

Abstract
Bile acids (BAs) are digestive secretions that are necessary for the emulsification and absorption of dietary fats. Given the episodic nature of BA secretion and intestinal re-absorption, the circulating and tissue levels of BAs, like those of the gut hormones, fluctuate in fasting and fed states, and BA levels and forms are markedly affected by disease. BAs exert widespread hormonal-like effects by activating receptors in the nucleus and at the plasma membrane. The nuclear steroid receptors mediate the genomic actions of BAs on BA, glucose and lipid homeostasis. GPBA (TGR5) is a G-protein coupled plasma membrane receptor for BAs that mediates many of the rapid, non-genomic actions of BAs. GPBA has been implicated in the control of glucose homeostasis, inflammation and liver functions. Recent observations have revealed an unexpected role for GPBA in the nervous system. GPBA is expressed by enteric neurons and enterochromaffin cells that control peristalsis, and GPBA mediates the prokinetic actions of BAs in the colon that have been known for millennia. GPBA is also present on primary spinal afferent and spinal neurons that are necessary for sensory transduction. BA-induced activation of GPBA in the sensory nervous system promotes scratching behaviours and analgesia, which may contribute to the pruritus and painless jaundice that are observed in some patients with chronic cholestatic disease, where circulating BA concentrations are markedly increased. Thus, GPBA has emerged as an intriguing target for diverse metabolic, inflammatory, digestive and sensory disorders, where agonists and antagonists may be of value.
Linked ArticlesThis article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-5
Keywords: TGR5, itch, analgesia, DCA, TRPA1, sensory neurons, pain, cholestasis, inflammation, DRG
Introduction
Bile acids (BAs), a major component of bile, are digestive secretions that are necessary for the emulsification and absorption of dietary fats. Remarkably, BAs are also signalling molecules. The levels of BAs fluctuate in the circulation under physiological conditions and during disease states, rather like the circulating levels of gut hormones, and BAs can regulate cells throughout the body by activating specific BA receptors in the nucleus and at the plasma membrane. These receptors mediate the effects of BAs on diverse physiological processes, ranging from the control of glucose homeostasis to peristaltic contractions of the digestive tract, and have been implicated in disorders as diverse as obesity and pruritus.
This review concerns GPBA (also called TGR5, GPBAR1, M-BAR, or GPR131), a recently identified GPCR for BAs. This nomenclature complies with the British Journal of Pharmacology's guidelines (Alexander et al., 2013). In the past decade, GPBA has been shown to mediate many of the non-genomic actions of BAs on thermogenesis, insulin secretion and glucose homeostasis, inflammation, digestive functions and sensory transduction. As such, GPBA has emerged as a major therapeutic target for metabolic, inflammatory, digestive and sensory disorders.
BAs are signalling molecules with hormonal-like actions
BAs are a large complex family of amphipathic molecules with a steroid backbone. They exhibit considerable structural diversity within different cells, body compartments and pathophysiological states, and there is considerable inter-species variability. The complexity and diversity of BAs is beyond the scope of this paper, but has been reviewed in detail (Hofmann and Hagey, 2008). BAs are synthesized and metabolized in mammalian cells and colonic bacteria by multiple and mostly well-characterized enzymatic pathways (reviewed in Lefebvre et al., 2009). BAs are synthesized in hepatocytes from cholesterol by an enzymatic pathway that includes hydroxylation and side-chain shortening, which confers BAs with detergent-like properties that are required for their principal digestive function, namely the emulsification and absorption of dietary fats and lipid-soluble vitamins (Figure 1). Primary BAs, the direct products of cholesterol metabolism in the liver, include chenodeoxycholic acid (CDCA) and cholic acid (CA) in humans. Before secretion into the biliary system, primary BAs are conjugated to taurine or glycine. However, BAs can be further modified in the liver by sulphation and glucuronidation. BAs are stored within the gall bladder and are secreted into the intestinal lumen after feeding in response to cholecystokinin, an intestinal hormone that is released by luminal fats and which stimulates contraction of the gall bladder. Primary BAs are actively and efficiently absorbed in the terminal ileum by the apical sodium-dependent BA transporter (ASBT) and the basolateral heterodimeric organic solute transporter (Ostα/Ostβ). Despite the efficiency of these transport processes, a small proportion of BAs escape ileal absorption and pass to the colon, where they are deconjugated, oxidized and dehydroxylated by bacterial enzymes to form secondary BAs, which include lithocholic acid (LCA) and deoxycholic acid (DCA) in humans (Figure 1). These reactions increase the hydrophobicity of BAs, which facilitates their passive flux across colonocytes. The absorbed primary and secondary BAs then enter the hepatic portal vein and recycle to the liver for reuse. This pathway of BA secretion, reabsorption and recycling comprises the enterohepatic circulation of the BA pool, which mostly comprises CA, CDCA and DCA.
Figure 1.
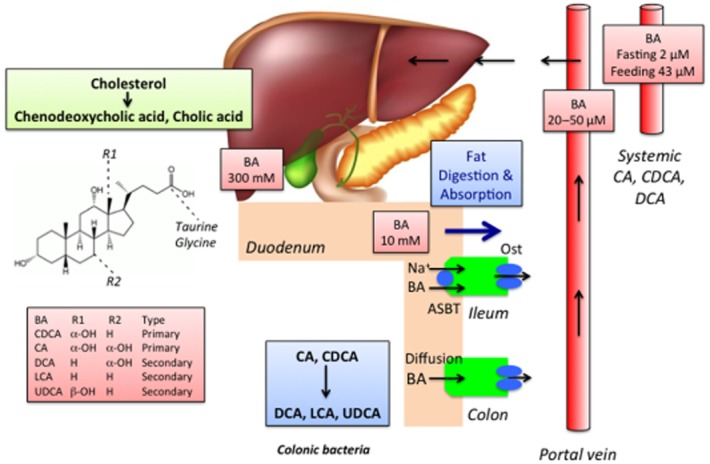
The synthesis, metabolism, and enterohepatic circulation of BAs.
