

Abstract
The blood–brain barrier (BBB) is a complex vascular structure consisting of microvascular endothelial cells that line the vessel wall, astrocyte end-feet, pericytes, as well as the basal lamina. BBB cells act in concert to maintain the characteristic impermeable and low paracellular flux of the brain vascular network, thus ensuring a homeostatic neuronal environment. Alterations in BBB stability that occur during injury have dire consequences on disease progression and it is clear that BBB cell-specific responses, positive or negative, must make a significant contribution to injury outcome. Reduced oxygenation, or hypoxia, is a characteristic of many brain diseases that significantly increases barrier permeability. Recent data suggest that hypoxia-inducible factor (HIF-1), the master regulator of the hypoxic response, probably mediates many hypoxic effects either directly or indirectly via its target genes. This review discusses current knowledge of physiological cell-specific regulation of barrier function, their responses to hypoxia as well as consequences of hypoxic-and HIF-1-mediated mechanisms on barrier integrity during select brain diseases. In the final sections, the potential of current advances in targeting HIF-1 as a therapeutic strategy will be overviewed.
Keywords: BBB cell-specific response, endothelial cells, astrocytes, pericytes, hypoxia, HIF-1, neurodegenerative diseases
The maintenance of CNS homeostasis is performed largely by the blood–brain barrier (BBB), which together with neurons and microglia form an organization referred to as the neurovascular unit (NVU). The BBB is dynamic performing both passive and active features of the brain endothelium essentially acting as a vascular gatekeeper that controls movement of substances from the circulating blood into the brain parenchyma – a role crucial for neuronal, and therefore CNS, homeostasis. Accumulating experimental evidence supports the hypothesis that opening of the BBB triggers a chain of events leading to neuronal dysfunction and damage resulting in neurological disease, and when coupled with previous insults BBB disruption could have serious detrimental consequences for patient outcome. Despite this knowledge, our understanding of physiological barrier function, as well as during disease, is very limited. In addition, the contribution of the perivascular cells that modulate barrier characteristics and their individual responses to injury is poorly characterized. This review will discuss the mechanisms through which hypoxia, a characteristic state of many brain diseases, disrupts barrier function and the importance of BBB cell-specific responses to barrier integrity. Additionally, consequences of hypoxia-mediated barrier modulation during brain disease and future therapeutic use of hypoxia-inducible factor-1 (HIF-1) modulators in the clinics will be reviewed.
Physiology
BBB organization and cell-specific function
The BBB is a complex structure consisting of microvascular endothelial cells (ECs) that line the vessel wall, astrocyte end-feet, pericytes, as well as the basal lamina (see Figure 1), which together with neurons and microglia form an organization referred to as the NVU. The basal lamina is an essential part of the BBB that surrounds the capillaries thereby anchoring the cells in place and providing a link with the resident brain cells. The brain ECs are in direct contact with brain pericytes (Abbott et al., 2010; Ogunshola and Al-Ahmad, 2012) and separated from the brain parenchyma by two layers of extracellular matrices (ECM). The inner layer is formed by the vascular basement membrane (BM) localized at the EC basolateral side and is shared with neighbouring pericytes (Hallmann et al., 2005). The outer layer is formed by the glia limitans as a result of glial end-feet processes that surrounds or ensheaths the cerebral vascular tree (Paolinelli et al., 2011). Perivascular glial end-feet form a direct interface between the vascular compartment and the brain parenchyma (see Figure 1) and are therefore thought to represent key checkpoints of brain metabolism and function (Wolburg et al., 2009a).
Figure 1.

The BBB protects neurons and glial cells from systemically circulating agents as brain microvessels form a very tight barrier clearly distinct from vessels in other organs. The barrier is formed by ECs (red), which line the blood vessels, surrounded by pericytes (light blue), BM (grey) and astrocytic (dark blue) end-feet. Astrocytes provide the cellular link to the adjacent neurons (pink). The illustration also shows microglial cells (grey) in contact with astrocytes. Inset (black box) shows the cell–cell contact that takes place between two adjacent ECs at the BBB and demonstrates the basic organization of the BBB tight and adherens junctional proteins. Note the interactions of the junctional proteins with the cytoskeleton.
BBB cells act in concert to maintain the characteristic impermeable and low paracellular flux of the vascular network that is chiefly mediated via high expression levels and localization of tight junction (TJ) proteins, namely, occludins, claudins, junctional adhesion molecules and their adaptor protein zonula occludens-1 (ZO-1; reviewed in Förster, 2008; Abbott et al., 2010). TJs are elaborate structures that function as both a ‘zipper’ that separates the apical and basolateral cell membranes thus enabling asymmetric distribution of membrane constituents, and a ‘fence’ that limits paracellular permeability (see Figure 1 inset). Thus, stringent regulation of CNS homeostasis by severe restriction of the paracellular diffusional pathway between the ECs and substances and/or cells within the circulating blood is ensured. TJs are highly dynamic structures that rapidly undergo subcellular redistribution, expression level alterations as well as post-translational modifications, all of which affect protein–protein interactions (reviewed in Förster, 2008; Ogunshola, 2011). Cellular interactions and local release of factors (discussed in detail later), in addition to signals from circulating substances, have marked effects on TJ expression and therefore barrier integrity. Brain ECs do not seem to spontaneously express high TJ levels but are induced by the surrounding perivascular cells, astrocytes and pericytes (Ballabh et al., 2004; Abbott et al., 2006; Ogunshola, 2011). Astrocytes are the most abundant cell in the brain and their long filamentous end-foot processes extend towards and envelope the vascular network. Astrocytes have long been known to play a central role in inducing expression of TJ proteins in microvascular ECs (Abbott, 2002; Ogunshola, 2011). Although pericytes have very close contact with ECs, lying juxtaposed to the capillary and sharing a BM, for a long time, the function of this cell in barrier maintenance was largely unknown. Scientific advances in tools and methodologies, however, have provided recent evidence from both in vitro and in vivo studies that pericytes also significantly contribute to barrier stability during development and adulthood.
Barrier effectiveness is directly related to the ability of the perivascular cells to maintain their normal functional activities. As such, alterations or modifications of perivascular cell properties per se, as a result of disease progression or signalling pathway activation, will compromise barrier integrity and increase permeability. Despite this fact, the highly coordinated signalling mechanisms and dynamic interactions that exist between individual cells of the BBB remain largely unknown. Such knowledge is key to better understand barrier function under physiological as well as pathological conditions. This implies possible future development of cell-specific therapies and targeted treatments that could indeed represent an important way to stabilize barrier function during injury. The following sections deal with some of the cell-specific responses that occur particularly during oxygen deprivation and diseases characterized by hypoxia in more detail. Notably, because of the complexity related to studying BBB in vivo, much of the data comes from in vitro model systems, however, where possible reference to in vivo studies will also be given.
Astrocytes at the BBB
Similar to neurons and other glial cells astrocytes originate from the neuroectoderm (Allen and Barres, 2009). To date, 11 different types of astrocytes are known of which eight are specifically associated with blood vessels (Abbott et al., 2006). Astrocytes are crucially involved in control of neuronal function through regulation of brain homeostasis, supply of neurons with energy and substrates for neurotransmission and recycling of neurotransmitters. Furthermore, they represent the connection between neurons and the brain vasculature, which they cover with their end-feet. Almost 50 years ago, the close proximity of the astrocytic end-feet to the brain endothelium raised the hypothesis that astrocytes contribute to BBB regulation (Davson and Oldendorf, 1967). Since then, a number of in vivo (Stewart and Wiley, 1981; Janzer and Raff, 1987; Willis et al., 2004) and particularly in vitro studies have demonstrated the importance of astrocytes to BBB induction and regulation. Model systems using astrocyte-endothelial co-cultures (Dehouck et al., 1990; Rist et al., 1997; Fischer et al., 2000; Al Ahmad et al., 2009) as well as astrocyte-conditioned media (ACM) (Rubin et al., 1991; Raub et al., 1992) demonstrated the inductive potential of astrocytes on barrier tightness by increased transendothelial electrical resistance (TEER) and reduced permeability to low molecular weight tracers (for detailed review, refer to the excellent article by Deli et al., 2005). There is now good evidence that barrier induction through astrocytes is mainly mediated via changes in the number, length and complexity of endothelial TJs (Tao-Cheng et al., 1987), expression levels of TJ and associated-proteins like occludin and ZO-1, respectively (Siddharthan et al., 2007; Colgan et al., 2008), and the redistribution of junctional proteins such as platelet endothelial cell adhesion molecule-1, ZO-1 and claudin-5 at endothelial junctions (Colgan et al., 2008; Al Ahmad et al., 2011). In addition, astrocytes modulate expression and polarized localization of endothelial transporters, such as P-glycoprotein (P-gp) or multi-drug resistance protein (MRP) (Berezowski et al., 2004; Al Ahmad et al., 2011) as well as BBB-specific enzyme-systems such as γ-glutamyl transpeptidase (Meyer et al., 1991; Hayashi et al., 1997) and thus crucially regulate EC function.
Major efforts have been made to unravel how astrocytes modulate TJs, transporters and enzyme systems. Astrocytes are known to secrete a large number of substances including peptides, growth factors and chemokines (Nico and Ribatti, 2012) several of which modulate barrier function, such as basic fibroblast growth factor (bFGF), TGF-β1, glial cell-derived neurotrophic factor and src-suppressed C-kinase substrate (SSeCKS) (Igarashi et al., 1999; Sobue et al., 1999; Lee et al., 2003; Reuss et al., 2003; Dohgu et al., 2004; Walshe et al., 2009; Shimizu et al., 2012). For instance, bFGF has demonstrated barrier-tightening effects in vitro by reducing barrier permeability (Sobue et al., 1999) and increasing endothelial γ-glutamyl-transpeptidase and alkaline phosphatase activity (Hafny et al., 1996). In vivo bFGF knockout increases BBB permeability to albumin and reduces the expression of ZO-1 and occludin, and coincides with reduced astrocyte differentiation (Reuss et al., 2003). The effect of astrocyte-secreted TGF-β1 on barrier function is controversial. While some studies observed reduced BBB permeability and increased TJ expression in the presence of TGF-β1/TGF-β (Dohgu et al., 2004; 20052004,2005; Walshe et al., 2009), others reported adverse effects (Shen et al., 2011). This discrepancy may be explained by the fact that TGF-β effects on the endothelium are highly dependent on the endothelial activation state and the tissue environment, especially the ECM as it mediates TGF-β activation (Cambier et al., 2005).
Over the past 20 years, our understanding of the importance of astrocytes as regulators of BBB physiology and how they modulate barrier characteristics has dramatically increased but, realistically, unravelling of the complex pathways has just begun.
