

Abstract
Although it has been known since the 1960s that trypsin and chymotrypsin can mimic hormone action in tissues, it took until the 1990s to discover that serine proteinases can regulate cells by cleaving and activating a unique four-member family of GPCRs known as proteinase-activated receptors (PARs). PAR activation involves the proteolytic exposure of its N-terminal receptor sequence that folds back to function as a ‘tethered’ receptor-activating ligand (TL). A key N-terminal arginine in each of PARs 1 to 4 has been singled out as a target for cleavage by thrombin (PARs 1, 3 and 4), trypsin (PARs 2 and 4) or other proteases to unmask the TL that activates signalling via Gq, Gi or G12/13. Similarly, synthetic receptor-activating peptides, corresponding to the exposed ‘TL sequences’ (e.g. SFLLRN—, for PAR1 or SLIGRL— for PAR2) can, like proteinase activation, also drive signalling via Gq, Gi and G12/13, without requiring receptor cleavage. Recent data show, however, that distinct proteinase-revealed ‘non-canonical’ PAR tethered-ligand sequences and PAR-activating agonist and antagonist peptide analogues can induce ‘biased’ PAR signalling, for example, via G12/13-MAPKinase instead of Gq-calcium. This overview summarizes implications of this ‘biased’ signalling by PAR agonists and antagonists for the recognized roles the PARs play in inflammatory settings.
Linked ArticlesThis article is part of a themed section on Molecular Pharmacology of GPCRs. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-5
Keywords: activated protein-C, biased signalling, GPCR, protease, proteinase-activated receptor, thrombin
Introduction
Proteinase-mediated signalling
One index of the importance of proteinases for regulating biological processes is that about 2% of the human genome has been found to code for either proteinases (commonly called proteases) or their inhibitors (Puente et al., 2005). Thus, in addition to their ‘classical roles’ as digestive protein-degrading enzymes, it comes as no surprise that proteinases are now recognized for their many roles in regulating tissue function by both receptor and non-receptor mechanisms (Figure 1A). For instance, the discovery of hypotensive urinary peptides that also have contractile activity in uterine smooth muscle (Werle and Erdos, 1954) heralded the work of Chrétien, Steiner and their colleagues showing that key hormones like adrenocorticotropic hormone and insulin were generated by the proteolytic processing of larger hormone precursors (Steiner et al., 1967; Steiner, 2011; Chrétien, 2012). The turnover of peptide agonists by proteolysis to reduce cell signalling, for example, by neutral endopeptidase cleavage of neurokinins, is of equal importance. Thus, proteinases can regulate cellular signalling events by cleaving a variety of targets, including pro-hormones, kininogens, chemokine and cytokine precursors and even growth factor receptors, like that for insulin. For instance, the ‘insulin-like’ actions of trypsin and other proteinases (Rieser and Rieser, 1964; Rieser, 1967; Kono and Barham, 1971) results from the cleavage of the extracellular α-subunit of the insulin receptor, thus abolishing inhibitory control of receptor function (Shoelson et al., 1988). At higher concentrations, trypsin was found to ‘disarm’ the insulin receptor thereby preventing its ability to bind insulin and signal (Cuatrecasas, 1969; Cuatrecasas, 1971). By the 1970s, it was also clear that, like insulin and epidermal growth factor, thrombin and trypsin could stimulate cell culture mitogenesis by a receptor-like mechanism (Burger, 1970; Sefton and Rubin, 1970; Chen and Buchanan, 1975; Carney and Cunningham, 1977; 1978). However, the mechanistic details of these actions of proteinases were not known at the time. As outlined in the following section, it was the search for the ‘thrombin’ receptor, responsible for stimulating human platelet aggregation and mitogenesis in cultured hamster cells that ultimately led to the discovery of the G-protein-coupled, proteinase-activated receptor (PAR; receptor nomenclature follows Alexander et al., 2013) responsible for these actions of thrombin (Rasmussen et al., 1991; Vu et al., 1991a). There are multiple mechanisms whereby proteinases can signal to cells, as outlined in Figure 1A, in addition to regulating PAR activity, which will be the main focus of this overview. The PAR- and non-PAR mechanisms of proteinase signalling (Figure 1A, B) can be seen as complementary ways in which tissue-derived proteinases can play a role in physiologically important processes involving inflammation.
Figure 1.
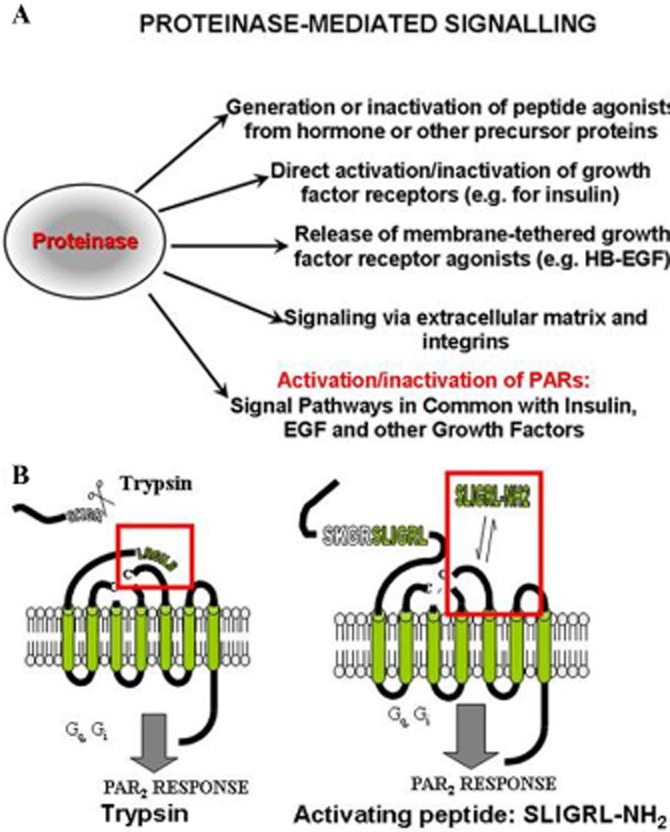
Proteinase-mediated signalling (A) and the mechanisms of PAR activation (B). (A, upper) The diagram shows five distinct ways by which proteinases can cause cell signals, ranging from the generation or degradation of active peptides (top) to the activation of PARs (bottom). (B, lower) The scheme shows the proteolytic activation of PARs either by the unmasking of a receptor-tethered activating ligand (left: SLIGRL— for PAR2) or by a synthetic peptide with a sequence in common with the revealed ‘TL’ (right: SLIGRL-NH2).
Signalling by thrombin and discovery of the four PAR GPCR family members
As mentioned above, two laboratories independently cloned a GPCR responsible for the ability of thrombin to regulate human platelet function and to stimulate cell replication in cultured hamster cells. A key feature of the cloned receptor observed in both laboratories was its ability, presumably by stimulating Gq, to mobilize intracellular calcium when activated by thrombin in an oocyte expression system (Rasmussen et al., 1991; Vu et al., 1991b1991a). It was also known at the time that thrombin signalling could inhibit adenylyl cyclase, presumably by triggering Gi (Vouret-Craviari et al., 1992). Thus, at the outset, it was realized that thrombin, via its cloned receptor (now termed PAR1, with its gene designated, F2R) could couple to multiple G-proteins in order to generate cell signals. The unusual mechanism that activates this unique class of GPCRs (Vu et al., 1991a,1991b,) involves the proteolytic unmasking of a cryptic N-terminal receptor sequence that, while remaining attached, folds back to act as a ‘tethered ligand’ (TL) and trigger signalling (Figure 1B, left panel). Surprisingly, synthetic peptides with sequences corresponding to the newly exposed TL sequence can stimulate receptor signalling without the need for proteolytic activation, albeit at 100–1000-fold higher concentrations than those of the proteinase agonists (Figure 1B, right-hand panel). Thus, on its own, the sequence, SFLLRNPNDKYEPF, was found to mimic the action of thrombin on human platelets (Vu et al., 1991a). However it rapidly became apparent from structure-activity studies of the actions of this peptide and its analogues that there was a distinct ‘thrombin receptor’ on rodent platelets that could not be activated by such peptides (Kinlough-Rathbone et al., 1993) and that a receptor different from the ‘thrombin receptor’ cloned by Vu and colleagues in 1991 was present in vascular and gastric tissue preparations (Hollenberg et al., 1993; Al-Ani et al., 1995). The fortuitous cloning of murine PAR2, found when searching a genomic library for a substance K receptor (Nystedt et al., 1994) and the subsequent cloning of human PAR2/F2RL1 (Bohm et al., 1996) paved the way for the additional cloning of the other two members of the PAR family, namely PAR3/F2RL2 (Connolly et al., 1996; Ishihara et al., 1997) and PAR4/F2RL3 (Kahn et al., 1998; Xu et al., 1998). All of the PARs have in their N-terminal sequences a principal serine proteinase-targeted arginine at which enzymic cleavage exposes a distinct ‘TL’ for each receptor, as summarized in Table 1. Unmasking of such TLs in PARs 1 and 4 by thrombin and in PAR2 by trypsin stimulates cell signalling that involves a number of G-proteins (Gq, Gi, G12/13). Further, the synthetic peptides based on the revealed TL sequences can also stimulate comparable signalling via the ‘partner’ G-proteins. As outlined in Table 1, it has been possible to synthesize PAR-selective activating peptides to evaluate the effects of signalling by PARs 1, 2 and 4 in a variety of cultured cell and in vivo contexts. PAR3 appears to function as a ‘co-factor’ for activation of PAR1 (Nakanishi-Matsui et al., 2000) and peptides derived from its TL sequence are able to activate both PARs 1 and 2 (Hansen et al., 2004). In certain circumstances, PAR3 is reported to signal on its own (Ostrowska and Reiser, 2008), but its general role in regulating tissue function remains to be fully elucidated.