The synthesis, transport and secretion of BAs are tightly regulated by multiple physiological mechanisms that can regulate the concentration of BAs in different compartments and tissues, and the overall composition of the BA pool. For example, given the episodic nature of bile secretion, the concentrations of BAs in the intestinal lumen and portal and systemic circulations wax and wane during feeding and fasting, reminiscent of the circulating concentrations of gut hormones. Moreover, disruption to the enterohepatic circulation of BAs, which can occur during disease or therapy, leads to marked alterations in the concentrations and composition of BAs in the intestinal lumen and the circulation. For example, cholestatic diseases that are characterized by diminished secretion of BAs from the gall bladder result in diminished delivery of BAs to the intestinal lumen and marked elevations in the circulating levels of BAs. These physiological and pathological alterations in the concentrations and composition of BAs are likely to be of direct relevance to the hormonal-like actions of BAs.
In addition to their role in the solubilization of dietary lipids and the fat-soluble vitamins A, K, D and E, BAs are signalling molecules that regulate many cell types by activating specific receptors in the nucleus and at the plasma membrane (Figure 2). Of these, the nuclear receptors have been most thoroughly characterized. An in-depth discussion of these receptors is beyond the scope of this paper, although they have been reviewed in detail (Lefebvre et al., 2009). Nuclear BA receptors include the farnesoid X receptor, the pregnane X receptor, and the vitamin D receptor, which mediate the transcriptional effects of BAs. The farnesoid X receptor is a ligand-activated transcription factor that is prominently expressed in the liver, intestine and kidney (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999). CDCA, LCA, DCA and CA can activate the farnesoid X receptor, which plays a major role in the regulation of BA synthesis, detoxification and transport, and mediates the actions of BAs on lipoprotein metabolism and glucose homeostasis. LCA can also activate the pregnane X and vitamin D receptors, which protect against the toxic actions of BAs (Staudinger et al., 2001; Xie et al., 2001; Makishima et al., 2002).
Figure 2.
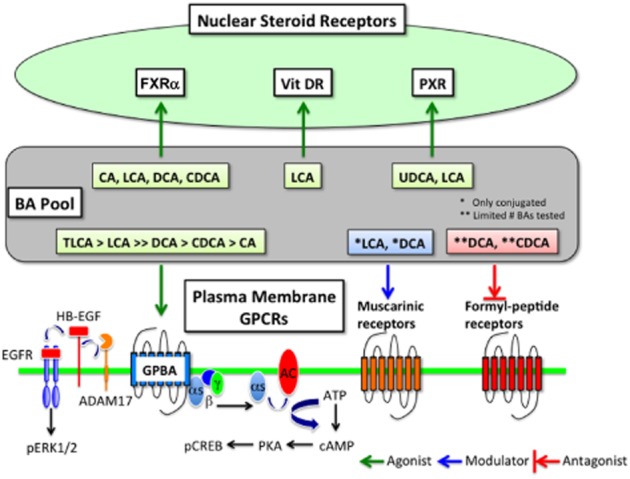
BA receptors in the nucleus and at the plasma membrane.
GPBA is a GPCR for BAs
BAs can regulate the activity of several GPCRs (Figure 2). Conjugated forms of LCA and DCA can modulate the activity of muscarinic receptors expressed in CHO and chief cells (Raufman et al., 1998; 2002), and DCA and CDCA are antagonists of the formyl-peptide receptor (Le et al., 2002). However, multiple BAs can activate GPBA (also known as Gpbar1, M-BAR and GPR131), which is recognized as a bona fide BA receptor (Maruyama et al., 2002; Kawamata et al., 2003). GPBA cDNA is encoded by a single exon gene located on mouse chromosome 1c3 and human chromosome 2q35. The open reading frame of the receptor encodes 330 amino acids and is predictive of the seven transmembranes spanning GPCR with 28% identity to human sphingosine-1-phosphate receptor (EDG-1), a member of the class A (rhodopsin-like) GPCR family (Maruyama et al., 2002). Several endogenous BAs activate GPBA, albeit with graded potencies. The rank order of potency with which the BAs stimulate cAMP generation in GPBA expressing CHO cells is tauro lithocholic acid (TLCA, 0.33 μM) > LCA (0.53 μM) >> DCA (1 μM) > CDCA (4.4 μM) > CA (7.7 μM), whereas ursodeoxycholic acid (UDCA) and cholesterol have little activity (Kawamata et al., 2003). Oleanolic acid (OA), an active component from the leaves of the European olive tree Olea europaea, activates GPBA with a similar potency to LCA, but does not activate the farnesoid X receptor (Sato et al., 2007). Multiple steroidal and non-steroidal GPBA ligands have been developed as potential treatments for metabolic and inflammatory disorders (Gioiello et al., 2012).
GPBA is widely expressed in various organs and cell types, although there are marked differences in the level of expression. GPBA mRNA is highly expressed in the human liver, gastrointestinal tract, gall bladder and immune tissues (Maruyama et al., 2002; Kawamata et al., 2003). GPBA mRNA and immunoreactivity have also been detected in the gall bladder epithelium (Vassileva et al., 2006; Keitel et al., 2009), monocytes (Kawamata et al., 2003), sinusoidal endothelial cells, Kupffer cells (Keitel et al., 2007; 2008), brown adipose tissue and skeletal muscle (Watanabe et al., 2006), and in neurons of the enteric nervous system, dorsal root ganglia and CNS (Keitel et al., 2010a,b2010b; Poole et al., 2010; Alemi et al., 2013b).