Pericytes at the BBB
The origin of CNS pericytes remains unclear with derivation from the mesoderm and neuroectoderm as well as from the monocyte lineage under general discussion (Sa-Pereira et al., 2012). Diverse functions within the brain have been attributed to pericytes including regulation of brain homeostasis, angiogenesis, blood flow, immune and phagocytic activity, as well as being a source of pluripotent stem cells (Sa-Pereira et al., 2012). The effect of pericytes on BBB induction and maintenance is less well characterized than astrocyte-induced responses. During development pericytes fulfil central roles in vessel stabilization through inhibition of EC proliferation and migration as well as regulation of vessel maturation (Antonelli-Orlidge et al., 1989; Hellström et al., 2001; Al Ahmad et al., 2011; Sa-Pereira et al., 2012). Recent in vivo studies using platelet-derived growth factor (PDGF) receptor β (PDGFRβ) knockout mice have significantly improved our understanding. During embryonic angiogenesis, pericytes are recruited to the vessels via EC-derived PDGF-β. The impaired recruitment of pericytes to the brain microvasculature caused by inhibition of PDGF-β signalling, induced either by PDGF-β or PDGFRβ knockout, resulted in severe vascular consequences such as increased vessel diameter, formation of microaneurysms, endothelial hyperplasia and increased vessel permeability (Lindahl et al., 1997; Hellström et al., 2001; Daneman et al., 2010). These alterations in vessel tightness were attributed to enhanced caveolae formation and transcytosis, abnormal TJ alignment and increased expression of permeability inducing factors like VEGF and angiopoietins (ANG) −2 (Hellström et al., 2001; Daneman et al., 2010). Corresponding studies in adult mice revealed that pericytes are also crucial for adult BBB maintenance, as reduced pericytic vessel coverage resulted in increased BBB permeability, altered endothelial gene expression and loss of astrocyte end-feet polarization (Armulik et al., 2010). Age-dependent pericyte loss in PDGFRβ+/− mice elevated parenchymal accumulation of blood proteins and leakage of neurotoxic and vasculotoxic substances into the brain parenchyma and a reduction in the expression of ZO-1 and occludin protein (Bell et al., 2010). In vitro studies using cells of human, murine, bovine and porcine origin further underlined the positive effect of pericytes on BBB tightness (Hayashi et al., 2004; Dohgu et al., 2005; Nakagawa et al., 2007; Al Ahmad et al., 2009; Daneman et al., 2010). Similar to astrocytes, pericytes are capable of secreting factors that modulate BBB permeability under normal conditions, like ANG-1 (Hori et al., 2004; Wang et al., 2007), TGF-β1 (Dohgu et al., 2005) and GDNF (Shimizu et al., 2012) that increase ZO-1, claudin-5 or occludin in vitro (Hori et al., 2004; Wang et al., 2007; Shimizu et al., 2011). Therefore, similar to astrocytes, pericytes seem capable of modulating TJs. In contrast, a few in vitro studies report that co-culture of ECs with pericytes reduces TEER via induction of matrix metalloproteinases (MMP)-2 and-9 activity and activation of VEGF-mediated signalling (Zozulya et al., 2008; Thanabalasundaram et al., 2010). Interestingly, this negative effect of pericytes seems to be dependent on their differentiation state (Thanabalasundaram et al., 2011).
Tricellular interactions – ECs, pericytes, astrocytes
How ECs, astrocytes and pericytes influence each other and concertedly modulate barrier function is a highly interesting but challenging question. Due to the complexity of the BBB in vivo data on this topic is limited, but some in vitro studies have investigated the effect of simultaneous astrocyte and pericyte co-culture on ECs. The majority of these studies report increased TEER in triple cultures compared with co-culture or monoculture models (Nakagawa et al., 2007; 20092007,2009; Al Ahmad et al., 2009); however, decreased TEER in triple cultures due to the presence of pericytes was also reported (Hatherell et al., 2011). Interestingly, only Hatherell et al. used human ECs. Importantly, the permeability to sodium-fluorescein, sucrose or 40 kDa FITC-dextran was not significantly altered in triple cultures compared with contact astrocyte co-cultures (Al Ahmad et al., 2009; Nakagawa et al., 2009). Nakagawa et al. indeed observed that claudin-5 and ZO-1 protein expression is increased and more restricted to cell–cell borders in triple cultures compared with endothelial monocultures (Nakagawa et al., 2009). Using a novel three-dimensional culture model, our group showed that co-culture of ECs with astrocytes and pericytes also induces polarized, luminal localization of the ABC transporters P-gp and MRP-2 and that astrocytes and pericytes are indispensable for P-gp activity (Al Ahmad et al., 2011). However, it is patently evident that many more studies and wider use of triple culture model systems is invaluable for understanding the multicellular crosstalk at the BBB.
Basement membrane
The BM represents an important but often neglected component of the BBB. Although the BM does not act as a diffusion barrier per se, it fulfils important functions for the BBB by providing structural support for the cells by anchoring them in place. Moreover, the BM provides an important platform for mediating signalling events that regulate cell differentiation, proliferation, migration and adhesion (Baeten and Akassoglou, 2011) and thus BBB function. The BM consists of structural matrix proteins that are secreted by the BBB cells with cell-specific differences (a detailed list is presented in Baeten and Akassoglou, 2011). BBB cells are anchored to the BM via ECM receptors, of which the best understood ones are the integrins and dystroglycan (Baeten and Akassoglou, 2011). More than just providing a physical link between the BM and the cells, the matrix receptors represent important modulators of signalling pathways that allow cellular adaptation to environmental changes. Perlecan, the dominating proteoglycan in the endothelial BM (Engelhardt and Sorokin, 2009), has been shown to interact with a number of different growth factors like VEGF, PDGF or TGF-β and retain them in the ECM thereby regulating cellular signal transduction to control cell responses and BBB maintenance (Roberts et al., 2012).
Composition of the BM can also regulate BBB tightness (Tilling et al., 1998). P-gp expression is increased when ECs are cultured on brain-derived ECM (Tatsuta et al., 1994). ECMs derived from pericytes or astrocytes exhibit differential effects on EC impedance in vitro compared with controls grown on their endogenous ECMs (Hartmann et al., 2007). Interestingly, ECs cultured on astrocyte-derived ECM displayed a higher resistance than those cultured on pericyte-derived ECM. Astrocyte-derived ECM has been shown to up-regulate the expression of endothelial-specific γ-glutamyl-transpeptidase and activity (Mizuguchi et al., 1994; Hayashi et al., 1997). Several BM-associated proteins have been shown to be important for BBB maintenance in vivo. Depletion of perlecan in vivo is lethal due to deterioration of brain vesicles and myocardial BM (Baeten and Akassoglou, 2011), whereas specific depletion of perlecan in the endothelial BM results in microvessel bleeding and endothelial dilations (Hallmann et al., 2005). Agrin, another proteoglycan, accumulates at brain microvessels at the time of BBB tightening (Wolburg et al., 2009b) and its depletion results in TJ disruption (Rascher et al., 2002) and depolarization of astrocytic end-feet mainly through redistribution of aquaporin 4 (AQP4; Wolburg et al., 2009b). Osada et al. showed that blocking of β1-integrin using a neutralizing antibody results in altered claudin-5 localization and increased endothelial permeability in vitro and in vivo (Osada et al., 2011).
Taken together, these data indicate that the BM composition participates in BBB regulation; however, the mechanisms are only poorly understood and need to be better addressed.
Hypoxia and the BBB
Mechanisms of hypoxic BBB disruption
Hypoxia, when the oxygen demand of tissues is not met, acts as an initial trigger for pathophysiological changes at the BBB such as altered distribution of water and ions, inflammatory events and oxidative stress, oedema formation, infiltration of peripheral immune cells and leakage of blood proteins into the brain. In addition, hypoxia induces major alterations in vessel structure as it stimulates proliferation of ECs leading to formation of new blood vessels and furthermore promotes activation and proliferation of astrocytes (reviewed by Ogunshola and Al-Ahmad, 2012; Stanimirovic and Friedman, 2012). A large number of in vivo and in vitro studies have demonstrated that hypoxia is a major stress factor inducing BBB disruption (Schoch et al., 2002; Kaur et al., 2006; Al Ahmad et al., 2009; Lochhead et al., 2010). Regarding the temporal course of hypoxic barrier opening, detailed in vivo studies are rare. Increased BBB permeability to Evans blue was observed in mice 6 h after onset of hypoxia (7% O2; Li et al., 2011). After 24 and 48 h of hypoxic exposure to 8% O2, increased BBB leakage to sodium fluorescein was demonstrated in mice (Schoch et al., 2002; Bauer et al., 2010). Another study by Witt and colleagues demonstrated that exposure to 6% O2 for 1 h with subsequent re-oxygenation in a rat model resulted in a biphasic opening of the BBB within the first hour and again after 6–24 h of re-oxygenation (Witt et al., 2008). For in vitro models, a generalized statement about the course of barrier opening is almost impossible due to different culture systems, cell sources, oxygen concentrations and read-outs. However, decreased endothelial tightness has been observed from the first 30 min of hypoxic exposure for up to 48 h (Abbruscato and Davis, 1999a; Fischer et al., 1999; Yamagata et al., 2004; Fleegal et al., 2005; Kuhlmann et al., 2007; Al Ahmad et al., 2009).
The mechanisms of hypoxic barrier disruption have been studied intensively and it is evident that disruption of the BBB occurs on many different molecular levels. TJ complexes are major targets of hypoxic BBB disruption. At the molecular level, hypoxia modulates protein expression levels and subcellular redistribution of occludin, ZO-1 and claudin-5 (Fischer et al., 2002; Mark and Davis, 2002; Koto et al., 2007; Bauer et al., 2010; Willis et al., 2010). The redistribution critically regulates TJ integrity and is probably mediated via phosphorylation changes at serine, threonine and tyrosine residues and by caveolae-mediated endocytosis that determines their localization at the plasma membrane and interaction with other proteins at the TJ (Luissint et al., 2012). PKC enzymes, myosin light chain kinase, and RhoA regulate TJ protein phosphorylation and contribute to hypoxic or inflammatory barrier disruption (for review, refer to Luissint et al., 2012). Increased transcellular and pinocytic activity of brain ECs represent additional events contributing to augmented barrier permeability during hypoxia (Plateel et al., 1997; Cipolla et al., 2004). Hypoxia-mediated alterations and breakdown of the BM also aggravate BBB opening and are particularly important during ischaemic events (Candelario-Jalil et al., 2009; Stanimirovic and Friedman, 2012). Direct and indirect contribution of pericytes and astrocytes to hypoxic-mediated BBB permeability is discussed below.
HIFs as mediators of the hypoxic response
Hypoxia requires an immediate response of the affected tissues/cells to sustain their function and prevent cell death. The most important tasks of the adaptation process are to decrease energy consumption and increase oxygenation. This is mainly achieved through metabolic adaptation and temporal cell cycle arrest as well as induction of angiogenic and erythropoietic genes to increase oxygen delivery to the hypoxic tissues (Wenger et al., 2005). Different signalling pathways are involved in these events including the unfolded protein response, mammalian target of rapamycin signalling, and HIF-mediated gene regulation (Majmundar et al., 2010). HIFs are considered master regulators of the hypoxic response and are heterodimeric transcription factors composed of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit. Under normoxia, the HIFα subunits are constitutively transcribed but constantly targeted for proteasomal degradation through a cascade of hydroxylation of conserved proline residues via prolyl hydroxylases (PHDs), subsequent recognition by the Von Hippel–Lindau (VHL) protein, ubiquitination and degradation by the proteasome. As oxygen tension drops, the PHD enzymes are inhibited and the lack of hydroxylation results in cytoplasmic stabilization of the α-subunits. After phosphorylation, HIFαs translocate to the nucleus and dimerize with ARNT (also known as HIF-β) and co-activators forming a functional HIF transcription factor (Wenger et al., 2005; Fandrey and Gassmann, 2009). By binding to hypoxia-responsive elements in promoter regions, HIFs induce the expression of target genes involved in cellular adaptation to hypoxic stress-regulating erythropoiesis, angiogenesis, proliferation and cellular metabolism (Ogunshola and Al-Ahmad, 2012; see Figure 2).
