MA, Sherman E, Kim S, Bycott P, Tortorici M, Shalinsky DR, Liau KF, Cohen RB. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26:4708–4713. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rini BI, Wilding G, Hudes G, Stadler WM, Kim S, Tarazi J, Rosbrook B, Trask PC, Wood L, Dutcher JP. Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27:4462–4468. doi: 10.1200/JCO.2008.21.7034. [DOI] [PubMed] [Google Scholar]
- 7.Schiller JH, Larson T, Ou SH, Limentani S, Sandler A, Vokes E, Kim S, Liau K, Bycott P, Olszanski AJ, von Pawel J. Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: results from a phase II study. J Clin Oncol. 2009;27:3836–3841. doi: 10.1200/JCO.2008.20.8355. [DOI] [PubMed] [Google Scholar]
- 8.Bhojani N, Jeldres C, Patard JJ, Perrotte P, Suardi N, Hutterer G, Patenaude F, Oudard S, Karakiewicz PI. Toxicities associated with the administration of sorafenib, sunitinib, and temsirolimus and their management in patients with metastatic renal cell carcinoma. Eur Urol. 2008;53:917–930. doi: 10.1016/j.eururo.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 9.Hutson TE, Figlin RA, Kuhn JG, Motzer RJ. Targeted therapies for metastatic renal cell carcinoma: an overview of toxicity and dosing strategies. Oncologist. 2008;13:1084–1096. doi: 10.1634/theoncologist.2008-0120. [DOI] [PubMed] [Google Scholar]
- 10.Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG, Negrier S, Ou Y-C, Castellano D, Lim HY, Uemura H, Tarazi J, Cella D, Chen C, Rosbrook B, Kim S, Motzer RJ. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 11.Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol. 2005;23:5474–5483. doi: 10.1200/JCO.2005.04.192. [DOI] [PubMed] [Google Scholar]
- 12.Zientek M, Kang P, Jiang Y, Smith B. 2010. In vitro kinetic characterization of axitinib metabolism to estimate the clinical implications of genetic polymorphisms Proceedings of the International Meeting of the International Society for the Study of Xenobiotics: Genetic Polymorphisms in Drug Disposition Workshop;April 11–13; Indianapolis, IN.
- 13.Center for Drug Evaluation and Research. 2011. Clinical pharmacology review: Inlyta In:Summary Basis for Approval, Clinical Pharmacology and Biopharmaceutics Review. Document # 202324Orig1s000. US Department Health and Human Services Food and Drug Administration,Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202324Orig1s000ClinPharmR.pdf(last accessed 9 October 2012)
- 14.Pithavala YK, Tong W, Mount J, Rahavendran SV, Garrett M, Hee B, Selaru P, Sarapa N, Klamerus KJ. Effect of ketoconazole on the pharmacokinetics of axitinib in healthy volunteers. Invest New Drugs. 2012;30:273–281. doi: 10.1007/s10637-010-9511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pithavala YK, Chen Y, Toh M, Selaru P, Labadie R, Garrett M, Hee B, Mount J, Ni G, Klamerus KJ, Tortorici MA. Evaluation of the effect of food on the pharmacokinetics of axitinib in healthy volunteers. Cancer Chemother Pharmacol. 2012;70:103–112. doi: 10.1007/s00280-012-1888-9. [DOI] [PubMed] [Google Scholar]
- 16.Pithavala YK, Tortorici M, Toh M, Garrett M, Hee B, Kuruganti U, Ni G, Klamerus KJ. Effect of rifampin on the pharmacokinetics of axitinib (AG-013736) in Japanese and Caucasian healthy volunteers. Cancer Chemother Pharmacol. 2010;65:563–570. doi: 10.1007/s00280-009-1065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Jiang J, Zhang J, Tortorici MA, Pithavala YK, Lu L, Ni G, Hu P. A Phase I study to evaluate the pharmacokinetics of axitinib (AG-13736) in healthy Chinese volunteers. Int J Clin Pharmacol Ther. 2011;49:679–687. doi: 10.5414/cp201570. [DOI] [PubMed] [Google Scholar]