Mechanisms of GPBA signalling
When expressed in model cell lines (e.g. CHO or HEK cells) GPBA couples to Gαs, leading to the stimulation of adenylyl cyclase, formation of cAMP, and subsequent activation of PKA (Maruyama et al., 2002; Kawamata et al., 2003). There has been considerable interest in characterizing the downstream pathways of GPBA signalling that control proliferation and apoptosis in oesophageal and gastric cancer cells in view of the potential role of BAs in cancer progression. In the human gastric carcinoma cell line ACS, DCA promotes the sustained activation of ERK1/2 by a mechanism that involves transactivation of the EGF receptor (Yasuda et al., 2007). siRNA knockdown indicated that DCA-stimulation of ERK1/2 required expression of GPBA and activation of the membrane protease ADAM17, which liberates membrane-tethered heparin-binding EGF-like growth factor, an agonist of the EGF receptor. GPBA is also highly expressed in metastatic gastric adenocarcinoma and taurodeoxycholic acid (TDCA)-induced proliferation of ACS cells requires expression of GPBA (Cao et al., 2013). In the oesophageal adenocarcinoma cell line FLO, GPBA mediates TDCA-stimulated expression of the NADPH oxidase NOX5-S, which is required for the generation of reactive oxygen species and cell proliferation (Hong et al., 2010). In gastric and oesophageal adenocarcinoma cell lines GPBA couples to Gαq, which appears to be necessary for BA-stimulated proliferation (Hong et al., 2010; Cao et al., 2013). GPBA signalling has also been implicated in BA-induced apoptosis of hepatocytes, which can occur during cholestasis. GPBA knockdown in a human hepatocyte cell line Huh-BAT, attenuates BA-induced activation of JNK and caspase 8, and suppresses BA-stimulated apoptosis, implicating GPBA in the hepatotoxic actions of BAs (Yang et al., 2007).
Because GPBA attenuates macrophage activity (Pols et al., 2011), there has been considerable interest in defining the signalling pathways that underlie GPBA's anti-inflammatory effects. GPBA activation inhibits NF-κB-mediated inflammatory signalling in activated B cells by a mechanism that involves GPBA-induced interaction of IκBα with β-arrestin2, a key regulator of GPCR signalling (Wang et al., 2011). The deletion of GPBA exacerbates lipopolysaccharide-induced hepatic inflammation, which highlights the importance of this anti-inflammatory signalling.
Despite the interest in developing GPBA agonists as therapies for metabolic, inflammatory and digestive diseases, very little is known about the mechanisms that regulate GPBA signalling. The signalling of most GPCRs is tightly regulated by processes that control the interaction of receptors with heterotrimeric G proteins, and by regulation of the subcellular location of the receptor. β-Arrestins play a major role in this regulation. β-Arrestins interact with agonist-occupied receptors and thereby sterically disrupt the interaction of receptors with G-proteins, which desensitizes signalling. β-Arrestins also couple GPCRs to clathrin and AP2, and thereby mediate receptor endocytosis. By recruiting GPCRs and downstream signalling proteins to endosomes, β-arrestins can also actively participate in signal transduction of internalized receptors. GPBA has been reported to internalize after activation (Kawamata et al., 2003), and β-arrestin2 has been shown to participate in GPBA signalling (Wang et al., 2011). However, a detailed analysis of GPBA trafficking and signalling failed to detect GPBA interaction with GPCR kinases or β-arrestins (Jensen et al., 2013). Consistent with lack of detectable interaction with β-arrestins, GPBA did not traffic to endosomes and GPBA-mediated cAMP signals were sustained, with no evidence of tachyphylaxis or desensitization. Instead, activated GPBA redistributed to lipid rafts, where it interacted with and transactivated the EGF receptor. The anti-inflammatory effects of GPBA, serving to partially protect against the development of atherosclerosis, are also β-arrestin independent (Pols et al., 2011). The lack of desensitization and endocytosis of GPBA (TGR5) may be relevant for the development of agonists for treatment of metabolic or inflammatory disorders.
GPBA and metabolic regulation
Energy homeostasis requires an intimate balance between energy input via caloric intake and energy expenditure. GPBA may control thermogenesis through activation of the type 2 iodothyronine deodinase (D2), which converts the minimally active thyroxine (T4) into active 3,5,3′-tri-iodothyronine (T3), a key regulator of metabolism (Watanabe et al., <2006; Figure 3). Dietary supplementation with CA reduced adiposity and increased energy expenditure and expression of D2 in brown adipose tissue in mice fed a high-fat diet. The effect of dietary CA on adiposity was not observed in D2 knockout mice, but was unaffected by antagonism of the farnesoid X receptor, which confirms the importance of D2 in mediating the anti-obesity actions of BAs and indicates that the mechanism is independent of nuclear BA receptors. Evidence for a role of GPBA in BA-induced energy expenditure is provided by the observations that the GPBA-selective agonist benzyl 2-keto-6-methyl-4-(2-thienyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate stimulated D2 activity in human skeletal muscle myoblasts (Watanabe et al., 2006). Furthermore, the synthetic GPBA agonist INT-777 (6α-ethyl-23(S)-methyl-CA) was shown to increase energy expenditure in mice fed a high-fat diet, as determined by indirect calorimetry (Thomas et al., 2009). INT-777 also attenuated body weight in GPBA wild-type and overexpressing transgenic mice, with no reduction seen in the GPBA knockout mice (Pols et al., 2011). When fed a high-fat diet, female GPBA knockout mice weigh more and have a higher body fat content than wild-type mice, although lean body fat is unaffected (Maruyama et al., 2006). However, another study failed to detect an effect of GPBA deletion on body weight (Vassileva et al., 2006). Thus, although several observations suggest the existence of a BA-GPBA-cAMP-D2 pathway, the contribution of GPBA to the regulation of energy expenditure and body weight requires further investigation.
Figure 3.
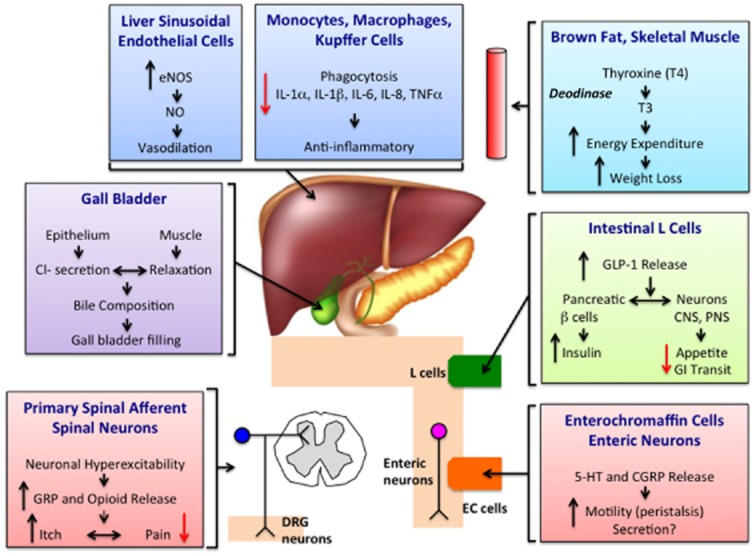
Proposed functions of GPBA.