Table 1.
The human PAR family, their canonical and non-canonical proteolytically unmasked tethered ligands (TLs) and their non-biased and biased peptide agonistsa
| Canonical and non-canonical PAR cleavage sites and TL sequences | ||||
|---|---|---|---|---|
| Receptor/gene designation | Canonical TL (human) | Receptor-selective PAR-activating peptides | Non-canonical TL | Biased PAR-activating peptides |
| PAR1/F2R | --//SFLLRN-- | TFLLR-NH2 | MMP1: --//PRSFLLRN-- APC: --//NPNDKYEPF-- NE: --// RNPNDKYEPF-- PR3: --//TLDPRSF-- |
PRSFLLRN; NPNDKYEPF; RNPNDKYEPF; TLDPRSF; YFLLRN |
| PAR2/F2RL1 | --//SLIGKV-- | SLIGRL-NH2; 2-furoyl-LIGRL-NH2 | NE: --//VLTGKL-- | SLAAAA-NH2 |
| PAR3/F2RL2 | --//TFRGAP-- | TL-derived peptides activate PARs 1 and 2 | Not known | Not known |
| PAR4/F2RL3 | --//GYPGQV-- | AYPGQV-NH2; AYPGKV-NH2 | Not known | Not known |
The ‘canonical’ PAR-TL resulting from cleavage activation of the PARs (cleavage site shown as: //) correspond to the sequences of the receptor-selective PAR-activating peptides. The sequences of synthetic receptor-selective ‘canonical’ PAR-activating peptides based on the thrombin (PARs 1 and 4) or trypsin (PAR2)-unmasked TL are shown along with ‘non-canonical’ TL unmasked (//) by neutrophil elastase (NE: PAR 2 and PAR1), neutrophil proteinase-3 (PR3: PAR1), MMP-1 (MMP1: PAR1) or activated protein-C (APC: PAR1). Further, biased synthetic PAR-activating peptides derived from either the ‘canonical’ or the ‘non-canonical’ enzyme-unmasked TL are shown in the last column on the right.
To sum up, the ability of serine proteinases to regulate tissue function can now be seen to include activation of PARs in addition to the other mechanisms illustrated in Figure 1A. The unusual features of the PARs are now well appreciated (Figure 1B) in terms of (i) their unique mechanism of activation, involving the exposure of a TL (left panel, Figure 1B), (ii) their ability to be activated selectively by receptor-specific synthetic activating peptides, based on the sequences of the revealed TL (right panel, Figure 1B; Table 1) and (3) their ability to be activated or inactivated/disarmed selectively by a variety of serine proteinases. For instance, thrombin activates PARs 1 and 4, but not PAR2; trypsin at low concentrations can activate PAR2 and PAR4, but disarms PAR1 (Kawabata et al., 1999); and Pseudomonas elastase disarms trypsin-mediated activation of PAR2 (Dulon et al., 2005). It is important to recognize that a proteinase that cleaves at residues C-terminal to the TL domain sequence, as shown in the left panel of Figure 1A (…SKGRSLIGRL…), will ‘disarm’ the PAR to prevent its activation by an enzyme such as trypsin, that acts via the ‘TL’. Thus, enzymically, the PARs can be seen to have their circulating proteinase ‘agonists’ that unmask the TL as well as circulating proteinases that act indirectly as ‘antagonists’ by cleaving downstream of the TL and removing it from the receptor.
The unanswered questions still are, however: (i) which are the endogenous proteinases that can regulate PAR function in vivo? and (ii) do the actions of the synthetic TL PAR-activating peptides stimulate the same signals as the proteinase-revealed TLs? As will be seen in the following sections, research aimed at answering these questions has revealed the ability of the PARs to signal in a ‘biased’ way.
Biased PAR signalling and the mobile or floating receptor model
Receptor plasticity and biased PAR signalling
Early on, from structure-activity studies with the PAR-activating peptides (PAR-APs), it was appreciated that signalling by the enzyme (e.g. thrombin for PAR1) was not completely reproduced by the receptor-activating peptides. For instance, the mitogenic-MAPKinase stimulating action of thrombin in hamster fibroblasts was not matched by the action of the receptor peptide agonist (Vouret-Craviari et al., 1992; 1993). Further, studies of PAR1 with mutations in its extracellular domains showed that the docking-activation mechanisms for the PAR1-activating peptides (most mutations affected PAR-activating peptides-induced responses) differed substantially from activation by the thrombin-revealed TL responses which were largely unaffected (Blackhart et al., 2000). Our own work with PAR2 then showed that mutations in the extracellular loop-2 domain (wild-type ‘EE’ to mutated ‘RR’) markedly affected calcium signalling by the PAR2-activating peptide, SLIGRL-amide (Figure 2A, panel A: large shift to the right to lower potency), but had little or no effect on calcium signalling by the trypsin-revealed TL (Figure 2A, panel B: no shift to the right for the mutant ‘RR’ vs. wild-type ‘EE’ receptor, activated by the TL sequence, /SLIGRLDT…; Al-Ani et al., 2002). Subsequently, we found that by inserting alanine mutations into the N-terminal ‘tethered’ ligand sequence of rat PAR2 (SLIGRL---), even the trypsin-revealed sequence, SLAAAA---, was sufficient to trigger calcium signalling when the receptor was activated by trypsin (Figure 2, lower panel, 2B, red arrow/red font); but the revealed sequence, AAIGRL--- failed to do so with any efficiency (Figure 2, lower panel, 2B, bottom, blue arrow/blue font). Of note, the trypsin-revealed TL with a mutated sequence, LSIGRL---failed to cause calcium signalling (Figure 2, lower panel 2B, blue font and arrow, X-axis). Quite surprisingly, the synthetic peptide based on the mutated-unmasked TL (SLAAAA-amide) failed to activate PAR2 calcium signalling (Figure 3A, top panel), but was found to activate PAR2 MAPKinase signalling (table 2 in Al-Ani et al., 2004, and Figure 3A, lower panel: Ramachandran et al., 2009). Thus, we concluded that for both PARs 1 and 2, the change in receptor conformation caused by the exposed TL differs from changes triggered by the synthetic PAR-activating peptides. Further, we had identified a PAR2 biased agonist (SLAAAA-amide) that could trigger MAPKinase activation but not calcium signalling. Previous work with a PAR1 agonist, YFLLRNP, pointed to its ability to be a ‘biased’ partial agonist for PAR1, able to trigger human platelet shape change but not aggregation (Rasmussen et al., 1993). Moreover, it was evident that like other GPCRs, the PARs were capable of ‘functional selectivity’ or ‘biased’ signalling, as shown schematically in Figure 3B (Kenakin and Miller, 2010; Kenakin, 2011). What was not determined was whether endogenous proteinase activation of the PARs might also activate the receptors in a ‘biased way’ like the synthetic peptide agonists. Further, the mechanism that could account for biased PAR signalling had not been determined.
Figure 2.