Figure 2.
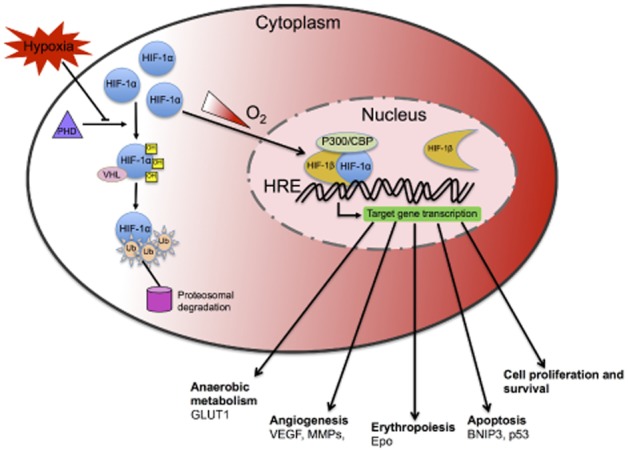
Schematic diagram illustrating the mechanism of HIF-1 regulation under normoxic and hypoxic conditions. HIFs are heterodimeric transcription factors composed of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit. Under normoxia (white gradient), the HIF-1α subunit is constitutively transcribed but constantly targeted degradation through hydroxylation of conserved proline residues by PHDs leading to recognition by VHL protein, ubiquitination and subsequent degradation by proteasome. As oxygen tension drops (red gradient), the PHD enzymes are inhibited and the lack of hydroxylation results in cytoplasmic stabilization of the α-subunits. After phosphorylation, HIF-1α translocates to the nucleus and dimerizes with HIF-1β (also known as ARNT) and co-activators such as p300/CBP forming a functional HIF-1 transcription factor. HIF-1 then binds to hypoxia-responsive elements (HREs) in the promoter regions of its many targets inducing expression of genes involved in cellular adaptation to hypoxic stress by regulating erythropoiesis, angiogenesis, proliferation and cellular metabolism in order to reduce O2 consumption and increase O2 delivery to tissues.
Three different HIFs have been identified, namely, HIF-1,-2 and-3 of which HIF-1 is the most widely studied. More than 100 HIF-1 target genes are known to date (Semenza, 2003). HIF-1 signalling is considered essential for cellular adaptation and survival during hypoxia. Several HIF-1 target genes are neuroprotective and act as pro-survival factors such as erythropoietin and VEGF. Moreover, HIF-1 signalling regulates the expression of several proteins implicated in glycolysis such as phosphofructokinase or enolase-1 and glucose transporter 1 (GLUT1), thus regulating metabolic adaptation to low oxygenation. Besides its positive effects, HIF-1 also accounts for detrimental effects observed during hypoxia/ischaemia by activation of prodeath genes, such as BNIP3, COX2 or p53 stabilization (reviewed in Singh et al., 2012). At present, it is not clear what determines whether the initiated HIF-1-mediated response is protective or detrimental. However, it is likely that the severity of the insult and its duration, as well as cell type-specific differences and paracrine signalling play a critical role. Thus, HIF-1 signalling is a key determinant of functional outcome.
HIF-1 regulates BBB permeability
Growing evidence suggests that HIF-1 could play a pivotal role in the changes that occur at the BBB during hypoxia. Good insights have been obtained from cerebral ischaemia and reperfusion studies as well as in vitro work (reviewed by Ogunshola and Al-Ahmad, 2012). The HIF-1 inhibitors 2-methoxyestradiol and YC-1 reduce oedema formation (directly correlating to BBB permeability) and infarct volumes after ischaemia or ischaemia reperfusion (Yeh et al., 2007; Chen et al., 2008a), whereas rapid DMOG (dimethyloxalylglycine)-induced HIF-1α stabilization increased oedema formation. In both studies, these effects were attributed to modulation of VEGF production (Chen et al., 2008a). Our own in vitro work using brain ECs also suggest that HIF-1 stabilization is directly linked to barrier disruption and that inhibition of HIF-1 can significantly improve barrier stability (Engelhardt et al. unpubl. data). Likewise, HIF-1 inhibition using siRNA in a rat focal ischaemia model showed less BBB disruption and a better outcome for the animals and is correlated with decreased VEGF levels, as well as reduction of caspase-3 and p53 expression (Chen et al., 2009). These studies suggest that HIF-1 stabilization is likely to trigger increased BBB permeability through activation of its multiple target genes and signalling cascade.
Contribution of astrocytes and pericytes to hypoxic BBB regulation
The importance of non-neuronal cells in brain diseases associated with hypoxia has long been neglected. The next section discusses the response of BBB cells to hypoxia/ischaemia and their subsequent effect on BBB function.
Astrocytes
Information on the basal responses of astrocytes to reduced oxygenation is limited and far from complete. We know that astrocytes are more resistant to hypoxia and ischaemia than neurons, which is probably due to their higher capacity to metabolically adapt to hypoxia/ischaemia by using alternative energy sources and switching to anaerobic glycolysis thereby ensuring maintenance of the ATP levels (Schmid-Brunclik et al., 2008; Turner and Adamson, 2011). Astrocytes are activated in vivo (Kaur et al., 2006) and in vitro (Schmid-Brunclik et al., 2008) following hypoxic/ischaemic injury and secrete elevated levels of a large number of factors that can be neurotoxic as well as neuroprotective (reviewed in Trendelenburg and Dirnagl, 2005; Vangeison and Rempe, 2009). Modulation of a variety of proteins suggests a highly complex tempering of barrier status. Indeed, a microarray study recorded more than 1100 hypoxia-responsive genes in human astrocytes with five times more genes up-regulated than suppressed. Notably, many of the up-regulated genes were glycolytic enzymes and angiogenic molecules (Mense et al., 2006). Astrocytes co-cultured with ECs results in mutual up-regulation and preservation of antioxidant enzymatic activity and reduced radical-induced EC injury (lipid peroxidation) compared with endothelial monocultures (Schroeter et al., 1999; Ogunshola and Al-Ahmad, 2012).
Different in vitro studies have shown that astrocyte co-culture or treatment of ECs with ACM improves EC performance and maintenance of barrier function during hypoxic insults (Fischer et al., 2000; Brown et al., 2003; Al Ahmad et al., 2009). Consistent with these observations astrocytes/ACM preserve the junctional localization of TJ proteins, like ZO-1 or claudin-5 (Fischer et al., 2000; Al Ahmad et al., 2011) and hypoxia/glycaemia-dependent down-regulation of the adherens junction protein E-cadherin is partially reversed in the presence of astrocytes (Abbruscato and Davis, 1999b). Media from hypoxic and re-oxygenated astrocytes contain ANG-1 that increases occludin expression and reduces endothelial proliferation thereby stabilizing the vasculature (Song et al., 2002). In addition, co-culture with astrocytes attenuates endothelial caspase-3 activation during hypoxia (Al Ahmad et al., 2009) thereby reducing cell death and caspase-3-mediated TJ disruption (Zehendner et al., 2011).
Astrocytes are the major source of VEGF in the brain and respond to hypoxic stimuli with increased induction and secretion of VEGF that acts as an endothelial survival factor (Chow et al., 2001; Kaur et al., 2006; Mense et al., 2006; Schmid-Brunclik et al., 2008). However, VEGF is also a prominent angiogenic molecule and thus a strong inducer of vascular permeability in vitro and in vivo (Fischer et al., 1999; Schoch et al., 2002). Our group has shown that despite a beneficial effect of astrocyte co-cultures on hypoxic barrier maintenance, inhibition of VEGF signalling (with the VEGF receptor inhibitor SU1498) further improved maintenance of barrier characteristics (Al Ahmad et al., 2011). In agreement with this result, astrocyte-derived VEGF was also shown to drive BBB disruption after hypoxic exposure (Kaur et al., 2006) and in CNS inflammatory disease (Argaw et al., 2012). Thus, the effect of VEGF secretion during insult is multifaceted. VEGF binds and activates two TK receptors, namely, VEGFR1 (also known as FLT1) and VEGFR2 (also known as KDR or FLK1). Hypoxia-induced endothelial hyper-permeability seems to be mediated predominantly by activation of VEGFR1 (Vogel et al., 2007), in agreement with the finding that hypoxia up-regulates both the expression of VEGFR1 and the binding of VEGF to VEGFR1 (Fischer et al., 1999). Excellent reviews on hypoxia-and HIF-1-mediated VEGF permeability have been published (Fan et al., 2009; Nakayama and Berger, 2013). However, it has also been shown that VEGF splicing in the terminal exon results in variants, termed VEGFxxxb (VEGF165b), that act as anti-angiogenic dominant negative splice isoforms (Ladomery et al., 2007; Nowak et al., 2008). Notably, high homology means these anti-angiogenic isoforms may have been mistakenly identified as the more canonical species in many studies. Although the ratio of the b-isoforms to the canonical species could be highly relevant, investigations on the effects of stimuli such as hypoxia on these splice variants are yet to be performed. Astrocytic secretion of MMPs during hypoxia also causes BM reorganization and weakens barrier function. Increased activity of MMP-2, MMP-9 and MMP-13 was detected in hypoxic astrocyte supernatants and treatment of ECs with those supernatants led to MMP-13-dependent delocalization and proteolysis of ZO-1 and disruption of VE-cadherin (Lu et al., 2009).
Astrocytes additionally express various cytokines in response to hypoxic stimulation. For example, IL-1β is a HIF-1 target gene (Zhang et al., 2006) that can activate HIF-1α and VEGF expression in a NFκB-dependent manner in astrocytes and cause down-regulation of the vessel-stabilizing factor SSeCKs (Argaw et al., 2006). Also, large amounts of the monocyte chemoattractant proteins (MCP) MCP-1 and MCP-5 are produced by hypoxic astrocytes in a HIF-1-dependent manner (Mojsilovic-Petrovic et al., 2007). Apart from its main function of recruiting leukocytes at sites of inflammation, MCP-1 was shown to increase the paracellular permeability of endothelial monolayers via TJ redistribution mediated by Rho signalling (Stamatovic et al., 2003) and is involved in formation of vasogenic oedema in vivo (Stamatovic et al., 2005). Clearly, hypoxic responses of astrocytes are critical during injury and disease and particularly affect the stability of the BBB.
Pericytes in hypoxic BBB regulation
Only a few studies have investigated the survival of pericytes after hypoxic and ischaemic insults. Our in vitro data suggest that pericytes have comparable sensitivity to astrocytes. We did not observe any impairment of mitochondrial activity in pericytes exposed for up to 48 h in 0.2% oxygen reflecting no loss of viability, whereas ischaemic conditions reduced mitochondrial function only after 24 h of exposure (Engelhardt et al., unpubl. obs.). Likewise, it was shown that ECs are much more susceptible to ischaemia than astrocytes and pericytes in vitro (Ceruti et al., 2011). In vivo pericytes were observed to migrate away from microvessels in response to traumatic brain injury (TBI; Dore-Duffy et al., 2000) and hypoxic stimuli already 2 h after onset of the insult and preceding any changes in vessel structure (Gonul et al., 2002). Furthermore, a reduction of pericyte-to-EC ratio was reported after 1 week of hypobaric hypoxia (Dore-Duffy et al., 2007). Although the cell status during these events remains unclear, the changes occurring during pathologies associated with increased BBB permeability suggest that pericyte loss per se could contribute to augmented barrier leakage. Indeed, in vitro models have shown that the presence of pericytes protects endothelial monolayers from hypoxic barrier disruption (Hayashi et al., 2004; Al Ahmad et al., 2009), particularly during prolonged and severe oxygen deprivation, by maintaining TJ protein localization and reducing endothelial caspase-3 activation (Al Ahmad et al., 2009; 20112009,2011). Interestingly, under severe conditions, co-culture of pericytes with ECs maintained TEER and reduced paracellular flux of labelled substances better than astrocyte co-cultures (Al Ahmad et al., 2009).