GPBA and glucose homeostasis
A profiling study of metabolites in the circulation of human subjects after glucose challenge revealed marked increases in the circulating levels of BAs after glucose that correlated with measures of insulin sensitivity that are beneficial to glucose metabolism (Shaham et al., 2008). OA extracted from olive leaves is a GPBA-selective agonist that does not activate the farnesoid X receptor (Sato et al., 2008). OA partially corrects the diet-induced insulin resistance in mice fed a high-fat diet as demonstrated by its ability to lower plasma glucose and insulin levels (Sato et al., 2007). At least two mechanisms may account for the anti-diabetic effect of GPBA agonists (Figure 3). Firstly, GPBA activation may activate D2 and thyroxine, thereby increasing mitochondrial energy expenditure in brown adipose tissue and skeletal muscle and improving glucose utilization (Watanabe et al., 2006; Sato et al., 2007). Secondly, GPBA agonists can promote secretion of glucagon-like peptide 1 (GLP-1; Katsuma et al., 2005; Thomas et al., 2009), a hormone derived from intestinal L-cells that stimulates insulin secretion and suppresses appetite and gastrointestinal transit (Lim and Brubaker, 2006). Collectively, these effects reduce circulating blood glucose. BAs and GPBA-selective agonists stimulate the release of GLP-1 from the murine enteroendocrine cell line STC-1 (Katsuma et al., 2005), and GPBA-stimulated GLP-1 release improves liver and pancreatic function and glucose tolerance in obese mice (Thomas et al., 2009). The effects of a GPBA-selective agonist on GLP-1 release from STC-1 cells is linked to an increased ATP/ADP ratio, which causes the closure of ATP-dependent potassium channels (KATP) and a subsequent calcium influx, leading to GLP-1 secretion (Thomas et al., 2009). Interestingly, the use of BA sequestrants, which complex with BAs in the intestine and prevent reabsorption into the enterohepatic circulation, has been associated with improvements in diabetes mellitus type II (Shang et al., 2010; Potthoff et al., 2013). The use of BA sequestrants correlates with increased GLP-1 release, which is known to improve glycaemic response (Shang et al., 2010; Potthoff et al., 2013). Additionally, the ability of BA sequestrants to induce GLP-1 release, and lower glycaemia was decreased in mice lacking GPBA (Harach et al., 2012; Potthoff et al., 2013) These anti-diabetic consequences of GPBA activation provide mechanistic insights into improved glycaemic control and have sparked interest into a possible avenue for the development of alternative drug therapy in type II diabetes mellitus.
Gastric bypass surgery can lead to a major improvement in glycaemic control and a complete resolution of type II diabetes mellitus. Gastric bypass can increase postprandial GLP-1 and insulin secretion (le Roux et al., 2006) and elevate total fasting serum BAs (Nakatani et al., 2009; Patti et al., 2009). Subfractionation analysis of the total serum BA revealed increases in multiple primary and secondary BAs, which correlated with the elevated postprandial GLP-1 levels (Patti et al., 2009). In addition, alteration of the gastrointestinal anatomy after gastric bypass affects the enterohepatic recirculation of BAs, which leads to undiluted delivery of BAs to the distal intestines (Pournaras et al., 2012; Kohli et al., 2013). Similarly, ileal transposition into the upper jejunum of rats also results in increased serum BAs and GLP-1, with similar metabolic benefits to those seen in human subjects after gastric bypass surgery (Kohli et al., 2010). It is possible that the increased delivery of undiluted BAs to the ileum, where most of the reabsorption occurs, results in enhanced enterohepatic circulation, resultant spillover of BAs into the serum, activation of GPBA in adipose tissue and skeletal muscle, and increased energy expenditure (Watanabe et al., 2006). However, one study showed that resting energy expenditure, insulin sensitivity and insulin response were all unaffected in patients after gastric bypass surgery (Kohli et al., 2013). Therefore, despite promising improvements of glycaemic control and energy metabolism, further studies are needed to substantiate whether bariatric surgery positively correlates with improved metabolic benefits.
GPBA, hepatic inflammation and injury
Hepatobiliary diseases, such as obstructive jaundice, viral hepatitis and cirrhosis, result in 10–20-fold increases in the circulating concentrations of BAs (normally < 10 μM). At high concentrations, BAs can exhibit immunosuppressive effects that may be mediated by GPBA (Figure 3). GPBA is prominently expressed by human monocytes and macrophages (Kawamata et al., 2003). In alveolar macrophages from rabbits, activation of GPBA by BAs stimulates the formation of cAMP and inhibits phagocytic activity and lipopolysaccharide-induced expression and secretion of cytokines including TNF-α, IL-1α, IL-1β, IL-6 and IL-8 (Kawamata et al., 2003). These effects may be GPBA-dependent, because GPBA expression in a human monocyte cell line (THP-1), which normally expresses low levels of GPBA, conferred on the cells the ability of BAs to suppress TNF-α secretion. BAs and the GPBA-selective agonist OA also suppress cytokine release from Kupffer cells, the resident macrophages of the liver (Keitel et al., 2008). Obstruction of the common bile duct up-regulates GPBA on Kupffer cells, where GPBA may serve to protect against liver injury (Keitel et al., 2008). Furthermore, activation of GPBA in macrophages by the synthetic BA agonists 6-EMCA and INT-777 attenuates pro-inflammatory cytokine production (Pols et al., 2011).
GPBA may also regulate the microcirculation of the enterohepatic blood flow (Figure 3). GPBA is localized to sinusoidal endothelial cells, which are exposed to varying concentrations of BAs from the portal vein (Keitel et al., 2007). BAs that activate GPBA induce cAMP production, expression of endothelial NOS and NO release, which dilates the hepatic microvasculature and reduces portal pressure. Insufficient NO is implicated in the pathogenesis of portal hypertension (Vallance and Moncada, 1991), and NO is overexpressed in cirrhotic patients with portal hypertension compared with non-cirrhotic patients (Yokomori et al., 2002). Thus, GPBA may play an important role in maintaining homeostasis within the hepatic microcirculation.