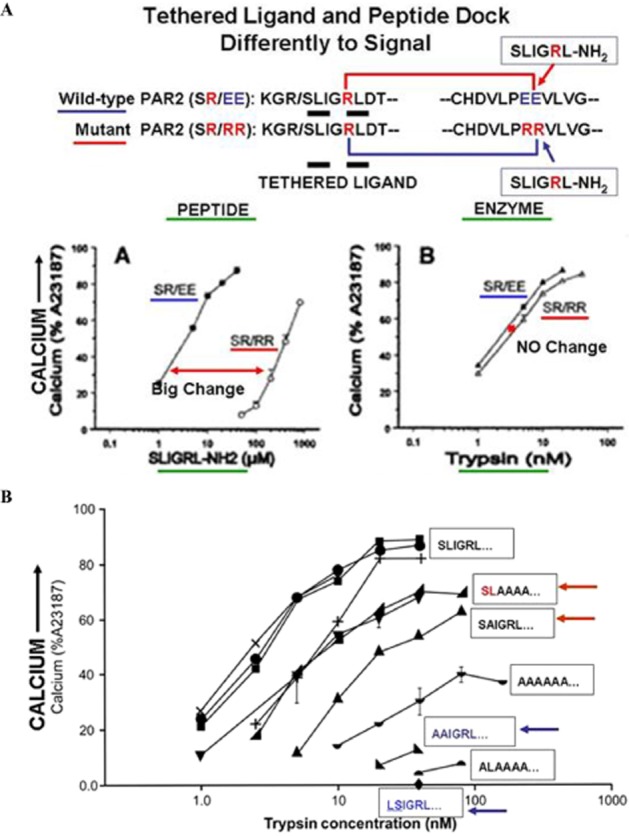
Differences in signalling by PAR2-activating peptide versus TL in a mutant PAR2 ‘SR/RR’ receptor (A, upper, panels A and B) and minimal TL sequence needed for calcium signalling (B, lower). (A, upper) The scheme at the top shows the wild-type receptor sequence (PAR2: SR/EE), with a TL sequence, ‘SLIGRL---) thought to interact with the ‘PEE’ sequence in extracellular loop-2. This wild-type receptor is compared with a mutant PAR2 having the same ‘TL’ (SLIGR---) but a mutated ‘PRR’ sequence in extracellular loop 2. The upper-left figure A shows calcium signalling stimulated by the PAR2-activating peptide, SLIGRL-NH2 in the wild-type (SR/EE), compared with the mutant (SR/RR) receptor, demonstrating a marked rightward shift in the concentration-effect curve for the mutant (SR/RR) versus the wild-type receptor (SR/EE). In contrast, the upper-right panel B shows there is no difference between the wild-type (SR/EE) versus mutant (SR/RR) receptor for activation by the trypsin-revealed TL. Thus, the receptor-docking sites for signalling by the synthetic PAR-activating peptide and the proteinase-unmasked TLs differ. Adapted from Al-Ani et al., 2002 with permission. (B, lower) A minimal PAR2-TL sequence (SL----) is required for calcium signalling. The proteinase-revealed ‘TL’ sequence of PAR2 was mutated so that trypsin cleavage unmasked different mutated ‘TL’ sequences with alanine substitutions (sequences shown in boxes) in the first six amino acids. When revealed, the mutated TL sequences, SLAAAA--- and SAIGRL--- were able to stimulate calcium signalling (red arrows, upper curves), whereas the sequences, AAIGRL--- and LSIGRL--- did not (blue arrows, bottom). Adapted from Al-Ani et al., 2004, with permission.
Figure 3.
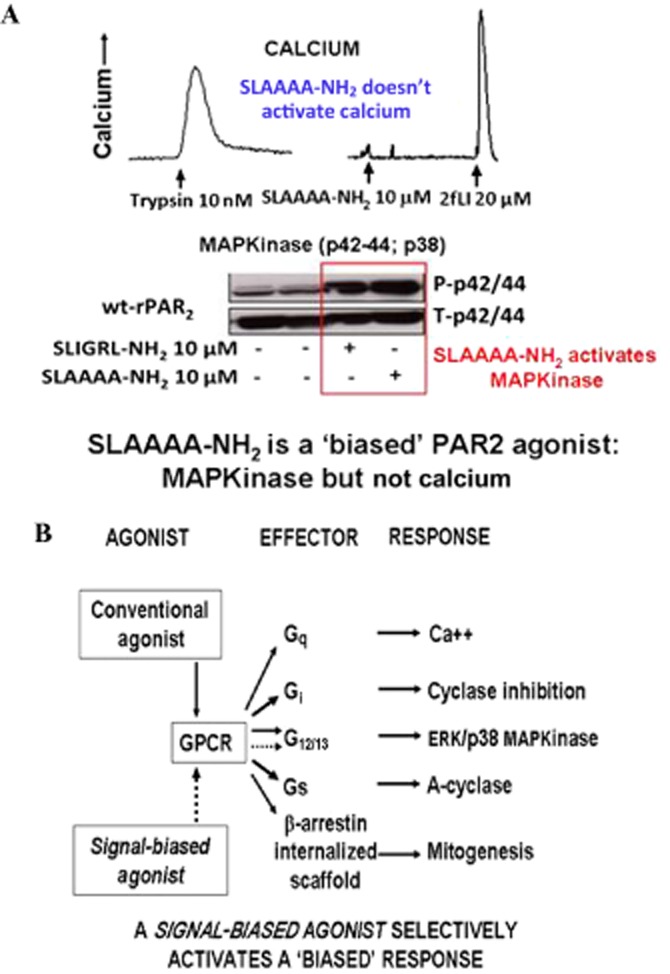
Biased PAR2 signalling by the PAR-activating peptide, SLAAAA-NH2 (A) and a scheme (B) that illustrates the ‘mobile’ or ‘floating’ receptor model of signal transduction. (A) Trypsin (A: left tracing) and the PAR2-activating peptide (2-furoyl-LIGRLO-NH2: right tracing, far-right) both stimulate PAR2 calcium signalling, but the activating peptide with multiple alanine substitutions (SLAAAA-NH2) fails to do so (right tracing, first agonist). However, SLAAAA-NH2, like SLIGRL-NH2, does nonetheless activate MAPKinase signalling, acting as a ‘biased’ PAR2 agonist (lower Western blot signal, red rectangle, panel A). Adapted from Ramachandran et al., 2009, with permission. (B) This ‘biased’ signalling by PAR2 is illustrated by the scheme (lower panel B). Thus, an individual GPCR capable of coupling to multiple G-proteins, as well as generating a β-arrestin internalized signalling scaffold, can in principle be activated selectively by a ‘biased’ agonist to signal via only one of the available G-proteins (dotted lines).
PAR dynamics and biased signalling
As illustrated in Figure 3B, the ability of an individual receptor to activate multiple signalling pathways, as described by the ‘mobile’ or ‘floating’ receptor model put forward some time ago (de Haën, 1976; Jacobs and Cuatrecasas, 1976), depends on its diffusion in the plane of the membrane to interact with multiple ‘effectors’ (e.g. different G-proteins); and in the case of GPCRs like PAR2, to interact with β-arrestin, forming an internalized G-protein-independent signalling scaffold (DeFea, 2008). Using PAR2 as a prototype for the PARs, we employed a C-terminally-yellow fluorescent protein-tagged receptor (i) to follow the dynamics of PAR2 upon activation by either trypsin or a PAR2-activating peptide (Figure 4, upper panel, 4A) and (ii) to monitor its interaction with rLuc-tagged β-arrestin, to generate a bioluminescence resonance energy transfer signal (BRET: Figure 4, lower panel 4B, red arrow, cartoon at bottom). Upon activation of PAR2 by either trypsin or the PAR-activating peptide, SLIGRL-NH2, the receptor internalizes (Figure 4A, right-hand panels ii and iii and histograms on left) in parallel with its interaction with β-arrestin that generates an increase in BRET emission (Figure 4B, panels A and B, red arrows). This activation of the wild-type receptor by either trypsin or SLIGRL-NH2 is accompanied by an increase in intracellular calcium and an activation of MAPKinase. In contrast, a mutated PAR2 TL with the trypsin-revealed sequence, /LSIGRL---, which does not signal via calcium (Figure 2, lower panel 2B, X-axis), can indeed activate MAPKinase (Figure 5, upper panel, 5A); but, activation of the mutant /LSIGRL--- receptor by trypsin did not result either in receptor internalization (Figure 5, lower panels, 5B and histograms) or in an interaction with β-arrestin (not shown: Ramachandran et al., 2009). Thus, the dynamics of PAR2 upon activation of the wild-type and mutant receptors we studied (internalization or not) can be correlated with ‘biased’ receptor signalling (internalization = calcium plus MAPKinase; non-internalization = MAPKinase only). The mechanisms of signalling by the receptor upon remaining at the plasma membrane instead of being internalized remain to be explored. The next step was to determine if endogenous PAR2 activating enzymes could trigger biased signalling, in step with a restricted receptor interaction with β-arrestin and a lack of internalization.
Figure 4.