Like astrocytes, pericytes respond to hypoxia by up-regulating various growth factors. Park et al. demonstrated that in retinal pericytes ANG-1, but not ANG-2, mRNA is significantly elevated (Park et al., 2003) again suggesting a vessel-stabilizing effect. Interestingly, the induction of ANG-1 could be mimicked through treatment of the pericytes with recombinant VEGF (Park et al., 2003). An in vivo study suggested that hypoxic pericytes rapidly increase VEGF levels within 24 h, whereas astrocytic VEGF production was observed after 4 days (Dore-Duffy et al., 2007). In our studies, we observed different outcomes when inhibiting VEGF signalling using SU1498; inhibition was only beneficial in EC pericyte co-cultures during 1% O2 but not during more severe oxygen deprivation (0.1% O2; Al Ahmad et al., 2009). Although temporal-spatial VEGF-mediated induction of ANG-1 may be a plausible explanation for some of the barrier-stabilizing effects of pericytes during hypoxia, it is unlikely that elevated VEGF expression does not largely contribute to hypoxic barrier disruption, indeed VEGF signalling by other brain cells may also have an effect. Like astrocytes, pericytes also secrete MMPs and their expression is augmented through pro-inflammatory cytokines like TNF-α (Takata et al., 2011). Additionally, pericytes can induce MMP expression in ECs, although the link between pericyte MMP expression and hypoxic barrier disruption is still to be properly demonstrated. Taken together, however, the data indicate that timing, severity and cellular crosstalk is likely to be very complex.
Overall, despite limited information, similar to astrocytes, pericytes appear to fulfil important barrier-stabilizing functions but in response to stress also secrete molecules that can cause BBB remodelling and increased permeability. Thus, more studies are required to understand the multiple roles of this elusive perivascular cell.
Pathophysiology
Hypoxia-mediated BBB dysfunction is associated with many neurological diseases such as stroke, TBI and Alzheimer's disease (AD) (Ballabh et al., 2004). As discussed earlier, paracrine interactions between brain ECs, astrocytes and pericytes play a crucial role in maintaining the BBB. However, their contribution to various brain diseases by secreting factors that modulate the barrier during injury is frequently overlooked. The next section of this review will focus on direct or indirect influences of hypoxic, and particularly HIF-1-mediated BBB cell-specific responses to progression of select diseases namely stroke, TBI and AD (see Figure 3).
Figure 3.
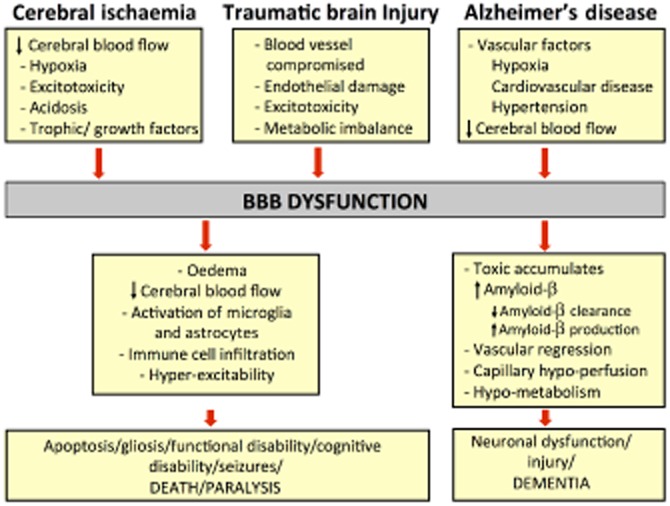
Flow chart of the events leading to BBB disruption and the following events occurring later in stroke, TBI and AD.
Stroke/cerebral ischaemia
Stroke is a major cause of morbidity and mortality across all industrialized countries. The most commonly occurring type of stroke is ischaemic (Bamford et al., 1990), which accounts for approximately 80% of the total incidences. Ischaemic stroke results from occlusion of a major cerebral artery either by thrombosis or an embolus. In either case, this restricts the delivery of substrates to the tissue, particularly oxygen and glucose, which leads to a cascade of ischaemic events (Dirnagl et al., 1999). Within the core of the ischaemic region, deficits in blood flow, decreased energy stores (ATP levels), metabolic failure and ionic imbalance are severe and cell death progresses within minutes. The region surrounding the compromised area, called the ischaemic penumbra, suffers milder insults and is salvageable if blood flow is restored quickly. However, the reperfusion of ischaemic tissue can also lead to secondary damage caused by the rapid re-exposure to oxygen and inflammatory responses in the damaged tissue. Restoration of blood flow also increases the pressure on the damaged ECs and their TJs (Sandoval and Witt, 2008). Additionally, reperfusion results in a fresh supply of leukocytes that translocate into the parenchyma triggering a cascade of cytokine release (Dirnagl et al., 1999). Activated astrocytes and pericytes modulate blood flow, maintain ion homeostasis and contribute to water homeostasis, as well as participate in the inflammatory cascade and release a number of substances that may be either neuroprotective or harmful during ischaemia. This wide variety of responses results in a number of effects including loss of TJ expression and localization, increase in permeability, degradation of BM, loss of integrins, oedema and further inflammation (Sandoval and Witt, 2008; Kwon et al., 2009), all of which contribute to BBB disruption (see Figure 3). Although the trigger for the pathological changes is hard to establish they are probably interdependent. Cerebral ischaemia induced by transient occlusion of middle cerebral artery evokes a marked biphasic opening of the BBB. Early opening of the BBB takes place within the first half hour of reperfusion and is followed by partial closing. This initial acute opening is described as a ‘haemodynamic’ BBB opening resulting in cytotoxic oedema (reviewed by Nagy et al., 1979; Kuroiwa et al., 1985). Subsequently, a second delayed, but progressive, opening occurs between 24 and 48 h post-reperfusion and results in vasogenic oedema (Huang et al., 1999). Notably, taking advantage of this later progressive BBB opening may aid the delivery of low permeability neuroprotective therapeutic agents.
Brain ECs maintain impermeability of the BBB largely through their TJs. Notably, ZO-1 and occludin expression were reduced in a microsphere-induced cerebral embolism model of cerebral ischaemia (Kago et al., 2003). Although not many studies have evaluated TJ changes post-ischaemia, some reports have demonstrated changes in BBB cell contacts partly due to BM degradation. Astrocyte end-feet are well known to swell post-ischaemia (Kimelberg, 2005) and degradation of BM also causes their detachment from the vessel wall leading to increased permeability (Kwon et al., 2009). In addition to swelling, surviving astrocytes in the vicinity of the ischaemic damage also undergo a process of hypertrophy referred to as reactive astrogliosis. Similarly, pericytes have also been shown to migrate away from vessels in animal models of ischaemia by breaking through the BM that ensheaths them (Dore-Duffy et al., 2000; Melgar et al., 2006). This indicates that in addition to changes in BBB permeability induced by TJ alterations, contact with BBB-supportive cells is also modified and probably enhances BBB disruption. It is reported that ischaemia followed by reperfusion in rats results in BBB disruption and breakdown of BM collagen IV (Hamann et al., 2002; Scholler et al., 2007). Likewise, ischaemic/reperfusion injury also results in a decrease in laminin and fibronectin (Hamann et al., 1995). Other BM proteins, such as agrin and perlecan are also degraded after ischaemia (Fukuda et al., 2004; Baumann et al., 2009). Fukuda and group were the first to provide direct evidence that active proteases, which are known to degrade the BM are generated in ischaemic cerebral tissue (Fukuda et al., 2004). The most widely studied proteases, MMPs, are released by astrocytes (Zhang et al., 2004) and microglia (Del Zoppo et al., 2007) post-ischaemic injury. MMP-9 and-2, in particular, are major contributors of ischaemic pathology. Many groups have demonstrated the detrimental role of MMP-9 in ischaemic models by using inhibitors of MMP-9 or MMP-9 knockout animals and reported reduction in BBB damage, brain water content, infarct size and neurological deficit (Romanic et al., 1998; Asahi et al., 2001; Jiang et al., 2001; Park et al., 2009; Cui et al., 2012). Therefore, inhibiting MMPs may be highly beneficial for treating cerebral ischaemia, as reviewed elsewhere (Cunningham et al., 2005; Morancho et al., 2010).
BBB disruption after ischaemia increases permeability to macromolecules allowing fluid movement from intravascular to extravascular spaces and leading to vasogenic oedema (Sandoval and Witt, 2008). Aquaporins (AQPs) are highly permeable water channels that play a crucial role in fluid balance. AQP4 in particular participates in the maintenance of brain water homeostasis and is highly concentrated on astrocyte end-feet (Nielsen et al., 1997). The role of AQP4 has been explored in various models of ischaemic stroke where it contributes to the formation of cerebral oedema. In disease models, such as acute cerebral ischaemia and water intoxication (Manley et al., 2000), water moves into the cell resulting in cytotoxic brain oedema. Deletion of AQP4 was shown to prevent cellular water uptake in these models, as demonstrated by reduced brain water content, lower intracranial pressure values, infarct size and lesion volume (Manley et al., 2000; Zeng et al., 2012). In chronic ischaemia or TBI models that result in vasogenic oedema due to BBB disruption, deletion of AQP4 exacerbated cerebral oedema formation assessed by water content measurements and intracranial pressure (Papadopoulos et al., 2004). Based on these findings it can be concluded that AQP4 plays a differential role in modulating brain water homeostasis and is dependent on the duration and type of insult.
VEGF, one of the most important factors induced by hypoxia, performs a major role in driving BBB disruption leading to oedema formation in the acute phase following ischaemia (Zhang et al., 2000; Chi et al., 2007; 20082007,2008). Elevated VEGF expression appears as early as 1 h after the onset of cerebral ischaemia (Abumiya et al., 1999; Plate et al., 1999b). Indeed, early administration of VEGF increases BBB permeability, increases infarct size and worsens neurological outcome following an ischaemic insult (Zhang et al., 2000). Similarly, inhibition of endogenous VEGF in the acute phase reduces BBB permeability (Mayhan, 1999; Valable et al., 2005; Yeh et al., 2007). This is in line with findings that suggest VEGF is not only an angiogenic factor but also a permeability factor (Plate, 1999a). Apart from its role at the vasculature, there are several studies that have also examined the effect of VEGF on cerebral infarct size. Van Bruggen et al. reported that antagonism of VEGF reduces ischaemia reperfusion-related brain oedema and injury in mice (van Bruggen et al., 1999). Similarly, VEGF administered i.v. increased infarct size when given early after ischaemia in rats but, in contrast, improved neurological function when given at later stages 2 days post-ischaemia (Zhang et al., 2000). These studies suggest that despite enhancing angiogenesis and reducing neurological deficits during late-stage stroke recovery, inhibition of VEGF at the acute stage of injury may reduce BBB compromise. The negative side of VEGF could perhaps be achieved partly by counteracting its permeabilizing effect in the acute phase of ischaemia. Indeed, systemic adenoviral gene delivery of ANG-1, a known anti-permeability factor that maintains and stabilizes mature vessels by promoting interaction between ECs and surrounding support cells (Hori et al., 2004), causes resistance to vascular leakage induced by VEGF in the brain after cerebral ischaemia (Zhang et al., 2002). Conversely, ANG-2 that leads to de-stabilization of vessels and dissociation of pericytes, is up-regulated by hypoxia and cytokines including VEGF (Mandriota and Pepper, 1998; Valable et al., 2005). Thus, the co-ordination of VEGF and the ANG is crucial for maintaining the integrity of existing vessels and modulating growing vessels for better outcome following ischaemia.