Consistent with the aforementioned anti-inflammatory properties of GPBA in the liver, GPBA may serve as a negative regulator of hepatic inflammation (Wang et al., 2011). GPBA knockout mice are more susceptible to lipopolysaccharide-induced activation of NF-κB, resulting in exacerbated hepatic inflammation compared with wild-type mice.
In contrast to the protective, anti-inflammatory actions of GPBA agonists, GPBA has also been linked to apoptosis. Chronic cholestasis is a well-known cause of cirrhosis and liver failure secondary to the intrahepatic accumulation of toxic BAs that induce hepatocyte injury and ultimately apoptosis (Guicciardi and Gores, 2002). GPBA activation of Huh-BAT cells (BA cotransporter transfected into human hepatocellular carcinoma cell line) and KMBC cells (human cholangiocarcinoma cell line) results in activation of JNK and recruitment of caspase 8 to the death-induced signalling complex (DISC), which results in apoptosis (Yang et al., 2007). Thus, GPBA agonists could have detrimental as well as protective effects.
GPBA and biliary functions
GPBA is highly expressed in the gall bladder, where GPBA regulates epithelial cells and smooth muscle functions (Figure 3). Immunoreactive GPBA is localized to the apical membrane and cilia of cholangiocytes and biliary epithelial cells (Keitel and Haussinger, 2011) as well as to gall bladder smooth muscle (Lavoie et al., 2010). In the epithelium, GPBA agonists promotes cAMP formation, which results in activation of the cAMP-dependent chloride channel cystic fibrosis transmembrane conductance regulator and resultant chloride secretion. BAs also stimulate the proliferation of cholangiocytes by a GPBA-dependent process. Thus, GPBA mediates the secretory and proliferative actions of BAs in the gall bladder, where GPBA may protect against the toxic effects of BAs.
GPBA activation of gall bladder smooth muscle promotes relaxation and gall bladder filling with bile (Li et al., 2011). Hydrophobic bile salts activate GPBA on gall bladder smooth muscle, leading to stimulation of cAMP/PKA pathway, opening of KATP channels, and membrane hyperpolarization that decreases contractility (Lavoie et al., 2010). The treatment of mice with BAs and a GPBA-selective agonist increases gall bladder volume, an effect that is absent from GPBA-deficient mice (Li et al., 2011). These agents also promote relaxation of gall bladder muscle, by a GPBA-mediated process.
GPBA is implicated in bile–lipid homeostasis in the gall bladder, with implications for gallstone formation. Gallstones are made up from cholesterol and other components of bile. GPBA deletion protects mice from formation of cholesterol gallstones when fed a CA-containing high-fat diet (Vassileva et al., 2006). Two genes that play a major role in cholesterol metabolism, cholesterol 7 α-hydroxylase (Cyp7a1) and sterol-27-hydroxylase (Cyp27a1), were markedly up-regulated in GPBA knockout mice, providing a possible mechanism for resistance to gallstone formation.
GPBA and intestinal functions
The small and large intestine are episodically exposed to high concentrations of BAs after feeding, and BAs have well known effects on intestinal motility and fluid and electrolyte secretion (reviewed in Bajor et al., 2010). Moreover, abnormal BA delivery to the intestine, which can occur due to disease or therapy, is associated with defects in digestive functions. For example, a reduced colonic delivery of BAs, which can occur during cholestatic disease or after administration of BA sequestrants to treat lipid disorders, is frequently associated with constipation (Knodel and Talbert, 1987; Ghaffari et al., 2004). Conversely, an increased colonic delivery of BAs, due to decreased ileal absorption that is a consequence of inflammatory bowel disease or surgical resection or due to continuous secretion in cholecystectomy patients, is often associated with diarrhoea (Miettinen, 1971; Arlow et al., 1987). Recent observations have implicated GPBA as a mediator of these well-known pathophysiological actions of BAs on colonic motility and secretion (Figure 3).
GPBA is localized to enteric neurons that regulate gastrointestinal motility and secretion (Poole et al., 2010). In a detailed survey of the localization and expression of GPBA in the mouse, GPBA immunoreactivity was detected in defined populations of myenteric and submucosal neurons, and GPBA mRNA was detected in nerve plexuses. Examination of the chemical coding of these neurons revealed that GPBA colocalized with NOS in the inhibitory motor neurons of the myenteric plexus of the large intestine. DCA, a GPBA agonist, inhibited spontaneous, phasic contractions of isolated segments of colonic longitudinal muscle by a neurogenic and nitrergic mechanism that is consistent with the GPBA localization in NOS-positive neurons. Notably, this inhibitory effect of DCA was not observed in tissues from GPBA-deficient mice, which indicates that DCA inhibits spontaneous contractions of mouse colonic longitudinal muscle by activating GPBA (Alemi et al., 2013b). Together, these findings suggest that GPBA activation of myenteric inhibitory motor neurons suppresses spontaneous contractility of the colon by a neurogenic and nitrergic pathway. Luminal administration of DCA also delays gastric emptying and small intestinal transit in mice (Poole et al., 2010), and BAs have variable effects on motility in different regions of the gut (Bajor et al., 2010), which may also depend on GPBA.
Studies of colonic peristalsis, transit and defecation in mice with a loss (knockout) or gain (transgenic) of GPBA function have revealed a pivotal role for GPBA in colonic motility and normal defecation (Alemi et al., 2013b). In addition to expression in inhibitory motor neurons, GPBA immunoreactivity in the mouse colon colocalizes with two transmitters of the afferent limb of the peristaltic reflex: 5-hydroxytryptamine (5-HT) in enterochromaffin cells and calcitonin gene-related peptide (CGRP) in intrinsic primary afferent neurons. The mucosal application of physiological concentrations of DCA and LCA (<100 μM) to a flat sheet preparation of mouse colon evokes an ascending contraction and descending relaxation of circular muscle, consistent with stimulation of peristalsis, and concomitantly stimulates release of the peristaltic transmitters 5-HT and CGRP. GPBA deletion abolishes these effects of BAs on peristalsis and transmitter release, and antagonism of 5-HT4 receptors and of CGRP receptors blocks BA-evoked peristalsis, consistent with the localization of GPBA to enterochromaffin cells and intrinsic primary afferent neurons. Further support for an involvement of GPBA in peristalsis is the finding that the GPBA-selective agonist OA also provokes GPBA-dependent peristalsis and release of 5-HT and CGRP. Considered together, these results suggest that luminal BAs trigger the afferent limb of the peristaltic reflex by activating GPBA on enterochromaffin cells and intrinsic primary afferent neurons to stimulate release of 5-HT and CGRP. The important contribution of GPBA to peristalsis has implications for transit and defecation (Alemi et al., 2013b). GPBA deletion delays colonic transit whereas GPBA overexpression has the opposite effect. Moreover, GPBA-deficient mice are constipated, excreting fewer pellets of lower water content. Thus, GPBA mediates the prokinetic actions of BAs and is required for normal defecation. Whether GPBA on submucosal neurons mediates the well-defined prosecretory actions of BAs in the colon (Bajor et al., 2010), which are also essential for normal defecation, remains to be determined.