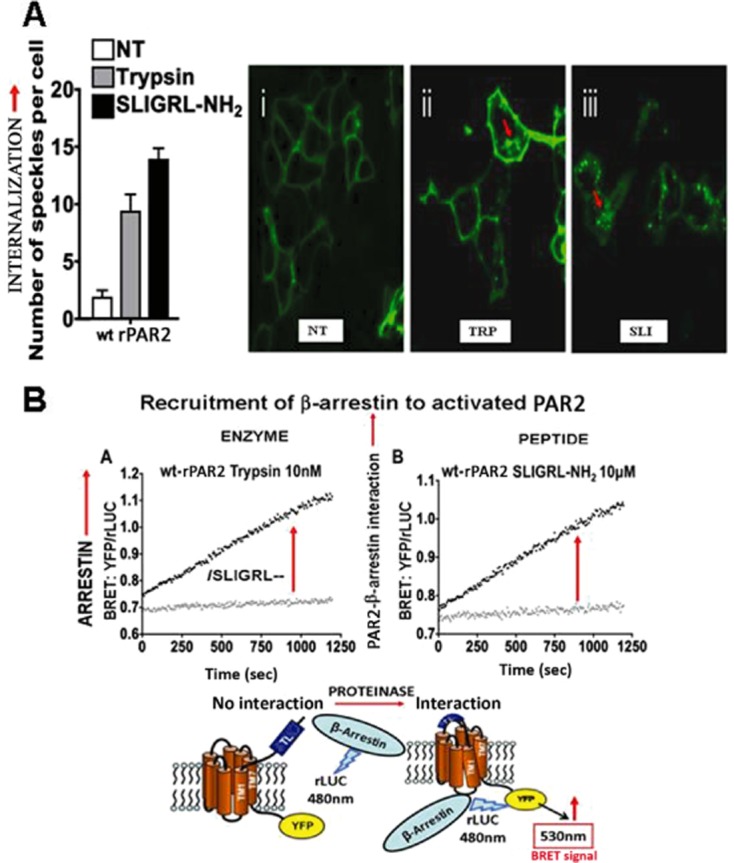
Monitoring PAR2 internalization and arrestin interaction upon activation. (A) PAR2 internalization (red arrows) after activation by either trypsin (TRP: middle micrograph) or PAR2-activating peptide (SLI: right-hand micrograph). Receptor in the untreated cells (NT) is membrane delimited. Internalization is quantified (left histograms) by morphometric analysis of internalized receptor clusters (green dots, red arrows, micrographs on the right, panel A). (B) Monitoring PAR2-β-arrestin interaction by BRET after activation by the TL revealed by trypsin (left) or by a synthetic PAR-activating peptide (right). PAR2-β-arrestin interactions stimulated by either trypsin (B, panel A) or PAR2-activating peptide (B, panel B) are quantified by measuring the yelllow fluorescent protein (YFP) emission at 540 nm relative to the rLUC signal (YFP/rLUC), caused by BRET (panel B), as shown by the diagram at the bottom of B. Adapted from Ramachandran et al., 2009, with permission.
Figure 5.
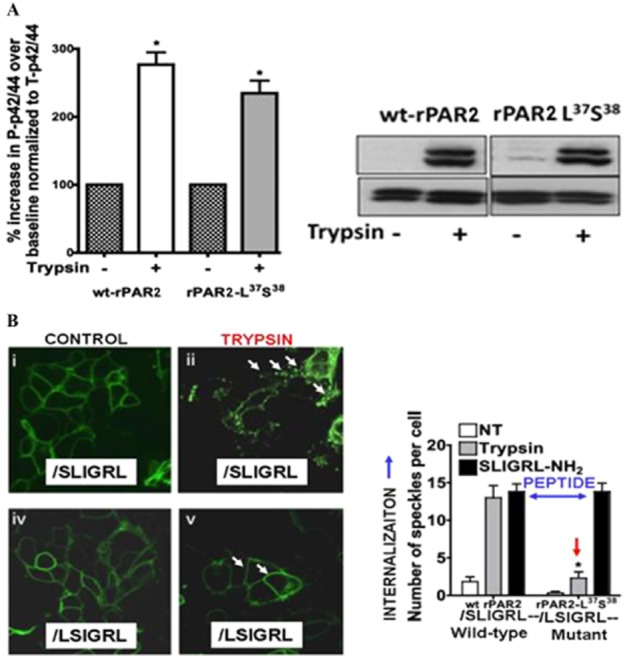
Biased MAPKinase signalling by a mutated rat PAR2-TL (A) without receptor internalization (B). (A, upper) Activation of PAR2 MAPKinase signalling by the trypsin-exposed mutated TL, //L37S38IGRL--- (rPAR2-L37S38). PAR2 was mutated so that trypsin cleavage (shown by //) at the R36//L37SIGRL--- sequence unmasks the TL sequence, L37S38IGRL--- (rPAR2-L37S38), which activates MAPKinase to the same level as for trypsin-activated wild-type receptor [A, histograms (left) and Western blot (right)]. However, the trypsin-unmasked mutant TL (rPAR2-L37S38) does not activate calcium signalling (Figure 2B, lower panel, blue arrow at the bottom, X-axis). (B) However, activation of the mutant PAR2 (rPAR2-L37S38) by the trypsin-exposed TL, //LSIGRL--- does not trigger receptor internalization (lower-right image, Figure 5B; red arrow, right histograms). In contrast, peptide-driven receptor activation causes internalization of both the wild-type and mutant rPAR2-L37S38 receptors (blue two-way arrow, right-hand histograms, Figure 5B). Adapted from Ramachandran et al., 2009.
Biased PAR activation by endogenous proteinases and distinct receptor dynamics: the discovery of non-canonical proteinase-revealed ‘TLs’ and signalling by non-canonical PAR1 TLs
The first clear evidence that PARs could signal by an enzyme-unmasked, ‘non-canonical’, TL generated by cleavage at sites other than the target arginine (e.g. R41/S42 in human PAR1) came from studies of the chemotactic properties of another type of proteinase, MMP-1 (Boire et al., 2005). By cleavage of human PAR1 at the D39/P40 site, which is located two amino acid residues to the N-terminal side of the thrombin cleavage site at R41-S42, thus revealing a TL sequence, PRSFLLR---, MMP-1 triggers PAR1 signalling (Boire et al., 2005; Trivedi et al., 2009). This ‘non-canonical’ PAR1 TL causes the receptor to signal in many ways like the classical TL unmasked by thrombin (TL = SFLLRN---); and the receptor-derived, ‘non-canonical’, TL peptide (PRSFLLRN) was a full agonist for PAR1-dependent Rho and p38 MAPK signalling in platelets (Trivedi et al., 2009). In our own hands, the peptide also triggers PAR1-mediated calcium signalling, but at much higher concentrations than for activating p42/44 MAPKinase. Thus, the peptide appears selective for MAPKinase and Rho signalling relative to calcium, but the ‘biased’ nature of MMP1-generated ‘non-canonical’ PAR1 TL remains to be fully characterized.
A ‘biased’ signalling pathway for PAR1 has also been observed for its stimulation by activated protein-C, (APC) compared with PAR1 activation by thrombin (Russo et al., 2009; Mosnier et al., 2012; Schuepbach et al., 2012). Thus, thrombin activation of PAR1 triggers RhoA, but not Rac1 signalling, along with an increase in endothelial barrier permeability. Conversely, APC activation of PAR1 causes the opposite: stimulation of Rac1 but not RhoA that results in a decrease in endothelial barrier permeability (Russo et al., 2009). This biased signalling by APC is dependent not only on a specific location of PAR1 in a caveolar environment (Russo et al., 2009), but also on the generation of a novel ‘non-canonical’ TL by APC cleavage at the R46/N47, position of human PAR1, to expose a TL with the sequence: N47PNDKYEPF--- (Mosnier et al., 2012; Schuepbach et al., 2012). It is of considerable interest that the ‘APC-generated’ TL sequence no longer contains the arginine at position 46, which had been proposed to interact with the glutamic acid at position 260 of the extracellular loop 2 of PAR1 (Nanevicz et al., 1995). Thus, one can conclude that the APC-generated non-canonical TL and its sequence-derived biased PAR1-activating peptide dock with the receptor differently than the thrombin-generated ‘canonical’ TL sequence, SFLLRN---. Indeed, APC-activated PAR1 in endothelial cells is retained on the cell surface, even when cells are subsequently exposed to thrombin (Schuepbach et al., 2008). Our new work indicates that proteinases other than MMP1 and APC can also generate distinct ‘biased’ ‘non-canonical’ TLs to regulate PAR1 function via ‘biased signalling’ (Mihara et al., 2013). Specifically, neutrophil proteinase-3 cleaves just upstream of the D39/P40 MMP1 cleavage site to unmask a TL, //TLDPRSF---; and neutrophil elastase (NE) cleaves just upstream of the APC cleavage site to reveal a non-canonical TL //RNPNDKYEPF---. As summarized in Table 1, the synthetic non-canonical PAR1-derived peptides derived from proteinase-3 and elastase cleavage, TLDPRSF-NH2 and RNPNDKYEPF-NH2 are biased agonists for PAR1 (Mihara et al., 2013).