In summary, the exact mechanisms causing brain vascular disruption are hard to establish but factors like VEGF and MMPs released from BBB-supportive cells are likely to play an instrumental role. Further confirmation of these conclusions using cell-specific conditional knockout animal experiments is necessary for better understanding of the role of these cells at the BBB post-ischaemia.
Traumatic brain injury
TBI results from an outside force causing instant mechanical disruption of brain tissue and delayed pathogenic events that collectively mediate widespread neurodegeneration (reviewed by Gaetz, 2004). Subsequently, the traumatized brain is highly susceptible to secondary brain injuries, which can be caused by hypoxia (Hellewell et al., 2010) and seizures (Bao et al., 2011). Reports from animal model studies and significant clinical data both suggest a central role for vascular integrity in TBI outcome, especially BBB breakdown which can last from minutes to several days to weeks, or even years after the acute event (Unterberg et al., 2004; Hawkins and Davis, 2005; Abbott et al., 2006; Tomkins et al., 2008). This kind of disruption might also influence the progression of long-term TBI complications, such as AD, which include delayed neuronal dysfunction and cell death (see Figure 3).
Studies of animal models of TBI have also demonstrated a biphasic increase in BBB permeability to albumin and other high molecular weight proteins peaking at 4 to 6 h as well as 2–3 days after injury (Başkaya et al., 1997; Hicks et al., 1997). The first peak in BBB permeability generally overlaps with increased production of a number of secretory factors that include chemokines, cytokines, MMPs and VEGF, which contribute to BBB dysfunction and also invasion of neutrophils. A thorough review on molecular pathogenesis of BBB breakdown in TBI has been recently published (Nag et al., 2011).
Chemokines not only play a key role in post-traumatic recruitment of leukocytes, but may also modify the permeability of the BBB. After injury recruitment of leukocytes into the brain parenchyma releases a number of inflammatory cytokines that worsen the outcome of the disease state. For example, as stated earlier MCP-1 is up-regulated post-hypoxia and very recently, Semple et al. reported its important role in reducing lesion size and secondary damage and improving functional outcome in mice following TBI (Semple et al., 2010). This improvement may be attributed to decreased leukocyte accumulation resulting in an environment more advantageous for neuronal survival. Increased levels of cytokines are correlated to poorer clinical outcomes. TNF-α mRNA and protein is elevated acutely after experimental TBI (Bermpohl et al., 2007) as well as in clinical settings (Csuka et al., 1999) and is known to exacerbate BBB disruption post-TBI (Bermpohl et al., 2007). Similarly, a rapid increase of IL-1β is observed early enough after experimental TBI (Wang and Shuaib, 2002). Patel et al., using intracerebral or intraventricular administration of exogenous IL-1β, demonstrated significant exacerbation of injury (Patel et al., 2003). In addition, transgenic mice, in which the IL-1β is overexpressed in astrocytes alone, were observed to have a leaky BBB (Shaftel et al., 2007). Thus, BBB dysfunction mediated via cytokines released from BBB cells can worsen injury outcome in the acute stages and may be hypoxia-dependent. In contrast, the mechanisms involved in the delayed (second phase) opening of the BBB are presently unclear and require further investigation.
Although the role of VEGF and its receptors has been studied extensively in ischaemia over the last years, only a few groups have performed studies elucidating the role of this molecule post-TBI. Using different models of TBI, it was reported that astrocytes as well as inflammatory cells react to injury by increasing VEGF expression (Papavassiliou et al., 1997; Sköld et al., 2002). In contrast, pericytes were shown to undergo cell death post-TBI using TUNEL assay (Dore-Duffy et al., 2007). Morgan et al. demonstrated a substantial increase in angiogenesis, based on BrdU-positive nuclei within the endothelium post-TBI via increased expression of VEGF and its receptor (Morgan et al., 2007). This induction of angiogenesis could be mediated by the up-regulation of astrocytic VEGF, which in turn increases both astrocyte proliferation and facilitates the expression of other growth factors, which support late-stage recovery (Krum et al., 2008). Corticosteroids that are usually used for treating early vasogenic oedema after acute BBB compromise have been demonstrated to stabilize the BBB by decreasing the expression of VEGF (Kim et al., 2008). Again, the data suggest that during disease early VEGF release from glial cells can be harmful to BBB function and further worsen the outcome post-TBI. Similarly, MMPs, in particular MMP-2, MMP-3 and MMP-9, are up-regulated following TBI (Shigemori et al., 2006; Vilalta et al., 2008; Hayashi et al., 2010). As found in cerebral ischaemia, genetic deletion of MMP-9 or pharmacological inhibition of the activity of MMPs using TIMPs significantly reduced the extent of tissue damage and improved functional outcome in murine models of TBI (Wang et al., 2000). In addition, mice overexpressing the human TIMP1 gene subjected to TBI showed reduced levels of MMP-9 synthesis and a less leaky vasculature accompanied by decreased brain tissue damage compared with control animals (Tejima et al., 2009). All these observations indicate that secretion of VEGF and MMPs by BBB cells could indeed represent a potential target for pharmacological intervention in TBI and ischaemia. Nonetheless, it should be emphasized that as these factors also play an important role in the repair process at later stages, delayed inhibition of their activity may actually have adverse therapeutic effects (Nag et al., 1997; Zhao et al., 2006).
AQP4 expression is markedly modified in both experimental and clinical TBI (Manley et al., 2000; Papadopoulos and Verkman, 2007; Tang et al., 2010; Fukuda and Badaut, 2012). Initial studies implied that induction of AQPs in a model of TBI promoted oedema formation (Manley et al., 2000) and that therapeutic inhibition of AQP4 could be beneficial in controlling oedema (Taya et al., 2008). Indeed, Fukuda et al. used siRNA against AQP4 to demonstrate improvements that were associated with decreased oedema formation, increased microglial activation and decreased BBB disruption post-TBI (Fukuda et al., 2013). However, it subsequently became apparent that the modifications in AQP4 expression are dependent on the type of oedema and its regional distinction (Sun et al., 2003; Papadopoulos et al., 2004). For instance, in an ischaemic model that results in cytotoxic oedema, inhibition of AQP4 expression was associated with decreased oedema, reduced infarct area and an improvement in functional outcome (Fazzina et al., 2010). In contrast, in a cold lesion injury model of vasogenic brain oedema, AQP4-deficient mice had a markedly worse clinical outcome (Papadopoulos et al., 2004). These findings suggest that AQP4 is mainly essential for the clearance of vasogenic oedema but its other effects depend on the type of injury and the time point being measured.
Finally, BBB disruption also triggers a chain of events that not only affects vascular cells but also neuronal cells by causing epilepsy (Tomkins et al., 2007; 20082007,2008). This theory seems plausible since the studies cited suggest that damage to the microvasculature might result in the extravasation of serum proteins such as albumin, which leads to the activation of astrocytes as the first step in the epileptogenic process. This interaction suggests a key functional role of activated astrocytes in neuronal excitability. Hardly any work has been performed on the specific role of pericytes in TBI, although endothelin-1-induced pericyte-mediated vasoconstriction was recently reported (Dore-Duffy et al., 2011). Many more studies on cell-specific responses to TBI are surely warranted.
Alzheimer's disease
AD is a progressive and irreversible neurodegenerative disorder characterized by the accumulation of amyloid β-peptide (Aβ) in the CNS, the presence of hyper-phosphorylated tau filaments and cerebrovascular changes that lead to cerebral amyloid angiopathy (Greenberg et al., 2004). AD is another neurodegenerative disease that is characterized by hypoxia, a state that is believed to only aggravate disease progression (Ogunshola and Antoniou, 2009; Peers et al., 2009; Figure 3).
At present, the cellular and molecular basis by which systemic hypoxia influences AD are not completely understood. However, the emerging evidence indicates that prolonged hypoxia induces formation of Aβ that accumulates over years and probably contributes to vascular alterations (Selkoe, 2001). Hypoxia induces Aβ processing through mechanisms that increase the activity of two enzymes crucial for Aβ formation, β-secretase and γ-secretase (Sun et al., 2006; Zhang et al., 2007; Li et al., 2009). HIF-1α mediates a transcriptional increase in β-secretase expression (Zhang et al., 2007) and presenilin-1 levels – presenilin being the major protein of the γ-secretase complex (Pluta, 2007). In addition, hypoxia also promotes down-regulation of neprilysin, an Aβ-degrading enzyme (Nalivaevaa et al., 2004), which can lead to alterations in the expression of vascular-specific genes in brain ECs (Wu et al., 2005). All of these effects can be considered pro-amyloidogenic, that is, would predispose to increased Aβ levels either through increased production or reduced degradation. Recently, Wilcock and colleagues used a transgenic mouse model to demonstrate that during progression of an AD-related pathology the numbers of astrocytic processes in apparent contact with cerebral vasculature were reduced. Furthermore, amyloid accumulation caused significant reductions in AQP4 and potassium channels associated with cerebral vessels in both the mouse model and individuals with AD (Wilcock et al., 2009). This observation seems to agree with studies performed in cerebral ischaemia or TBI models during the vasogenic phase suggesting that the presence of AQP4 is beneficial for functional outcome. Clearly, AQ4 plays a significant role in controlling brain water homeostasis; however, many more studies are required to define how its modulation will affect functional outcome of disease progression in different pathologies. An excellent review on aquaporins and AD has been published (Foglio and Rodella, 2010). Emerging evidence suggests that the Aβ accumulation that causes vascular alterations ultimately leads to hypo-perfusion (Sagare et al., 2012). Pericytes could probably influence this hypo-perfusion in conditions of severe ischaemia and oxidative stress, as they contract not only during the hypoxic insult but remain contracted even after reperfusion (Yemisci et al., 2009). Hypoxia also promotes phosphorylation of tau through the MAPK pathway (Fang et al., 2010). Tau is a microtubule-associated protein that plays a major role in stabilizing microtubules, found mainly in neurons but has also been shown in low levels in astrocytes (Lee et al., 2001). Aβ and/or hypo-perfusion can induce hyper-phosphorylation of tau, leading to the protein structural changes found in AD patients, which affects its binding with tubulin and causes destabilization of the cytoskeleton (Michaelis et al., 2002). This leads to formation of neurofibrillary tangles and suggests that tau pathology develops secondary to Aβ injury and can be modulated via hypoxia-mediated signalling.