In line with the immunosuppressive properties of GPBA in the immune system, GPBA serves to protect against colonic inflammation in mice by maintaining integrity of the colonic epithelial barrier (Cipriani et al., 2011). GPBA-deficient mice exhibit altered architecture of the colonic mucosa at 12 months of age that is characterized by disruption of epithelial tight junctions and a redistribution of zonulin-1 from tight junctions, which results in increased mucosal permeability, a recognized feature of intestinal inflammatory disorders. Following challenge with disodium sulphate, a barrier-breaking agent, GPBA-deficient mice exhibit increased susceptibility to development of colitis, as exemplified by worsened macroscopic and histopathological damage scores compared with wild-type littermates. There is an influx of GPBA-expressing mononuclear cells into the inflamed colon of mice with 2,4,6-trinitrobenzene sulphonic acid-induced colitis and of patients with Crohn's disease. Ciprofloxacin, an antibiotic that appears to activate GPBA, and OA, a naturally occurring GPBA-selective agonist, protect mice against trinitrobenzene sulphonic acid-induced colitis. Whether BAs and GPBA agonists protect against inflammatory bowel disease in patients remains to be determined.
Given the role of GPBA in intestinal motility and inflammation, genetic variations in this receptor could lead to digestive abnormalities. Genetic variations in GPBA, particularly single nucleotide polymorphism (SNP rs11554825), have been proposed to contribute to altered small-bowel transit and colonic transit (Camilleri et al., 2011). An analysis of the association between GPBA SNP rs11554825 and symptom phenotypes suggests that genetic variations of GPBA could alter small-bowel and colonic transit in patients with lower functional gastrointestinal disorders, in particular, the diarrhoea-predominant variant of irritable bowel syndrome.
The involvement of GPBA in bile homeostasis and inflammation suggests a potential role in primary sclerosing cholangitis, a chronic inflammatory disease of the bile duct that is associated with ulcerative colitis (Hov et al., 2010; 2011). Moreover, the GPBA gene is localized at chromosome 2q35, in close proximity to a genetic variant associated with primary sclerosing cholangitis and ulcerative colitis. An analysis of the sequence of GPBA in primary sclerosing cholangitis patients and healthy control subjects identified six non-synonymous mutations, five of which resulted in a loss of GPBA function when expressed in epithelial cell lines. These studies reveal possible associations between GPBA mutations and biliary and intestinal inflammatory diseases that warrant further investigation.
GPBA and sensory transduction
Recent observations have implicated GPBA in the defects in sensation that can accompany cholestatic diseases (Figure 3). Cholestatic liver diseases are characterized by diminished delivery of bile into the intestine, which leads to a 10–20-fold increase in the circulating and tissue concentrations of BAs. Cholestatic patients can experience a pruritus that is so severe and intractable pruritus that it is an indication for liver transplantation (Bergasa, 2011). Moreover, cholestatic patients can also exhibit abnormal pain perception. In contrast to acute obstruction of the bile duct with gallstones, which is painful, the gradual malignant obstruction of the biliary tract does not cause pain (‘painless jaundice’), as recognized by Courvoisier a century ago (Fitzgerald et al., 2009). Patients with cholestatic diseases undergoing liver transplantation have lower post-operative pain than patients undergoing liver resection (Moretti et al., 2002), and bile duct ligation in mice also induces a mechanical analgesia (Nelson et al., 2006). Recent observations indicate that GPBA mediates BA-evoked itch and analgesia in mice (Alemi et al., 2013b). GPBA immunoreactivity and mRNA are expressed by a subpopulation of small-diameter neurons of the dorsal root ganglia that participate in itch and pain transmission. These neurons coexpress gastrin-releasing peptide, an itch-selective transmitter, and the transient receptor potential vanilloid 1 and ankyrin 1 ion channels, which participate in pain and itch transmission. GPBA is also present in spinal neurons and dermal macrophages that are known to contain opioids. Patch clamp recordings of dorsal root ganglia neurons indicate that BAs and OA, a GPBA-selective agonist, evoke an increased excitability and stimulate action potential discharge by a mechanism that requires expression of GPBA. GPBA agonists also stimulate the release of gastrin-releasing peptide and leucine-enkephalin, transmitters of itch and analgesia, from segment of spinal cord with attached dorsal root ganglia fibres. Behavioural studies of scratching in mice indicate that i.d. injections of BAs and OA to the neck stimulate a robust site-directed scratching that is attenuated in GPBA knockout mice yet amplified in GPBA transgenic mice, which also exhibit spontaneous pruritus. Administration of antagonists of gastrin-releasing peptide and μ-opioid receptors inhibit DCA-evoked scratching, supporting an involvement of these transmitters. Of note, the intrathecal administration of DCA does not induce scratching behaviour, suggesting that activation of GPBA mediates itch in the periphery, but not in the spinal cord. In contrast, the intraplantar or intrathecal injection of BAs and GPBA agonists induces analgesia to mechanical stimulation of the planter surface of the paw with von Frey filaments. GPBA deletion and antagonism of μ-opioid receptors inhibit this analgesia, which indicates that BAs cause analgesia by peripheral and central mechanisms that require activation of GPBA and release of opioids that activate μ-opioid receptors. Considered together, these results reveal a new role for GPBA in mediating the pruritogenic and analgesic actions of BAs in mice. Further studies are required to assess the contributions of BAs and GPBA to cholestatic itch and painless jaundice in patients with cholestatic liver diseases.