Biased PAR1 signalling by cockroach allergen proteinase E1
In common with many insect allergens, cockroach allergen has been found to contain serine proteinase activity that can activate PAR2 (Arizmendi et al., 2011). As PAR2 plays a role in the allergen sensitization process (Arizmendi et al., 2011), we wondered if the three serine proteinases present in cockroach allergen, as detected by their tagging with a biotinylated serine proteinase activity-based probe (ABP; Figure 6A), might also activate PAR1. As shown by the ABP biotin blots in Figure 6A, we have separated the three cockroach enzymes chromatographically, as indicated by the presence of only one ABP-labelled enzyme in each isolated fraction (E1, E2 and E3; Figure 6A, upper left). In a human PAR1-expressing cell line (Kirsten-virus-transformed normal rat kidney cells; KNRK), thrombin was able to activate both calcium signalling and MAPKinase (Figure 6B, second lane from left; Figure 6C, left panel). However, cockroach enzyme E1, at a concentration that activated PAR1 MAPKinase (4 U·mL−1; Figure 6B, red outline, lane on far right), was not able to activate calcium signalling (Figure 6C, right panel). Thus, like APC, the cockroach allergen serine proteinase E1 is able to cause biased signalling via PAR1. The presumed ‘non-canonical’ TL responsible for PAR1 biased signalling triggered by the cockroach enzyme E1 remains to be determined.
Figure 6.
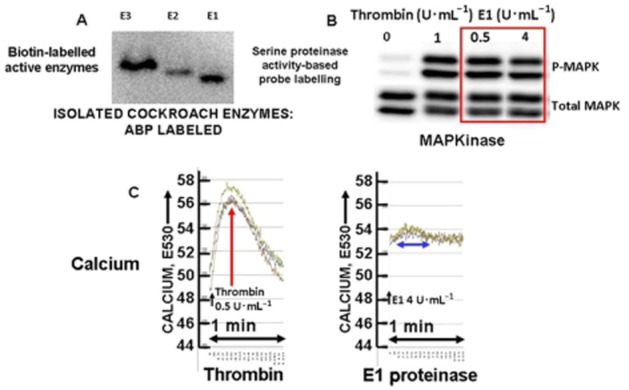
Biased PAR1 signalling (MAPKinase, but not calcium) by purified cockroach allergen serine proteinase E1. (A) Biotin-labelling of isolated cockroach proteinases E1, E2 and E3 with the biotin-serine proteinase activity-based probe (ABP). (B) Activation of MAPKinase in PAR1-expressing cells (Western blot signal: P-MAPK) by cockroach proteinase E1 (0.5 and 4 U·mL−1 enzyme activity: red outline) compared with thrombin (1 U·mL−1). (C) Calcium signalling stimulated by thrombin (0.5 U·mL−1: red arrow, left panel E530) but not by E1 proteinase (4 U·mL−1: blue two-way arrow, right panel, E530) at concentrations that nonetheless trigger MAPKinase activation (Figure 6B, red outline and data not shown).
Signalling by a non-canonical PAR2 TL
Our interest in the potential roles of neutrophil or pathogen-derived proteinases in inflammatory settings like cystic fibrosis led us to the observation that Pseudomonas aeruginosa elastase can ‘disarm’ PAR2, thereby preventing its activation by trypsin (Dulon et al., 2005). We next assessed the effects of human NE on PAR2 function. As expected, this enzyme ‘dis-armed’ PAR2, so that trypsin was no longer able to activate it (Figure 7A, panel A). To our surprise, however, we found that NE could activate PAR2 MAPKinase signalling (Figure 7A, panels B and C) without triggering calcium signalling (left-hand tracing, Figure 7A, panel A). This result echoed the data we had obtained previously for trypsin-mediated activation of the mutant PAR2 with a TL sequence (‘//LSIGRL---’) that does not activate calcium signalling (Figure 2, panel B, X-axis) but triggers MAPKinase (Figure 5, panel A). Further, PAR2 activation by human NE did not stimulate its interaction with β-arrestin, unlike activation by either trypsin or a PAR-activating peptide (Figure 7B). In keeping with the distinct receptor dynamics we observed for the PAR2 ‘//LSIGRL---’ TL mutant (no internalization; Figure 5B), elastase activation of the receptor did not cause PAR2 internalization, whereas trypsin did (Figure 8). Thus, the biased PAR2 signalling triggered by the NE ‘non-canonical’ TL, which involves a selective activation of MAPKinase via G12/13, is both spatially and kinetically distinct from signalling activated by the ‘canonical’ TL revealed by trypsin (Ramachandran et al., 2011) and is independent of an interaction with β-arrestin. Using synthetic peptides representing the N-terminal extracellular sequence of rat PAR2, we were able to predict the likely site at which NE generated its ‘non-canonical’ TL (underlined: DEFSAS// VLTGKLTTVFL---; Ramachandran et al., 2011). However, a synthetic peptide based on this sequence (VLTGKLTTVFL-NH2) did not activate PAR2 either for calcium or MAPKinase signalling. Thus, both PAR2 and PAR1 can lead to biased signalling when activated by endogenous proteinases; and this biased signalling generated by the unmasking of ‘non-canonical’ TLs in PARs 1 and 2 is caused by mechanisms that are distinct from signalling triggered by the ‘canonical’ TLs in terms of receptor dynamics. Whether proteinase activation can cause biased signalling by PAR4 remains to be determined, but appears highly likely.
Figure 7.
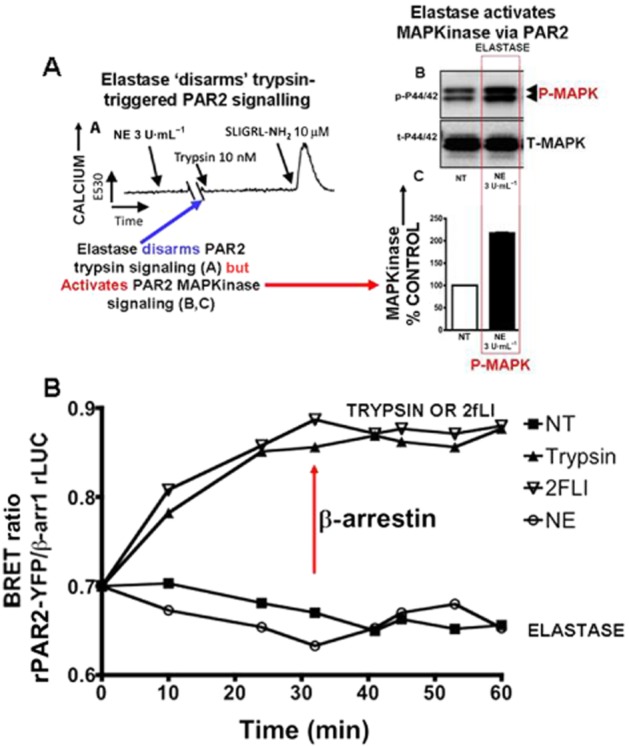
Neutrophil elastase (NE) stimulates ‘biased’ PAR2 signalling (MAPKinase (A, panel B); but not calcium (A, panel A) without triggering a PAR2-β-arrestin interaction (B). (A, panel A) NE, which does not cause a calcium signal on its own, disarms PAR2 preventing trypsin-stimulated (blue arrow) but not peptide-stimulated PAR2 calcium signalling (increase in E530). (A, sections B and C) NE activates PAR2 MAPKinase signalling. (B, bottom panel) Elastase activation of PAR2 does not trigger a β-arrestin interaction (bottom curve:), whereas trypsin and a PAR-activating peptide (2-furoyl-LIGRLO-NH2:) do (red arrow: upper curves). Adapted from Ramachandran R et al., 2011, with permission.
Figure 8.
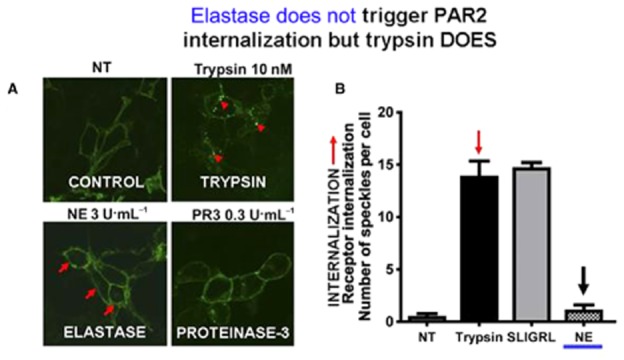
Neutrophil elastase (NE) does not stimulate PAR2 internalization whereas trypsin does. Trypsin-stimulated PAR2 internalization (upper-right micrograph, panel A, red arrows) was quantified by morphometric analysis of internalized receptor clusters (green dots: arrows, quantified in the right filled histogram: panel B, red arrow). No internalization was observed when cells were treated with NE (lower left quadrant image, panel A; right-hand histogram, NE, black arrow, panel B). Adapted from Ramachandran R et al., 2011 with permission.