Alterations in vascular permeability and BBB disruption are detected in the brains of AD patients (Claudio, 1996). However, deposition of Aβ aggregates in cerebrovasculature and the brain is less understood and the mechanisms that cause changes in permeability are not clear. The BBB regulates the entry of plasma-derived Aβ into the brain and clears brain-derived Aβ through the receptor for advanced glycation end-products (RAGE) and low-density lipoprotein receptor-related protein respectively (Shibata et al., 2000; Deane et al., 2003). Previous reports showed increased levels of free Aβ in plasma of AD patients or mouse models (Kawarabayashi et al., 2001; Zhou et al., 2012). Through these studies, one can speculate that Aβ may disrupt the TJs of BBB via interaction with RAGE as a specific mediator. In agreement with this hypothesis, a recent study performed by Kook et al. using cultured ECs showed Aβ-induced structural alterations and reduction in protein levels of ZO-1 as well as increased permeability. Furthermore, a neutralizing antibody against the extracellular domain of RAGE effectively blocked Aβ-induced alterations in ZO-1 distribution suggesting that Aβ–RAGE interactions are critical for TJ integrity (Kook et al., 2012). It is well known that calcium influx is induced by Aβ in the cells (Kawahara et al., 2000) and increased intracellular calcium also leads to TJ alterations as well as inducing MMP expression (Stuart et al., 1996; Rosenberg, 2009; Kook et al., 2012). Thus, MMP activation could further accelerate degradation of the BM as observed post-ischaemia and worsen the outcome of AD.
Thus, although the amyloid hypothesis for the pathogenesis of AD suggests this peptide initiates a cascade of events leading to neuronal injury and loss and eventually dementia, from the studies mentioned above, vascular alteration may significantly contribute to the disease as well. Recently, Zlokovic presented an alternative, two-step vascular hypothesis as discussed in the review (Zlokovic, 2011). The hypothesis suggests that in the first step, the primary damage to brain vasculature initiates a non-amyloidogenic pathway of neuronal dysfunction and injury, which is mediated by BBB dysfunction. This is correlated with leakage and secretion of multiple toxic molecules and/or reduced blood flow, which causes multiple focal ischaemic or hypoxic injuries. In the second step, BBB dysfunction also leads to increased Aβ generation and impairment of Aβ clearance. Both these processes contribute to accumulation of Aβ species in the brain and toxicity. It is clear from this hypothesis that phenomena like vascular regression and hypo-metabolism occur secondary to vascular and/or Aβ injury but also that BBB alterations probably make a sizeable contribution to disease progression. Temporal-spatial BBB disruption in senescence-accelerated mouse prone 8 mice, considered by some researchers as a model of AD, was recently reported (Del Valle et al., 2009). However, the role of the perivascular cells, astrocytes and pericytes, and the mechanisms that caused the changes in permeability are not clear and require further investigation.
Vascular regression is a phenomenon observed in AD patients (Zlokovic, 2005; Grammas, 2011) as well as in APP transgenic mice – a model for late-stage AD (Paris et al., 2004). For example, reduction in brain capillary length in the hippocampus correlated well with increased clinical dementia rating scores in AD patients (Bouras et al., 2006). The reason for this phenomenon is unclear but could be explained partly by the anti-angiogenic activity of Aβ (Thomas et al., 1996). Aβ counteracts the pro-angiogenic effects of VEGF and bFGF in ECs by sequestering VEGF in the plaques (Yang et al., 2004). Reduced angiogenic effects in AD are also caused by TGF-β1 (Tesseur and Wyss-Coray, 2006). Transgenic overexpression of TGF-β1 in astrocytes stimulates Aβ deposition in brain vessels, along with alterations in regional cerebral blood flow and AD-like vascular degeneration and brain metabolic activity. Furthermore, elevated levels of TGF-β1 in human AD correlate with increased Aβ deposition in brain vessels (Wyss-Coray et al., 2001). However, it is noteworthy that TGF-β1 can also be neuroprotective and promote Aβ clearance as shown in microglia (Wyss-Coray et al., 2001). These findings raise an interesting question of whether vascular regression and/or degeneration in AD results from unsuccessful vascular repair and/or remodelling.
Another phenomenon that occurs during AD progression that may be modulated by BBB cells is hypo-metabolism. Neurons are incapable of synthesizing or storing glucose and are dependent on glucose transport across the BBB, a process that is mediated by glucose transporters (GLUTs). GLUT1 is also located on cerebrovascular ECs and astrocytes (Vannucci et al., 1997). In AD, dysfunctional cerebral ECs express less GLUT1 as well as HIF-1, a major regulator of GLUT1, thereby reducing glucose uptake in the brain (Liu et al., 2008). Furthermore, studies using 18F-fluorodeoxyglucose PET have identified reductions in glucose uptake in individuals with a high risk of dementia (Herholz, 2010). Decreased glucose uptake across the BBB, as seen by PET, may also precede brain atrophy (Herholz, 2010). This suggests that hypo-metabolism due to BBB dysfunction is a contributing factor for disease progression and occurs at a later stage after vascular and/or Aβ injury. Whether swelling and retraction of astrocyte end-feet inhibits glucose transport to neurons and thereby facilitates neuronal hypo-metabolism needs further study.
It is clear that many more studies are needed to fully understand the effect of hypoxia-mediated changes on neurodegenerative pathologies with respect to BBB function and contribution of astrocytes and pericytes. And the question remains, are we missing important mechanistic roles of the BBB under pathophysiological conditions by studying the nervous system in isolation from the influence of the vascular system? The likely answer is yes. Thus, more research based on the BBB and the NVU as a whole is warranted to provide important insights in the future.
Pharmacology
HIF modulators as therapeutic targets
The important role for HIF-1 in the mediation of the adaptive processes to hypoxia means that it could potentially represent an important target to prevent cellular damage during disease progression. Targeting HIF-1 now represents a potential therapeutic strategy in numerous physiological including myocardial ischaemia and cerebrovascular diseases. In this regard, a wide variety of PHD inhibitors that cause stabilization of HIF-1 have been developed as erythropoiesis stimulating agents and neuroprotective drugs. Additionally, activity relating to development of HIF-1 inhibitors to prevent solid tumour angiogenesis has also exponentially increased in the last years but may also be useful to prevent BBB alterations during injury conditions. Some promising advances in these areas are discussed below.
HIF stabilizers
The therapeutic potential of development and use of small molecule HIF stabilizers to improve cell survival after injury is gaining popularity in many different fields. In particular PHDs, the enzymes that regulate HIF-1 stabilization, are now being recognized as important targets for future medical intervention. PHD enzymes require iron and 2-oxyglutarate (2-OG) in order to catalyse HIF prolyl hydroxylation (Jaakkola et al., 2001). hus iron chelators and competitive inhibitors impair the activity of PHD enzymes and other iron-dependent enzymes. deferoxamine, an iron chelator, and cobalt chloride (CoCl2), a competitive inhibitor of iron, are routinely used both in vitro and in vivo to inhibit PHD enzyme activity and thus stabilize HIF. Both seem to be protective in preconditioning preclinical models of cerebral ischaemia (Prass et al., 2002; Siddiq et al., 2005; Aminova et al., 2008; Jones et al., 2008). Indeed, in vivo high concentrations of iron, stored in the cytoplasmic protein ferritin, are released during ischaemia (Harten et al., 2010) and could lead to radical-mediated damage of cellular components (Sorond and Ratan, 2000). Thus, sequestration of iron in general may be beneficial in certain injuries as well as making a major contribution to the neuroprotective action of iron chelators such as CoCl2. The 2-OG analogues L-mimosine, DMOG and 3,4-dihydroxybenzoate can also be used to inhibit PHD enzymes (reviewed by Harten et al., 2010). Some small-molecule activators have also been identified. A potent and effective activator of the HIF pathway is tilorone. Tilorone appears to be able to cross the BBB, stabilize HIF-1α protein and confer significant resistance to stroke and spinal cord injury by an as yet unknown mechanism (Ratan et al., 2008). However, as its use is associated with the accumulation glycosaminoglycans, its future use in humans is debatable (Harten et al., 2010). Finally, the race is on for development of novel PHD inhibitors, particularly for the treatment of ischaemic diseases such as stroke, and a number of compounds has been developed by Fibrogen (reviewed by Harten et al., 2010). A selection of the most promising PHD inhibitors currently being considered for therapeutic strategies has recently been reviewed (Nagel et al., 2010; Karuppagounder and Ratan, 2012). However, it must be emphasized that, although HIF stabilizers mimic the neuroprotective effects of preconditioning in both in vitro and in vivo ischaemic models, the use of PHD inhibition as a post-injury treatment remains somewhat controversial. In mice, post-ischaemic PHD inhibition offered less protection than pre-ischaemic treatments (Baranova et al., 2007) and early activation of HIF-1α using DMOG accentuated brain oedema and BBB disruption compared with ischaemia alone (Chen et al., 2008b). Importantly, in both cases, these effects were attributed to increased VEGF levels with astrocytes being the most likely culprits (Vangeison et al., 2008).
HIF inhibitors
Although identification of agents that specifically increase hydroxylation activity of PHDs – and thus HIF-1 degradation – is relatively unexplored, the fact that HIF-1 induction is closely linked to BBB permeability means putative inhibitors may have a relevant application preventing barrier changes associated with hypoxic and/or ischaemic-induced injury progression. Indeed, in the cancer field, targeting of HIF-1 in hypoxic cells is an attractive therapeutic strategy that is already being actively pursued, since HIF-1 is tightly linked to the metastatic potential of many tumours, treatment resistance and poor prognosis (Hu et al., 2012; Meijer et al., 2012; Xia et al., 2012). To date, a number of molecules that inhibit the HIF-1 pathway have been identified predominantly using cell-based screening systems and research of HIF-1 activity in cancer cell lines. Supraphysiological supplementation of iron and ascorbate enhances PHD activity and results in HIF-1 degradation (Harten et al., 2010) and has already been used in the clinics. In addition, both are considered relatively safe, inexpensive and readily available. 2-OG and its derivatives, the rate-limiting co-substrate for PHDs, stimulate PHD activity and reduce basal HIF-1α protein levels suggesting they may also have a therapeutic role (Ban et al., 2011). Moreover, targeted screening recently identified a compound, KRH102053, as a PHD2 activator and showed it reduced levels of HIF-1 and its downstream target genes, leading to inhibition of hypoxia-induced responses including metastasis and glucose metabolism in vitro (Choi et al., 2008). A number of other inhibitors have also been developed – for excellent overviews of putative HIF-1 inhibitors in development as well as clinical and preclinical trials see Ban et al. (2011); Xia et al. (2012). Although various small molecules have been developed as HIF-1 inhibitors, the mechanisms by which they work are still unclear in many cases. The fact that none of them seem to exclusively target HIF-1α signalling suggests they can also have multiple off-target effects. Perhaps an exception is EZN-2968, an RNA antagonist that targets HIF-1α mRNA and thereby directly inhibits HIF-1α expression (Greenberger et al., 2008). Tumour reduction was found in nude mice implanted with DU145 human prostate cancer cells treated with EZN-2968 and a phase I clinical study in patients with advanced malignancies revealed that EZN-2968 was well tolerated (Ban et al., 2011). EZN-2968 is currently discussed as the best potential for specific HIF-1α inhibition and promising results of the clinical studies are expected (Ban et al., 2011).
In conclusion, the future looks bright for the use of HIF inhibitors as therapeutics, but overall, it is apparent that future design of more specific agents is required for better drugs to be developed. Perhaps such drugs will also be useful to prevent BBB disruption particularly during the acute phase after injury in the future.