- 18.Tortorici MA, Toh M, Rahavendran SV, Labadie RR, Alvey CW, Marbury T, Fuentes E, Green M, Ni G, Hee B, Pithavala YK. Influence of mild and moderate hepatic impairment on axitinib pharmacokinetics. Invest New Drugs. 2011;29:1370–1380. doi: 10.1007/s10637-010-9477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin LP, Kozloff MF, Herbst RS, Samuel TA, Kim S, Rosbrook B, Tortorici M, Chen Y, Tarazi J, Olszanski AJ, Rado T, Starr A, Cohen RB. Phase I study of axitinib combined with paclitaxel, docetaxel or capecitabine in patients with advanced solid tumours. Br J Cancer. 2012;107:1268–1276. doi: 10.1038/bjc.2012.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukohara T, Nakajima H, Mukai H, Nagai S, Itoh K, Umeyama Y, Hashimoto J, Minami H. Effect of axitinib (AG-013736) on fatigue, thyroid-stimulating hormone, and biomarkers: a phase I study in Japanese patients. Cancer Sci. 2010;101:963–968. doi: 10.1111/j.1349-7006.2009.01465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiwara Y, Kiyota N, Chayahara N, Suzuki A, Umeyama Y, Mukohara T, Minami H. Management of axitinib (AG-013736)-induced fatigue and thyroid dysfunction, and predictive biomarkers of axitinib exposure: results from phase I studies in Japanese patients. Invest New Drugs. 2012;30:1055–1064. doi: 10.1007/s10637-011-9637-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozloff MF, Martin LP, Krzakowski M, Samuel TA, Rado TA, Arriola E, De Castro Carpeno J, Herbst RS, Tarazi J, Kim S, Rosbrook B, Tortorici M, Olszanski AJ, Cohen RB. Phase I trial of axitinib combined with platinum doublets in patients with advanced non-small cell lung cancer and other solid tumours. Br J Cancer. 2012;107:1277–1285. doi: 10.1038/bjc.2012.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demetri GD, Wang Y, Wehrle E, Racine A, Nikolova Z, Blanke CD, Joensuu H, von Mehren M. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141–3147. doi: 10.1200/JCO.2008.20.4818. [DOI] [PubMed] [Google Scholar]
- 24.Houk BE, Bello CL, Kang D, Amantea M. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res. 2009;15:2497–2506. doi: 10.1158/1078-0432.CCR-08-1893. [DOI] [PubMed] [Google Scholar]
- 25.Jain L, Woo S, Gardner ER, Dahut WL, Kohn EC, Kummar S, Mould DR, Giaccone G, Yarchoan R, Venitz J, Figg WD. Population pharmacokinetic analysis of sorafenib in patients with solid tumors. Br J Clin Pharmacol. 2011;72:294–305. doi: 10.1111/j.1365-2125.2011.03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyamichev V, Brow MA, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 27.Rousseau A, Leger F, Le Meur Y, Saint-Marcoux F, Paintaud G, Buchler M, Marquet P. Population pharmacokinetic modeling of oral cyclosporin using NONMEM: comparison of absorption pharmacokinetic models and design of a Bayesian estimator. Ther Drug Monit. 2004;26:23–30. doi: 10.1097/00007691-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Duval V, Karlsson MO. Impact of omission or replacement of data below the limit of quantification on parameter estimates in a two-compartment model. Pharm Res. 2002;19:1835–1840. doi: 10.1023/a:1021441407898. [DOI] [PubMed] [Google Scholar]
- 29.Garrett M, Amantea MA, Pithavala YK, Ruiz A. Evaluation of the impact of omitting drug concentraion data below the lower limit of quantification (LLOQ) on the pharmacokinetics of axitinib (AG-013736), an anti-angiogenic agent. Clin Pharmacol Ther. 2010;87(Suppl. 1):S78–79. abstr PIII-33): [Google Scholar]
- 30.Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–569. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rini BI, Garrett M, Poland B, Dutcher JP, Rixe O, Wilding G, Stadler WM, Pithavala YK, Kim S, Tarazi J, Motzer RJ. Axitinib in metastatic renal cell carcinoma: results of a pharmacokinetic and pharmacodynamic analysis. J Clin Pharmacol. 2013;53:491–504. doi: 10.1002/jcph.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Erp N, Gelderblom H, van Glabbeke M, Van Oosterom A, Verweij J, Guchelaar HJ, Debiec-Rychter M, Peng B, Blay JY, Judson I. Effect of cigarette smoking on imatinib in patients in the soft tissue and bone sarcoma group of the EORTC. Clin Cancer Res. 2008;14:8308–8313. doi: 10.1158/1078-0432.CCR-08-1303. [DOI] [PubMed] [Google Scholar]