Biased PAR antagonists and their therapeutic potential for inflammatory diseases
Biased PAR antagonists
As outlined in more detail elsewhere (Ramachandran et al., 2012), the development of low MW antagonists for PAR1 has been successful (e.g. Maryanoff et al., 2003; Chackalamannil et al., 2008), while the emergence of useful antagonists for either PAR2 or PAR4 has lagged behind. Indeed, compared with PAR1, there are regrettably very few ‘antagonist tools’ available for PARs 2, 3 and 4, to dissect receptor function, apart from siRNA and antibodies that block proteinase-mediated receptor activation (Ramachandran et al., 2012). For the PAR1 antagonists, screening that resulted in the identification of ‘lead’ compounds involved measurements of either human platelet aggregation or calcium signalling. Thus, despite the effectiveness of the PAR1 antagonists to block thrombin-induced platelet aggregation, the ability of those compounds to affect the receptor in a biased way has not been evaluated in detail. Similarly, the PAR2 antagonists developed successfully to date have been evaluated primarily for their block of agonist-induced calcium signalling (Sevigny et al., 2011; Suen et al., 2012), but not for their ‘biased’ antagonist or agonist properties. Given the prime focus of our laboratory on PAR2-induced inflammation, we studied the properties of GB88 (5-isoxazoyl-Cha-Ile-spiro[indene-1,4′-piperidine]) and P2-pal-18S-pepducin in terms of their potential for biased antagonist activities.
As shown in Figure 9A, GB88 does indeed block agonist-stimulated PAR2 calcium signalling in keeping with previous observations (Lohman et al., 2012a,b; Suen et al., 2012); but on its own, GB88 activates MAPKinase at levels equivalent to those achieved by either trypsin or the PAR2 peptide agonist, 2-furoyl-LIGRLO-NH2 (Figure 9B; Han, 2008; Suen, Fairlie et al., unpublished, 2013). In accord with its ability to activate MAPKinase via PAR2, GB88 promotes the interaction of the receptor with β-arrestin (Figure 10A). However, GB88 does not itself trigger receptor internalization (Figure 10B, left panel) nor does it prevent internalization of PAR2 activated by either trypsin or 2-furoyl-LIGRLO-NH2 (Figure 10B, middle and right-hand panels). Thus, GB88 behaves as a ‘biased’ PAR2 partial-agonist-antagonist, blocking calcium signalling stimulated by either trypsin or a PAR2-activating peptide, but simultaneously activating MAPKinase and other signalling pathways (Figures 9 and 10, and Suen, Fairlie et al., unpublished, 2013). Further work evaluating the full extent of signalling induced by GB88 and its ability to promote the interaction of PAR2 with β-arrestin merits attention. The ability of GB88 to act as a ‘full’ or ‘partial’ agonist for its ability to activate MAPKinase has not yet been reported in detail.
Figure 9.
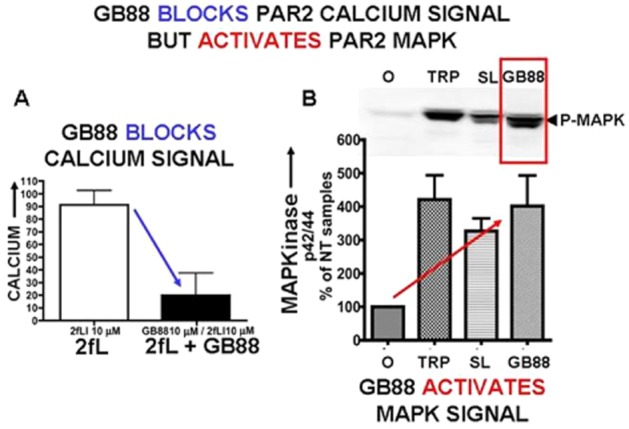
The PAR2 antagonist GB88 (10 μM) blocks calcium signalling in PAR2-expressing KNRK cells (A, left) but activates PAR2 MAPKinase signalling (B, right). (A) Pretreatment of cells with GB88 (10 μM) blocked PAR-agonist (2-furoyl-LIGRLO-NH2, 10 μM)-stimulated calcium signalling (histograms, panel A), but (B) GB88 (3 μM) stimulated MAPKinase signalling (phospho-p42/44; P-MAPK) relative to ‘no-treatment’ (NT) cells [% p42/44 relative to untreated (0) cells: panel B, red arrow] in PAR2-expressing KNRK cells. The level of the GB88-stimulated MAPKinase was equivalent to that triggered by either trypsin (TRP 1 U mL−1; 10 nM) or a PAR2-activating peptide (SL: SLIGRL-NH2: 10 μM). Data from Ramachandran R et al. unpublished experiments, 2013.
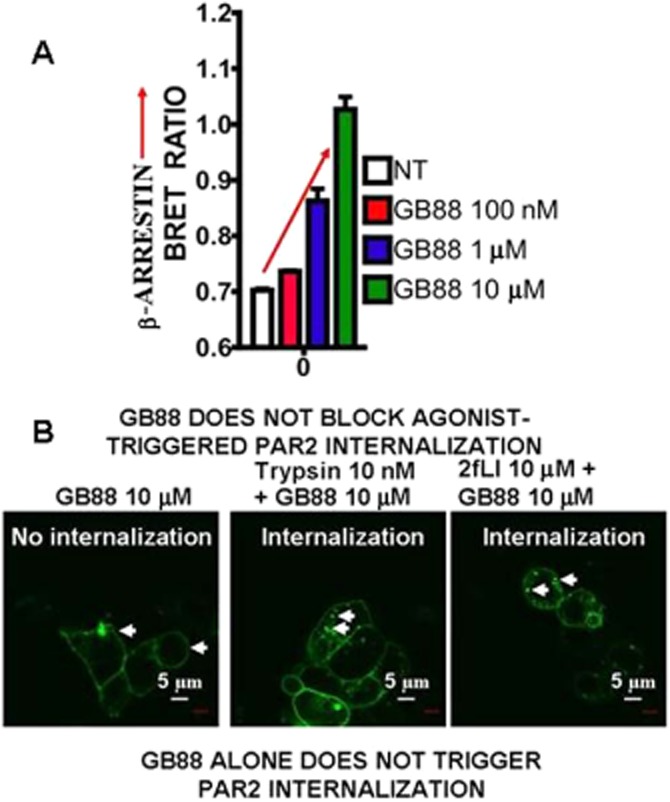
The PAR2 ligand, GB88 (0.1–10 μM), that stimulates MAPKinase, promotes β-arrestin interactions (A), but does not either cause receptor internalization on its own (B, left panel; GB88 10 μM) or block agonist-triggered PAR2 internalization (B, middle and right panels). (A) The concentration-dependence of GB88-stimulated PAR2 interaction with β-arrestin is shown in panel A (upper). PAR2 internalization caused by either trypsin (10 nM, middle panel, Figure 10B) or a PAR2 peptide agonist (2-furoyl-LIGRLO-NH2: 2fL; 10 μM, right panel, Figure 10B) is shown by the white arrows. The scale marker (white bar) represents 5 microns (μm). Data from Ramachandran R et al. unpublished experiments, 2013.
Like GB88, the pepducin PAR2 antagonist, P2pal-18S-pepducin, does not trigger calcium signalling on its own and is able to block PAR2-mediated calcium signalling (Sevigny et al., 2011; Figure 11A). However, this ‘biased antagonist’ does not prevent the interaction of agonist-triggered PAR2 with β-arrestin (Figure 11B). Therefore, one can predict that, like GB88, P2pal-18S-pepducin will block only a subset of PAR2 responses, thus functioning as a ‘biased’ antagonist. This issue clearly merits further study, as does the possibility that P2pal-18S-pepducin may stimulate a subset of PAR2 signalling pathways, comparable to the action of GB88.
Figure 11.