Targeted BBB cell-specific treatment
The major focus of therapeutic targets has always been neuronal mechanisms of injury. Notably, attention to dysfunction or loss of non-neuronal cell types, that is, the cells that maintain the neuronal homeostatic environment, are avenues not well explored even though they may significantly increase the chances of success. As discussed above, HIF stabilization, and/or PHD inhibition, compromises barrier stability and as such targeting HIF-1 may be a way to maintain barrier stability during injury. Using therapeutic targets that activate a broad programme of genes in different cell types, as discussed above, may stimulate biological effects better than the separate application of growth factors or other repair proteins (reviewed by (Ratan et al., 2007), especially in this case as an endogenous programme of adaptation is augmented. However, the wide range of HIF targets could have the caveat that modulation of multiple processes could potentially have a negative outcome on whole body physiology. For example, clinical syndromes associated with excessive activation of the HIF pathway exist (Gassmann et al., 2003) and whereas some studies indicate that HIF-1 is neuroprotective, others demonstrate negative outcomes. Clearly, the effects on the NVU in particular must be better understood to ascertain the general widespread application of such therapeutics. Since different cell types of the BBB and CNS show diverse and sometimes opposing responses to HIF-1 modulation, targeting the pathway in a cell-specific manner will circumvent adverse side effects that can be expected of general HIF-1 modulators. Cell-specific therapeutics (e.g. endothelial-, astrocyte-or pericyte-specific drugs) may also result in more efficacious treatment and better tailoring to the injury in question. Targeting and augmenting vascular EC function directly will provide the significant advantage that such drugs would not be required to cross the BBB, that is, be easier targets than current neuron-directed research. Thus, detailed knowledge of cell-specific responses during injury is required to support development of more specific therapeutic agents. Additionally, in-depth knowledge of the side effects of long-term or high-dose treatments must be well defined prior to their clinical implementation. It must also be emphasized that the consequence of increased levels of HIF-1 in different cell types is highly divergent and dependent on the type of injury. Indeed, it is now becoming clear that the PHDs may also have specific endogenous substrates as well as divergent cell-specific roles (Smirnova et al., 2012). Thus, an additional challenge will be whether development of isoform-specific inhibitors will be more efficacious and safe than a more global approach. Clearly, BBB cell-specific responses and their contribution to injury progression are instrumental factors that need to be carefully considered, and the balance of HIF-1 neuroprotective effects with the putative negative outcome on BBB function must be weighed before implementing any treatment strategy.
Conclusion
Overall, hypoxia and HIF-1 significantly contribute to BBB dysfunction in various neurological diseases. The influence of BBB cell specificity to outcome remains very unclear but deserves significantly more investigation since this knowledge will enable development of more effective therapeutic strategies to combat disease progression. Clearly, balancing HIF-1 beneficial and deleterious effects with cell-specific responses, disease profiles, and windows of opportunity will be very difficult.
Conflict of interest
No conflict of interest.
Abbreviations
- BBB
blood–brain barrier
- HIF-1
hypoxia-inducible factor-1
- TJ
tight junction
- ECs
endothelial cells
- TEER
transendothelial electrical resistance
References
- Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abbruscato TJ, Davis TP. Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J Pharmacol Exp Ther. 1999a;289:668–675. [PubMed] [Google Scholar]
- Abbruscato TJ, Davis TP. Protein expression of brain endothelial cell E-cadherin after hypoxia/aglycemia: influence of astrocyte contact. Brain Res. 1999b;842:277–286. doi: 10.1016/s0006-8993(99)01778-3. [DOI] [PubMed] [Google Scholar]
- Abumiya T, Lucero J, Heo JH, Tagaya M, Koziol JA, Copeland BR, et al. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–1050. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Al Ahmad A, Gassmann M, Ogunshola OO. Maintaining blood-brain barrier integrity: pericytes perform better than astrocytes during prolonged oxygen deprivation. J Cell Physiol. 2009;218:612–622. doi: 10.1002/jcp.21638. [DOI] [PubMed] [Google Scholar]
- Al Ahmad A, Taboada CB, Gassmann M, Ogunshola OO. Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab. 2011;31:693–705. doi: 10.1038/jcbfm.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Neuroscience: glia –more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Aminova LR, Siddiq A, Ratan RR. Antioxidants, HIF prolyl hydroxylase inhibitors or short interfering RNAs to BNIP3 or PUMA, can prevent prodeath effects of the transcriptional activator, HIF-1alpha, in a mouse hippocampal neuronal line. Antioxid Redox Signal. 2008;10:1989–1998. doi: 10.1089/ars.2008.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli-Orlidge A, Saunders KB, Smith SR, D'Amore PA. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006;177:5574–5584. doi: 10.4049/jimmunol.177.8.5574. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. 2011;71:1018–1039. doi: 10.1002/dneu.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Bamford J, Sandercock P, Dennis M, Burn J, Warlow C. A prospective study of acute cerebrovascular disease in the community: the Oxfordshire Community Stroke Project – 1981–86. 2. Incidence, case fatality rates and overall outcome at one year of cerebral infarction, primary intracerebral and subarachnoid haem. J Neurol Neurosurg Psychiatry. 1990;53:16–22. doi: 10.1136/jnnp.53.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban HS, Uto Y, Nakamura H. Hypoxia-inducible factor inhibitors: a survey of recent patented compounds (2004–2010) Expert Opin Ther Pat. 2011;21:131–146. doi: 10.1517/13543776.2011.547477. [DOI] [PubMed] [Google Scholar]
- Bao Y, Bramlett HM, Atkins CM, Truettner JS, Lotocki G, Alonso OF, et al. Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma. 2011;28:35–42. doi: 10.1089/neu.2010.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Başkaya MK, Rao AM, Doğan A, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett. 1997;226:33–36. doi: 10.1016/s0304-3940(97)00239-5. [DOI] [PubMed] [Google Scholar]
- Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30:837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann E, Preston E, Slinn J, Stanimirovic D. Post-ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and SPARC, and the blood-brain barrier disruption after global cerebral ischemia. Brain Res. 2009;1269:185–197. doi: 10.1016/j.brainres.2009.02.062. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezowski V, Landry C, Dehouck MP, Cecchelli R, Fenart L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Res. 2004;1018:1–9. doi: 10.1016/j.brainres.2004.05.092. [DOI] [PubMed] [Google Scholar]
- Bermpohl D, You Z, Lo EH, Kim H-H, Whalen MJ. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2007;27:1806–1818. doi: 10.1038/sj.jcbfm.9600487. [DOI] [PubMed] [Google Scholar]
- Bouras C, Kövari E, Herrmann FR, Rivara C-B, Bailey TL, Gunten AV, et al. Stereologic analysis of microvascular morphology in the elderly: Alzheimer disease pathology and cognitive status. J Neuropathol Exp Neurol. 2006;65:235–244. doi: 10.1097/01.jnen.0000203077.53080.2c. [DOI] [PubMed] [Google Scholar]
- Brown RC, Mark KS, Egleton RD, Huber JD, Burroughs AR, Davis TP. Protection against hypoxia-induced increase in blood-brain barrier permeability: role of tight junction proteins and NFkappaB. J Cell Sci. 2003;116:693–700. doi: 10.1242/jcs.00264. [DOI] [PubMed] [Google Scholar]
- Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, et al. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Colombo L, Magni G, Viganò F, Boccazzi M, Deli MA, et al. Oxygen-glucose deprivation increases the enzymatic activity and the microvesicle-mediated release of ectonucleotidases in the cells composing the blood-brain barrier. Neurochem Int. 2011;59:259–271. doi: 10.1016/j.neuint.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Chen C, Hu Q, Yan J, Yang X, Shi X, Lei J, et al. Early inhibition of HIF-1alpha with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol Dis. 2009;33:509–517. doi: 10.1016/j.nbd.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Chen W, Jadhav V, Tang J, Zhang JH. HIF-1 alpha inhibition ameliorates neonatal brain damage after hypoxic-ischemic injury. Acta Neurochir Suppl. 2008a;102:395–399. doi: 10.1007/978-3-211-85578-2_77. [DOI] [PubMed] [Google Scholar]
- Chen W, Jadhav V, Tang J, Zhang JH. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008b;31:433–441. doi: 10.1016/j.nbd.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi OZ, Hunter C, Liu X, Weiss HR. Effects of anti-VEGF antibody on blood-brain barrier disruption in focal cerebral ischemia. Exp Neurol. 2007;204:283–287. doi: 10.1016/j.expneurol.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Chi OZ, Hunter C, Liu X, Weiss HR. Effects of deferoxamine on blood–brain barrier disruption and VEGF in focal cerebral ischemia. Neurol Res. 2008;30:288–293. doi: 10.1179/016164107X230135. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Song B-J, Gong Y-D, Gwak WJ, Soh Y. Rapid degradation of hypoxia-inducible factor-1alpha by KRH102053, a new activator of prolyl hydroxylase 2. Br J Pharmacol. 2008;154:114–125. doi: 10.1038/bjp.2008.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J, Ogunshola O, Fan SY, Li Y, Ment LR, Madri JA. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Brain Res Dev Brain Res. 2001;130:123–132. doi: 10.1016/s0165-3806(01)00220-6. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Crete R, Vitullo L, Rix RD. Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front Biosci. 2004;9:777–785. doi: 10.2741/1282. [DOI] [PubMed] [Google Scholar]
- Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol. 1996;91:6–14. doi: 10.1007/s004010050386. [DOI] [PubMed] [Google Scholar]
- Colgan OC, Collins NT, Ferguson G, Murphy RP, Birney YA, Cahill PA, et al. Influence of basolateral condition on the regulation of brain microvascular endothelial tight junction properties and barrier function. Brain Res. 2008;1193:84–92. doi: 10.1016/j.brainres.2007.11.072. [DOI] [PubMed] [Google Scholar]
- Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J Neuroimmunol. 1999;101:211–221. doi: 10.1016/s0165-5728(99)00148-4. [DOI] [PubMed] [Google Scholar]
- Cui J, Chen S, Zhang C, Meng F, Wu W, Hu R, et al. Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol Neurodegener. 2012;7:21. doi: 10.1186/1750-1326-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davson H, Oldendorf WH. Symposium on membrane transport. Transport in the central nervous system. Proc R Soc Med. 1967;60:326–329. [PMC free article] [PubMed] [Google Scholar]
- Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Dehouck MP, Meresse S, Delorme P, Fruchart JC, Cecchelli R. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem. 1990;54:1798–1801. doi: 10.1111/j.1471-4159.1990.tb01236.x. [DOI] [PubMed] [Google Scholar]