Neurosteroids as GPBA agonists in the nervous system
Most studies have focused on defining the role of GPBA in cells and tissues that are normally exposed to high concentrations of BAs, such as the liver and gastrointestinal tract. However, GPBA is also expressed by primary spinal (Keitel et al., 2010a) afferent and spinal neurons (Alemi et al., 2013a). Although sensory nerve endings in peripheral tissues are likely to be exposed to fluctuating levels of BAs during feeding and fasting, it remains to be determined whether BAs are the physiological agonists of GPBA in the CNS, or whether there are other physiological agonists in these locations. One possibility is that neurosteroids, which are structurally related to BAs, are the physiological agonists of GPBA in the nervous system.
Neurosteroids are steroids transmitters that are synthesized in the brain, where they participate in the regulation of neuronal development and myelination, and have been implicated in epilepsy, anxiety, stress, depression, learning and pain transmission (reviewed in Reddy, 2010). Neurosteroids rapidly regulate neuronal excitability by non-genomic mechanisms that are independent of activation of nuclear steroid receptors and are dependent instead on modulation of neuronal membrane receptors and channels. Abundant evidence indicates that neurosteroids are positive allosteric modulators of GABAA receptors, although neurosteroids can also modulate NMDA-type glutamate receptors. Certain neurosteroids can also activate GPBA (Sato et al., 2008; Keitel et al., 2010a). For example, 5β-pregnan-3α-ol-20-one, 5β-pregnan-3α-17α-21-triol-20-one and 5α-pregnan-3α-ol-20-one (allopregnanolone) can activate GPBA-expressing HEK cells to stimulate the formation of cAMP, and 5β-pregnan-3α-ol-20-one stimulates cAMP formation and increases intracellular calcium levels in astrocytes and cortical neurons, possibly by a GPBA-dependent mechanism. GPBA agonists, including 5β-pregnan-3α-ol-20-one, also stimulate the generation of reactive oxygen species by cultured astrocytes. Further studies are required to determine whether GPBA contributes to the well-defined pathophysiological actions of neurosteroids in the CNS.
Conclusions and future perspectives
GPBA is a target for metabolic diseases and disorders of digestion and sensation. GPBA is expressed in various organs including the nervous system and has pleiotropic effects involving anti-inflammatory, digestive, and sensory sensation pathways. Future studies in GPBA receptor biology may yield identification of additional ligands (agonists or antagonists) giving us a better insight into its pharmacology. Therefore, elucidation of the biological and physiological functions of GPBA holds great potential promise for the intervention of many pathophysiological diseases.
Acknowledgments
Supported by NHMRC 63303, 103188, and Monash University (N.W.B.).
Glossary
- ADAM
A disintegrin and metalloproteinase domain-containing protein
- ASBT
apical sodium-dependent BA transporter
- BA
bile acid
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CGRP
calcitonin gene-related peptide
- D2
type 2 iodothyronine deodinase
- DCA
deoxycholic acid
- GLP-1
glucagon-like peptide 1
- GPBA
bile acid receptor
- KATP
ATP-dependent potassium channels
- LCA
lithocholic acid
- OA
oleanolic acid
- TDCA
taurodeoxycholic acid
- TLCA
taurolithocholic acid
- UDCA
ursodeoxycholic acid
Conflict of interest
The authors declare that no conflict of interest exists.
References
- Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest. 2013a;123:1513–1530. doi: 10.1172/JCI64551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemi F, Poole DP, Chiu J, Schoonjans K, Cattaruzza F, Grider JR, et al. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology. 2013b;144:145–154. doi: 10.1053/j.gastro.2012.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlow FL, Dekovich AA, Priest RJ, Beher WT. Bile acid-mediated postcholecystectomy diarrhea. Arch Intern Med. 1987;147:1327–1329. [PubMed] [Google Scholar]
- Bajor A, Gillberg PG, Abrahamsson H. Bile acids: short and long term effects in the intestine. Scand J Gastroenterol. 2010;45:645–664. doi: 10.3109/00365521003702734. [DOI] [PubMed] [Google Scholar]
- Bergasa NV. The itch of liver disease. Semin Cutan Med Surg. 2011;30:93–98. doi: 10.1016/j.sder.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Camilleri M, Vazquez-Roque MI, Carlson P, Burton D, Wong BS, Zinsmeister AR. Association of bile acid receptor TGR5 variation and transit in health and lower functional gastrointestinal disorders. Neurogastroenterol Motil. 2011;23:995–999. doi: 10.1111/j.1365-2982.2011.01772.x. e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Tian W, Hong J, Li D, Tavares R, Noble L, et al. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol. 2013;304:G322–G327. doi: 10.1152/ajpgi.00263.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JE, White MJ, Lobo DN. Courvoisier's gallbladder: law or sign? World J Surg. 2009;33:886–891. doi: 10.1007/s00268-008-9908-y. [DOI] [PubMed] [Google Scholar]
- Ghaffari K, Savadkuhi ST, Honar H, Riazi K, Shafaroodi H, Moezi L, et al. Obstructive cholestasis alters intestinal transit in mice: role of opioid system. Life Sci. 2004;76:397–406. doi: 10.1016/j.lfs.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Gioiello A, Rosatelli E, Nuti R, Macchiarulo A, Pellicciari R. Patented TGR5 modulators: a review (2006–present) Expert Opin Ther Pat. 2012;22:1399–1414. doi: 10.1517/13543776.2012.733000. [DOI] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ. Bile acid-mediated hepatocyte apoptosis and cholestatic liver disease. Dig Liver Dis. 2002;34:387–392. doi: 10.1016/s1590-8658(02)80033-0. [DOI] [PubMed] [Google Scholar]
- Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep. 2012;2:430. doi: 10.1038/srep00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Behar J, Wands J, Resnick M, Wang LJ, DeLellis RA, et al. Role of a novel bile acid receptor TGR5 in the development of oesophageal adenocarcinoma. Gut. 2010;59:170–180. doi: 10.1136/gut.2009.188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hov JR, Keitel V, Laerdahl JK, Spomer L, Ellinghaus E, ElSharawy A, et al. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS ONE. 2010;5:e12403. doi: 10.1371/journal.pone.0012403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hov JR, Keitel V, Schrumpf E, Haussinger D, Karlsen TH. TGR5 sequence variation in primary sclerosing cholangitis. Dig Dis. 2011;29:78–84. doi: 10.1159/000324138. [DOI] [PubMed] [Google Scholar]
- Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, et al. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem. 2013;288:22942–22960. doi: 10.1074/jbc.M113.455774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- Keitel V, Haussinger D. TGR5 in the biliary tree. Dig Dis. 2011;29:45–47. doi: 10.1159/000324127. [DOI] [PubMed] [Google Scholar]
- Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50:861–870. doi: 10.1002/hep.23032. [DOI] [PubMed] [Google Scholar]
- Keitel V, Gorg B, Bidmon HJ, Zemtsova I, Spomer L, Zilles K, et al. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia. 2010a;58:1794–1805. doi: 10.1002/glia.21049. [DOI] [PubMed] [Google Scholar]
- Keitel V, Gorg B, Haussinger D. The bile acid receptor TGR5 is expressed in brain and is responsive to neurosteroids. Hepatology. 2010b;52:599A–599A. [Google Scholar]
- Knodel LC, Talbert RL. Adverse effects of hypolipidaemic drugs. Med Toxicol. 1987;2:10–32. doi: 10.1007/BF03259858. [DOI] [PubMed] [Google Scholar]
- Kohli R, Kirby M, Setchell KD, Jha P, Klustaitis K, Woollett LA, et al. Intestinal adaptation after ileal interposition surgery increases bile acid recycling and protects against obesity-related comorbidities. Am J Physiol. 2010;299:G652–G660. doi: 10.1152/ajpgi.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R, Bradley D, Setchell KD, Eagon JC, Abumrad N, Klein S. Weight loss induced by Roux-en-Y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J Clin Endocrinol Metab. 2013;98:E708–E712. doi: 10.1210/jc.2012-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie B, Balemba OB, Godfrey C, Watson CA, Vassileva G, Corvera CU, et al. Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of K(ATP) channels. J Physiol. 2010;588:3295–3305. doi: 10.1113/jphysiol.2010.192146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, et al. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25:1066–1071. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GE, Brubaker PL. Glucagon-like peptide 1 secretion by the L-cell – the view from within. Diabetes. 2006;55:S70–S77. [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- Miettinen TA. The role of bile salts in diarrhoea of patients with ulcerative colitis. Gut. 1971;12:632–635. doi: 10.1136/gut.12.8.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti EW, Robertson KM, Tuttle-Newhall JE, Clavien PA, Gan TJ. Orthotopic liver transplant patients require less postoperative morphine than do patients undergoing hepatic resection. J Clin Anesth. 2002;14:416–420. doi: 10.1016/s0952-8180(02)00390-2. [DOI] [PubMed] [Google Scholar]
- Nakatani H, Kasama K, Oshiro T, Watanabe M, Hirose H, Itoh H. Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metabolism. 2009;58:1400–1407. doi: 10.1016/j.metabol.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Nelson L, Vergnolle N, D'Mello C, Chapman K, Le T, Swain MG. Endogenous opioid-mediated antinociception in cholestatic mice is peripherally, not centrally, mediated. J Hepatol. 2006;44:1141–1149. doi: 10.1016/j.jhep.2005.11.043. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity. 2009;17:1671–1677. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010;22:814–825. doi: 10.1111/j.1365-2982.2010.01487.x. e227–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, et al. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol. 2013;304:G371–G380. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pournaras DJ, Aasheim ET, Sovik TT, Andrews R, Mahon D, Welbourn R, et al. Effect of the definition of type II diabetes remission in the evaluation of bariatric surgery for metabolic disorders. Br J Surg. 2012;99:100–103. doi: 10.1002/bjs.7704. [DOI] [PubMed] [Google Scholar]
- Raufman JP, Zimniak P, Bartoszko-Malik A. Lithocholyltaurine interacts with cholinergic receptors on dispersed chief cells from guinea pig stomach. Am J Physiol. 1998;274(6 Pt 1):G997–1004. doi: 10.1152/ajpgi.1998.274.6.G997. [DOI] [PubMed] [Google Scholar]
- Raufman JP, Chen Y, Zimniak P, Cheng K. Deoxycholic acid conjugates are muscarinic cholinergic receptor antagonists. Pharmacology. 2002;65:215–221. doi: 10.1159/000064347. [DOI] [PubMed] [Google Scholar]
- Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113–137. doi: 10.1016/B978-0-444-53630-3.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Roux CW, Aylwin SJ, Batterham RL, Borg CM, Coyle F, Prasad V, et al. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg. 2006;243:108–114. doi: 10.1097/01.sla.0000183349.16877.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A, et al. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun. 2007;362:793–798. doi: 10.1016/j.bbrc.2007.06.130. [DOI] [PubMed] [Google Scholar]
- Sato H, Macchiarulo A, Thomas C, Gioiello A, Une M, Hofmann AF, et al. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. 2008;51:1831–1841. doi: 10.1021/jm7015864. [DOI] [PubMed] [Google Scholar]
- Shaham O, Wei R, Wang TJ, Ricciardi C, Lewis GD, Vasan RS, et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol Syst Biol. 2008;4:214. doi: 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol. 2010;298:G419–G424. doi: 10.1152/ajpgi.00362.2009. [DOI] [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337:776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JI, Yoon JH, Myung SJ, Gwak GY, Kim W, Chung GE, et al. Bile acid-induced TGR5-dependent c-Jun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007;361:156–161. doi: 10.1016/j.bbrc.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007;354:154–159. doi: 10.1016/j.bbrc.2006.12.168. [DOI] [PubMed] [Google Scholar]
- Yokomori H, Oda M, Ogi M, Sakai K, Ishii H. Enhanced expression of endothelial nitric oxide synthase and caveolin-1 in human cirrhosis. Liver. 2002;22:150–158. doi: 10.1034/j.1600-0676.2002.01588.x. [DOI] [PubMed] [Google Scholar]