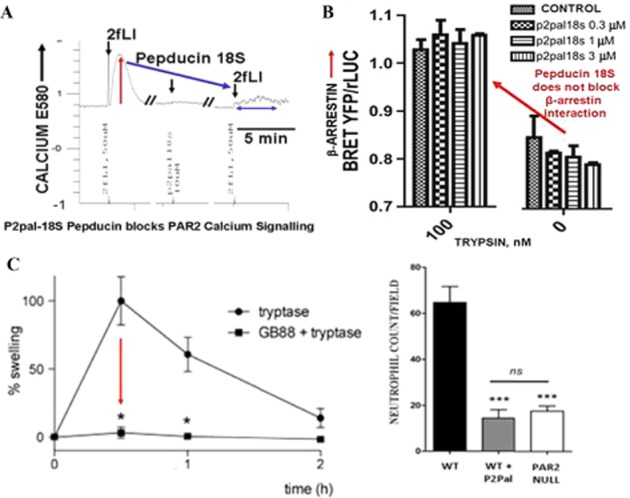
Pepducin P2pal-18S blocks calcium signalling (A) but does not affect agonist-stimulated PAR2-β-arrestin interactions (B). However, both P2pal-18S and GB88 inhibit inflammation in vivo, despite their ‘biased antagonist action’ (C). (A) Calcium signalling (E580) stimulated by 2-furoyl-LIGRLO-NH2 (2fLI: 50 μM) in PAR2-expressing HEK cells (upward deflection of tracing) is blocked by pretreatment of the cells with P2pal-18S-pepducin (10 μM, arrow on right) (Data from Ramachandran et al. unpublished experiments, 2013). (B) However, treatment of cells with concentrations of pepducin P2pal-18S that block calcium signalling (A, upper) does not affect the trypsin-stimulated interaction of PAR2 with β-arrestin monitored by BRET (panel B, red arrow: histograms on right (Data from Ramachandran et al. unpublished experiments, 2013). (C) Nonetheless, P2pal-18S (C, right panel histogram, WT + P2PAL) blocks paw inflammation neutrophil influx in vivo to the level found in PAR2 knockout mice; and GB88 (10 mg·kg−1 p.o.) also blocks paw oedema in a tryptase model (20 μg per paw) of peripheral paw inflammation (11C, left panel). Recalculated from Sevigny et al., 2011 and adapted from Lohman et al. 2012a, with permission.
Anti-inflammatory actions of ‘biased’ PAR2 antagonists
Despite the ‘biased’ block of PAR2 action caused by either GB88 or the P2pal-18S-pepducin outlined in the previous paragraph, both PAR2 antagonists have been encouragingly successful in attenuating inflammation in vivo in a number of model systems (Lohman et al., 2012a, 2012b; Sevigny et al., 2011; Suen et al., 2012). In particular, both antagonists can block agonist-induced paw oedema (Figure 11C, GB88, left panel) and neutrophil infiltration (Figure 11C, P2pal-18S, right panel) in a murine model of paw oedema (Sevigny et al., 2011; Lohman et al., 2012a,b; Suen et al., 2012). In the past, we have shown that this inflammatory response involves a neurogenic component (Vergnolle et al., 1999; Steinhoff et al., 2000). Thus, it is likely that in the paw oedema model, the antagonists are affecting PAR2 calcium signalling both on the neuronal elements as well as on the endothelial and inflammatory cells. However, our data indicate that these antagonists would not block the β-arrestin signalling mechanisms responsible for the migration of eosinophils into the site(s) of airway inflammation in an in vivo asthma model (Nichols et al., 2012). Thus, the biased nature of the pepducin and GB88 antagonists predict that they will block only a subset of the inflammatory responses due to PAR2 activation. This situation may be an important advantage, in that a role for PAR2 in the resolution of inflammation is likely; and the selective block of the calcium signalling arm of PAR2, whilst retaining the β-arrestin and other pathways triggered by PAR2 internalization, may fortuitously accelerate the ‘healing process’ in which PAR2 may participate. Thus, the therapeutic potential of biased PAR antagonists should not be overlooked. Indeed, the differential nature of the pathways being affected by each biased ligand (either agonist or antagonist) will dictate which disease context will benefit most from their therapeutic use.
Summary
Given the ‘hormone-like’ role of proteinases, their inflammatory and pathophysiological effects will be mediated by a number of mechanisms quite apart from the regulation of PAR activity (Figure 1). Nonetheless, PARs will undoubtedly be found to play important roles in a variety of settings including cardiovascular, inflammatory and neurodegenerative diseases as well as cancer. The unusual mechanism of PAR regulation by proteolysis makes these receptor systems unique in having not only a number of molecularly distinct circulating full and biased agonists, such as trypsin and elastase for PAR2, but also multiple circulating proteinase antagonists that can silence the receptors by disarming. The importance of the PARs can be seen in certain inflammatory settings like arthritis, colitis, tumour invasion and CNS neurodegeneration, where mouse knockout studies suggest that the PARs can play key roles and may therefore be attractive therapeutic targets (see Ramachandran and Hollenberg, 2008; Adams et al., 2011; Ramachandran, 2012; Ramachandran et al., 2012). Thus, the development of therapeutically useful antagonists for PARs 1 and 2 deserves to be pursued, despite the somewhat limited success to date for the clinical use of PAR1 antagonists in the setting of vascular disease (see Ramachandran et al., 2012 for discussion). To continue this search for PAR-targeted drugs, we suggest considering seriously the potential utility of biased antagonists as they may be able to diminish acute inflammatory responses, but also retain a PAR-mediated impact on the resolution of inflammation following the initial inflammatory insult. Further, biased PAR agonists may be of value in certain settings, where the localized activation of PARs may be advantageous, for example, in the bronchi, where PAR2 activation can cause bronchodilation (Chow et al., 2000). Overall, it is hoped that this overview highlighting the ‘biased’ properties of both endogenous and synthetic PAR agonist and antagonists will stimulate further the development of therapeutic agents in this field.
Acknowledgments
Work described in this overview was supported in large part by an operating grant from the Canadian Institutes of Health Research (to M. D. H.) and also by funds from the Australian National Health and Medical Research Council (to D. P. F.: grants 1047759; 1027369). R.R. was supported in part by an Alberta Heritage Foundation for Medical Research (now termed, Alberta Innovates Health Solutions) postdoctoral fellowship and D.P. was supported in part by an Alberta Lung Association/Canadian Thoracic Society.
Glossary
- ABP
activity-based probe
- APC
activated protein-C
- GB88
5-isoxazoyl-Cha-Ile-spiro[indene-1,4′-piperidine]
- KNRK
Kirsten-virus-transformed normal rat kidney cells
- MAPKinase
MAPK or ERK
- NE
neutrophil elastase
- PAR
proteinase-activated receptor (PAR1, PAR2, PAR3, PAR4)
- TL
tethered ligand (receptor-activating peptide unmasked by proteolytic cleavage of an extracellular PAR N-terminal sequence)
Conflict of interests
J. Y. Suen and D. P. Fairlie are inventors on a patent AU20109033378 covering PAR2 agonists and antagonists that is owned by The University of Queensland. The authors have no other conflicts of interest.
References
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Al-Ani B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol. 1995;73:1203–1207. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- Al-Ani B, Wijesuriya SJ, Hollenberg MD. Proteinase-activated receptor 2: differential activation of the receptor by tethered ligand and soluble peptide analogs. J Pharmacol Exp Ther. 2002;302:1046–1054. doi: 10.1124/jpet.302.3.1046. [DOI] [PubMed] [Google Scholar]
- Al-Ani B, Hansen KK, Hollenberg MD. Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharmacol. 2004;65:149–156. doi: 10.1124/mol.65.1.149. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, et al. Mucosal allergic sensitization to cockroach allergens is dependent on proteinase activity and proteinase-activated receptor-2 activation. J Immunol. 2011;186:3164–3172. doi: 10.4049/jimmunol.0903812. [DOI] [PubMed] [Google Scholar]
- Blackhart BD, Ruslim-Litrus L, Lu CC, Alves VL, Teng W, Scarborough RM, et al. Extracellular mutations of protease-activated receptor-1 result in differential activation by thrombin and thrombin receptor agonist peptide. Mol Pharmacol. 2000;58:1178–1187. doi: 10.1124/mol.58.6.1178. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314(Pt 3):1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Burger MM. Proteolytic enzymes initiating cell division and escape from contact inhibition of growth. Nature. 1970;227:170–171. doi: 10.1038/227170a0. [DOI] [PubMed] [Google Scholar]
- Carney DH, Cunningham DD. Initiation of check cell division by trypsin action at the cell surface. Nature. 1977;268:602–606. doi: 10.1038/268602a0. [DOI] [PubMed] [Google Scholar]
- Carney DH, Cunningham DD. Transmembrane action of thrombin initiates chick cell division. J Supramol Struct. 1978;9:337–350. doi: 10.1002/jss.400090305. [DOI] [PubMed] [Google Scholar]
- Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- Chen LB, Buchanan JM. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc Natl Acad Sci U S A. 1975;72:131–135. doi: 10.1073/pnas.72.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JM, Moffatt JD, Cocks TM. Effect of protease-activated receptor (PAR)-1, -2 and -4-activating peptides, thrombin and trypsin in rat isolated airways. Br J Pharmacol. 2000;131:1584–1591. doi: 10.1038/sj.bjp.0703738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrétien M. My road to Damascus: how I converted to the prohormone theory and the proprotein convertases. Biochem Cell Biol. 2012;90:750–768. doi: 10.1139/o2012-031. [DOI] [PubMed] [Google Scholar]
- Connolly AJ, Ishihara H, Kahn ML, Farese RV, Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- Cuatrecasas P. Interaction of insulin with the cell membrane: the primary action of insulin. Proc Natl Acad Sci U S A. 1969;63:450–457. doi: 10.1073/pnas.63.2.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuatrecasas P. Properties of the insulin receptor of isolated fat cell membranes. J Biol Chem. 1971;246:7265–7274. [PubMed] [Google Scholar]
- Defea K. Beta-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br J Pharmacol. 2008;153(Suppl. 1):S298–S309. doi: 10.1038/sj.bjp.0707508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, et al. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2005;32:411–419. doi: 10.1165/rcmb.2004-0274OC. [DOI] [PubMed] [Google Scholar]
- de Haën C. The non-stoichiometric floating receptor model for hormone sensitive adenylyl cyclase. J Theor Biol. 1976;58:383–400. doi: 10.1016/s0022-5193(76)80126-9. [DOI] [PubMed] [Google Scholar]
- Han A. 2008. GB88 is a biased ligand by inhibiting Ca2+ release but inducing ERK and MAPK activation without inducing PAR2 internalisation, Honours thesis, University of Queensland.