- 33.Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54:1271–1294. doi: 10.1016/s0169-409x(02)00066-2. [DOI] [PubMed] [Google Scholar]
- 34.Dai D, Tang J, Rose R, Hodgson E, Bienstock RJ, Mohrenweiser HW, Goldstein JA. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J Pharmacol Exp Ther. 2001;299:825–831. [PubMed] [Google Scholar]
- 35.Wojnowski L, Kamdem LK. Clinical implications of CYP3A polymorphisms. Expert Opin Drug Metab Toxicol. 2006;2:171–182. doi: 10.1517/17425255.2.2.171. [DOI] [PubMed] [Google Scholar]
- 36.Zhao W, Elie V, Roussey G, Brochard K, Niaudet P, Leroy V, Loirat C, Cochat P, Cloarec S, Andre JL, Garaix F, Bensman A, Fakhoury M, Jacqz-Aigrain E. Population pharmacokinetics and pharmacogenetics of tacrolimus in de novo pediatric kidney transplant recipients. Clin Pharmacol Ther. 2009;86:609–618. doi: 10.1038/clpt.2009.210. [DOI] [PubMed] [Google Scholar]
- 37.Ando Y, Hasegawa Y. Clinical pharmacogenetics of irinotecan (CPT-11) Drug Metab Rev. 2005;37:565–574. doi: 10.1080/03602530500316254. [DOI] [PubMed] [Google Scholar]
- 38.Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Jang IJ, Lee DH, Lee JS. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. doi: 10.1200/JCO.2005.03.0239. [DOI] [PubMed] [Google Scholar]
- 39.Maeda A, Ando H, Asai T, Ishiguro H, Umemoto N, Ohta M, Morishima M, Sumida A, Kobayashi T, Hosohata K, Ushijima K, Fujimura A. Differential impacts of CYP2C19 gene polymorphisms on the antiplatelet effects of clopidogrel and ticlopidine. Clin Pharmacol Ther. 2011;89:229–233. doi: 10.1038/clpt.2010.268. [DOI] [PubMed] [Google Scholar]
- 40.Hunfeld NG, Mathot RA, Touw DJ, van Schaik RH, Mulder PG, Franck PF, Kuipers EJ, Geus WP. Effect of CYP2C19*2 and *17 mutations on pharmacodynamics and kinetics of proton pump inhibitors in Caucasians. Br J Clin Pharmacol. 2008;65:752–760. doi: 10.1111/j.1365-2125.2007.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaniwa N, Kurose K, Jinno H, Tanaka-Kagawa T, Saito Y, Saeki M, Sawada J, Tohkin M, Hasegawa R. Racial variability in haplotype frequencies of UGT1A1 and glucuronidation activity of a novel single nucleotide polymorphism 686C> T (P229L) found in an African-American. Drug Metab Dispos. 2005;33:458–465. doi: 10.1124/dmd.104.001800. [DOI] [PubMed] [Google Scholar]
- 42.Liu JY, Qu K, Sferruzza AD, Bender RA. Distribution of the UGT1A1*28 polymorphism in Caucasian and Asian populations in the US: a genomic analysis of 138 healthy individuals. Anticancer Drugs. 2007;18:693–696. doi: 10.1097/CAD.0b013e32803a46fe. [DOI] [PubMed] [Google Scholar]
- 43.Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP, Chowdhury NR. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med. 1995;333:1171–1175. doi: 10.1056/NEJM199511023331802. [DOI] [PubMed] [Google Scholar]
- 44.Brennan M, Williams JA, Chen Y, Tortorici M, Pithavala Y, Liu YC. Meta-analysis of contribution of genetic polymorphisms in drug-metabolizing enzymes or transporters to axitinib pharmacokinetics. Eur J Clin Pharmacol. 2012;68:645–655. doi: 10.1007/s00228-011-1171-8. [DOI] [PubMed] [Google Scholar]
- 45.Center for Drug Evaluation and Research. 2005. Clinical pharmacology review: Sutent In:Summary Basis for Approval, Clinical Pharmacology and Biopharmaceutics Review. Document # NDA 21-938 (GIST), NDA 21-968 (MRCC). US Department of Health and Human Services Food and Drug Administration,Available at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021938_S000_Sutent_BioPharmR.pdf(last accessed 9 October 2012)
- 46.Center for Drug Evaluation and Research. 2008. Clinical pharmacology review: Votrient In:Summary Basis for Approval, Clinical Pharmacology and Biopharmaceutics Review. Document # 22-465. US Department of Health and Human Services Food and Drug Administration,Avaialble at http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_ClinPharmR.pdf(last accessed 9 October 2012)