- Hansen KK, Saifeddine M, Hollenberg MD. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology. 2004;112:183–190. doi: 10.1111/j.1365-2567.2004.01870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg MD, Laniyonu AA, Saifeddine M, Moore GJ. Role of the amino- and carboxyl-terminal domains of thrombin receptor-derived polypeptides in biological activity in vascular endothelium and gastric smooth muscle: evidence for receptor subtypes. Mol Pharmacol. 1993;43:921–930. [PubMed] [Google Scholar]
- Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Cuatrecasas P. The mobile receptor hypothesis and ‘cooperativity’ of hormone binding. Application to insulin. Biochim Biophys Acta. 1976;433:482–495. doi: 10.1016/0005-2736(76)90275-3. [DOI] [PubMed] [Google Scholar]
- Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinlough-Rathbone RL, Perry DW, Guccione MA, Rand ML, Packham MA. Degranulation of human platelets by the thrombin receptor peptide SFLLRN: comparison with degranulation by thrombin. Thromb Haemost. 1993;70:1019–1023. [PubMed] [Google Scholar]
- Kono T, Barham FW. Insulin-like effects of trypsin on fat cells. Localization of the metabolic steps and the cellular site affected by the enzyme. J Biol Chem. 1971;246:6204–6209. [PubMed] [Google Scholar]
- Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, et al. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012a;26:2877–2887. doi: 10.1096/fj.11-201004. [DOI] [PubMed] [Google Scholar]
- Lohman RJ, Cotterell AJ, Suen JY, Liu L, Do TA, Vesey DA, et al. Antagonism of protease activated receptor 2 protects against experimental colitis. J Pharmacol Exp Ther. 2012b;340:256–265. doi: 10.1124/jpet.111.187062. [DOI] [PubMed] [Google Scholar]
- Maryanoff BE, Zhang HC, Andrade-Gordon P, Derian CK. Discovery of potent peptide-mimetic antagonists for the human thrombin receptor, protease-activated receptor-1 (PAR-1) Curr Med Chem Cardiovasc Hematol Agents. 2003;1:13–36. doi: 10.2174/1568016033356724. [DOI] [PubMed] [Google Scholar]
- Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD. Neutrophil elastase and proteinase-3 trigger G-protein biased signaling through proteinase activated receptor-1 (PAR1) J Biol Chem. 2013;288:32979–32990. doi: 10.1074/jbc.M113.483123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012;120:5237–5246. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- Nanevicz T, Ishii M, Wang L, Chen M, Chen J, Turck CW, et al. Mechanisms of thrombin receptor agonist specificity. Chimeric receptors and complementary mutations identify an agonist recognition site. J Biol Chem. 1995;270:21619–21625. doi: 10.1074/jbc.270.37.21619. [DOI] [PubMed] [Google Scholar]
- Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, et al. Beta-arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A. 2012;109:16660–16665. doi: 10.1073/pnas.1208881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowska E, Reiser G. The protease-activated receptor-3 (PAR-3) can signal autonomously to induce interleukin-8 release. Cell Mol Life Sci. 2008;65:970–981. doi: 10.1007/s00018-008-7555-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente XS, Sanchez LM, Gutierrez-Fernandez A, Velasco G, Lopez-Otin C. A genomic view of the complexity of mammalian proteolytic systems. Biochem Soc Trans. 2005;33(Pt 2):331–334. doi: 10.1042/BST0330331. [DOI] [PubMed] [Google Scholar]
- Ramachandran R. Developing PAR1 antagonists: minding the endothelial gap. Discovery Medicine. 2012;13:425–431. [PubMed] [Google Scholar]
- Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153(Suppl. 1):S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, et al. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol. 2009;76:791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Defea KA, et al. Neutrophil elastase acts as a biased agonist for proteinase activated receptor-2 (PAR2) J Biol Chem. 2011;286:24638–24648. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86. doi: 10.1038/nrd3615. [DOI] [PubMed] [Google Scholar]
- Rasmussen UB, Vouret-Craviari V, Jallat S, Schlesinger Y, Pages G, Pavirani A, et al. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 1991;288:123–128. doi: 10.1016/0014-5793(91)81017-3. [DOI] [PubMed] [Google Scholar]
- Rasmussen UB, Gachet C, Schlesinger Y, Hanau D, Ohlmann P, Van Obberghen-Schilling E, et al. A peptide ligand of the human thrombin receptor antagonizes alpha-thrombin and partially activates platelets. J Biol Chem. 1993;268:14322–14328. [PubMed] [Google Scholar]
- Rieser P. The insulin-like action of pepsin and pepsinogen. Acta Endocrinol (Copenh) 1967;54:375–379. doi: 10.1530/acta.0.0540375. [DOI] [PubMed] [Google Scholar]
- Rieser P, Rieser CH. Anabolic responses of diaphragm muscle to insulin and to other pancreatic proteins. Proc Soc Exp Biol Med. 1964;116:669–671. doi: 10.3181/00379727-116-29339. [DOI] [PubMed] [Google Scholar]
- Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A. 2009;106:6393–6397. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuepbach RA, Feistritzer C, Brass LF, Riewald M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood. 2008;111:2667–2673. doi: 10.1182/blood-2007-09-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. Protease activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost. 2012;10:1675–1684. doi: 10.1111/j.1538-7836.2012.04825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefton BM, Rubin H. Release from density dependent growth inhibition by proteolytic enzymes. Nature. 1970;227:843–845. doi: 10.1038/227843a0. [DOI] [PubMed] [Google Scholar]
- Sevigny LM, Zhang P, Bohm A, Lazarides K, Perides G, Covic L, et al. Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc Natl Acad Sci U S A. 2011;108:8491–8496. doi: 10.1073/pnas.1017091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, White MF, Kahn CR. Tryptic activation of the insulin receptor. Proteolytic truncation of the alpha-subunit releases the beta-subunit from inhibitory control. J Biol Chem. 1988;263:4852–4860. [PubMed] [Google Scholar]
- Steiner DF. On the discovery of precursor processing. Methods Mol Biol. 2011;768:3–11. doi: 10.1007/978-1-61779-204-5_1. [DOI] [PubMed] [Google Scholar]
- Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157:697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, et al. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110) Br J Pharmacol. 2012;165:1413–1423. doi: 10.1111/j.1476-5381.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol. 1999;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouret-Craviari V, Van Obberghen-Schilling E, Rasmussen UB, Pavirani A, Lecocq JP, Pouyssegur J. Synthetic alpha-thrombin receptor peptides activate G protein-coupled signaling pathways but are unable to induce mitogenesis. Mol Biol Cell. 1992;3:95–102. doi: 10.1091/mbc.3.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouret-Craviari V, Van Obberghen-Schilling E, Scimeca JC, Van Obberghen E, Pouyssegur J. Differential activation of p44mapk (ERK1) by alpha-thrombin and thrombin-receptor peptide agonist. Biochem J. 1993;289(Pt 1):209–214. doi: 10.1042/bj2890209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991a;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991b;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- Werle E, Erdos EG. [A new hypotensive, enterotropic and hysterotropic substance in human urine.] Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1954;223:234–243. [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]