- Del Valle J, Duran-Vilaregut J, Manich G, Camins A, Pallàs M, Vilaplana J, et al. Time-course of blood-brain barrier disruption in senescence-accelerated mouse prone 8 (SAMP8) mice. Int J Dev Neurosci. 2009;27:47–52. doi: 10.1016/j.ijdevneu.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dohgu S, Yamauchi A, Takata F, Naito M, Tsuruo T, Higuchi S, et al. Transforming growth factor-beta1 upregulates the tight junction and P-glycoprotein of brain microvascular endothelial cells. Cell Mol Neurobiol. 2004;24:491–497. doi: 10.1023/B:CEMN.0000022776.47302.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, et al. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 2005;1038:208–215. doi: 10.1016/j.brainres.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60:55–69. doi: 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Wang X, Mehedi A, Kreipke CW, Rafols JA. Differential expression of capillary VEGF isoforms following traumatic brain injury. Neurol Res. 2007;29:395–403. doi: 10.1179/016164107X204729. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Wang S, Mehedi A, Katyshev V, Cleary K, Tapper A, et al. Pericyte-mediated vasoconstriction underlies TBI-induced hypoperfusion. Neurol Res. 2011;33:176–186. doi: 10.1179/016164111X12881719352372. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- Fan X, Heijnen CJ, van der Kooij MA, Groenendaal F, Van Bel F. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. 2009;62:99–108. doi: 10.1016/j.brainresrev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Fandrey J, Gassmann M. Oxygen sensing and the activation of the hypoxia inducible factor 1 (HIF-1) – invited article. Adv Exp Med Biol. 2009;648:197–206. doi: 10.1007/978-90-481-2259-2_23. [DOI] [PubMed] [Google Scholar]
- Fang H, Zhang L-F, Meng F-T, Du X, Zhou J-N. Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci Lett. 2010;474:173–177. doi: 10.1016/j.neulet.2010.03.037. [DOI] [PubMed] [Google Scholar]
- Fazzina G, Amorini AM, Marmarou CR, Fukui S, Okuno K, Dunbar JG, et al. The protein kinase C activator phorbol myristate acetate decreases brain edema by aquaporin 4 downregulation after middle cerebral artery occlusion in the rat. J Neurotrauma. 2010;27:453–461. doi: 10.1089/neu.2008.0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Clauss M, Wiesnet M, Renz D, Schaper W, Karliczek GF. Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. Am J Physiol. 1999;276:C812–C820. doi: 10.1152/ajpcell.1999.276.4.C812. [DOI] [PubMed] [Google Scholar]
- Fischer S, Wobben M, Kleinstuck J, Renz D, Schaper W. Effect of astroglial cells on hypoxia-induced permeability in PBMEC cells. Am J Physiol Cell Physiol. 2000;279:C935–C944. doi: 10.1152/ajpcell.2000.279.4.C935. [DOI] [PubMed] [Google Scholar]
- Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63:70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005;289:H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- Foglio E, Rodella LF. Aquaporins and neurodegenerative diseases. Curr Neuropharmacol. 2010;8:112–121. doi: 10.2174/157015910791233150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Badaut J. Aquaporin 4: a player in cerebral edema and neuroinflammation. J Neuroinflammation. 2012;9:279. doi: 10.1186/1742-2094-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Adami A, Pop V, Bellone JA, Coats JS, Hartman RE, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33:1–12. doi: 10.1038/jcbfm.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston Jr LL, Zoppo G. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004;35:998–1004. doi: 10.1161/01.STR.0000119383.76447.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- Gassmann M, Heinicke K, Soliz J, Ogunshola OO. Non-erythroid functions of erythropoietin. Adv Exp Med Biol. 2003;543:323–330. doi: 10.1007/978-1-4419-8997-0_22. [DOI] [PubMed] [Google Scholar]
- Gonul E, Duz B, Kahraman S, Kayali H, Kubar A, Timurkaynak E. Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvasc Res. 2002;64:116–119. doi: 10.1006/mvre.2002.2413. [DOI] [PubMed] [Google Scholar]
- Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflammation. 2011;8:26. doi: 10.1186/1742-2094-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7:3598–3608. doi: 10.1158/1535-7163.MCT-08-0510. [DOI] [PubMed] [Google Scholar]
- Hafny B, Bourre JM, Roux F. Synergistic stimulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities by retinoic acid and astroglial factors in immortalized rat brain microvessel endothelial cells. J Cell Physiol. 1996;167:451–460. doi: 10.1002/(SICI)1097-4652(199606)167:3<451::AID-JCP9>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85:979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- Hamann GF, Okada Y, Fitridge R, Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- Hamann GF, Liebetrau M, Martens H, Burggraf D, Kloss CUA, Bültemeier G, et al. Microvascular basal lamina injury after experimental focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2002;22:526–533. doi: 10.1097/00004647-200205000-00004. [DOI] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Zozulya A, Wegener J, Galla HJ. The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: an in vitro study. Exp Cell Res. 2007;313:1318–1325. doi: 10.1016/j.yexcr.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation Transwell models. J Neurosci Methods. 2011;199:223–229. doi: 10.1016/j.jneumeth.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Nakao S, Nakaoke R, Nakagawa S, Kitagawa N, Niwa M. Effects of hypoxia on endothelial/pericytic co-culture model of the blood-brain barrier. Regul Pept. 2004;123:77–83. doi: 10.1016/j.regpep.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Kaneko Y, Yu S, Bae E, Stahl CE, Kawase T, et al. Quantitative analyses of matrix metalloproteinase activity after traumatic brain injury in adult rats. Brain Res. 2010;1280:177–192. doi: 10.1016/j.brainres.2009.05.040. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. [PubMed] [Google Scholar]
- Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: analysis of axonal injury and glial responses. J Neurotrauma. 2010;27:1997–2010. doi: 10.1089/neu.2009.1245. [DOI] [PubMed] [Google Scholar]
- Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, Wolburg H, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz K. Cerebral glucose metabolism in preclinical and prodromal Alzheimer's disease. Expert Rev Neurother. 2010;10:1667–1673. doi: 10.1586/ern.10.136. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Baldwin SA, Scheff SW. Serum extravasation and cytoskeletal alterations following traumatic brain injury in rats. Comparison of lateral fluid percussion and cortical impact models. Mol Chem Neuropathol. 1997;32:1–16. doi: 10.1007/BF02815164. [DOI] [PubMed] [Google Scholar]
- Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem. 2004;89:503–513. doi: 10.1111/j.1471-4159.2004.02343.x. [DOI] [PubMed] [Google Scholar]
- Hu Y, Liu J, Huang H. Recent agents targeting HIF-1α for cancer therapy. J Cell Biochem. 2012;114:1–28. doi: 10.1002/jcb.24390. [DOI] [PubMed] [Google Scholar]
- Huang ZG, Xue D, Preston E, Karbalai H, Buchan AM. Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci. 1999;26:298–304. doi: 10.1017/s0317167100000421. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- Jiang X, Namura S, Nagata I. Matrix metalloproteinase inhibitor KB-R7785 attenuates brain damage resulting from permanent focal cerebral ischemia in mice. Neurosci Lett. 2001;305:41–44. doi: 10.1016/s0304-3940(01)01800-6. [DOI] [PubMed] [Google Scholar]
- Jones NM, Kardashyan L, Callaway JK, Lee EM, Beart PM. Long-term functional and protective actions of preconditioning with hypoxia, cobalt chloride, and desferrioxamine against hypoxic-ischemic injury in neonatal rats. Pediatr Res. 2008;63:620–624. doi: 10.1203/PDR.0b013e31816d9117. [DOI] [PubMed] [Google Scholar]
- Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2003;339:341–346. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32:1347–1361. doi: 10.1038/jcbfm.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Zhang Y, Ling EA. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia. 2006;54:826–839. doi: 10.1002/glia.20420. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee JM, Park JS, Jo SA, Kim Y-O, Kim C-W, et al. Dexamethasone coordinately regulates angiopoietin-1 and VEGF: a mechanism of glucocorticoid-induced stabilization of blood-brain barrier. Biochem Biophys Res Commun. 2008;372:243–248. doi: 10.1016/j.bbrc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- Kook S-Y, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I. Aβ1-42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. J Neurosci. 2012;32:8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. 2007;170:1389–1397. doi: 10.2353/ajpath.2007.060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum JM, Mani N, Rosenstein JM. Roles of the endogenous VEGF receptors flt-1 and flk-1 in astroglial and vascular remodeling after brain injury. Exp Neurol. 2008;212:108–117. doi: 10.1016/j.expneurol.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann CRW, Tamaki R, Gamerdinger M, Lessmann V, Behl C, Kempski OS, et al. Inhibition of the myosin light chain kinase prevents hypoxia-induced blood-brain barrier disruption. J Neurochem. 2007;102:501–507. doi: 10.1111/j.1471-4159.2007.04506.x. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Ting P, Martinez H, Klatzo I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985;68:122–129. doi: 10.1007/BF00688633. [DOI] [PubMed] [Google Scholar]
- Kwon I, Kim EH, Del Zoppo GJ, Heo JH. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J Neurosci Res. 2009;87:668–676. doi: 10.1002/jnr.21877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladomery MR, Harper SJ, Bates DO. Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 2007;249:133–142. doi: 10.1016/j.canlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang X, Yang D, Luo G, Chen S, Le W. Hypoxia increases Abeta generation by altering beta-and gamma-cleavage of APP. Neurobiol Aging. 2009;30:1091–1098. doi: 10.1016/j.neurobiolaging.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Li M-M, Wu L-Y, Zhao T, Wu K-W, Xiong L, Zhu L-L, et al. The protective role of 5-hydroxymethyl-2-furfural (5-HMF) against acute hypobaric hypoxia. Cell Stress Chaperones. 2011;16:529–537. doi: 10.1007/s12192-011-0264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong C-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D-YY, Yu W-HH, Yeh W-LL, Tang C-HH, Leung Y-MM, Wong K-LL, et al. Hypoxia-induced matrix metalloproteinase-13 expression in astrocytes enhances permeability of brain endothelial cells. J Cell Physiol. 2009;220:163–173. doi: 10.1002/jcp.21746. [DOI] [PubMed] [Google Scholar]
- Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota SJ, Pepper MS. Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res. 1998;83:852–859. doi: 10.1161/01.res.83.8.852. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H1485–H1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhan WG. VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. Am J Physiol. 1999;276:C1148–C1153. doi: 10.1152/ajpcell.1999.276.5.C1148. [DOI] [PubMed] [Google Scholar]
- Meijer TWH, Kaanders JHAM, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18:5585–5594. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- Melgar MA, Rafols J, Gloss D, Diaz FG. Postischemic reperfusion: ultrastructural blood-brain barrier and hemodynamic correlative changes in an awake model of transient forebrain ischemia. Neurosurgery. 2006;59:E1152. doi: 10.1227/01.neu.0000154702.23664.3d. [DOI] [PubMed] [Google Scholar]
- Mense SM, Sengupta A, Zhou M, Lan C, Bentsman G, Volsky DJ, et al. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol Genomics. 2006;25:435–449. doi: 10.1152/physiolgenomics.00315.2005. [DOI] [PubMed] [Google Scholar]
- Meyer J, Rauh J, Galla HJ. The susceptibility of cerebral endothelial cells to astroglial induction of blood-brain barrier enzymes depends on their proliferative state. J Neurochem. 1991;57:1971–1977. doi: 10.1111/j.1471-4159.1991.tb06411.x. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Dobrowsky RT, Li G. Tau neurofibrillary pathology and microtubule stability. J Mol Neurosci. 2002;19:289–293. doi: 10.1385/JMN:19:3:289. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Hashioka Y, Fujii A, Utoguchi N, Kubo K, Nakagawa S, et al. Glial extracellular matrix modulates gamma-glutamyl transpeptidase activity in cultured bovine brain capillary and bovine aortic endothelial cells. Brain Res. 1994;651:155–159. doi: 10.1016/0006-8993(94)90692-0. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Callaghan D, Cui H, Dean C, Stanimirovic DB, Zhang W. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. J Neuroinflammation. 2007;4:12. doi: 10.1186/1742-2094-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morancho A, Rosell A, Garcia-Bonilla L, Montaner J, García-Bonilla L. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. 2010;1207:123–133. doi: 10.1111/j.1749-6632.2010.05734.x. [DOI] [PubMed] [Google Scholar]
- Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res. 2007;29:375–381. doi: 10.1179/016164107X204693. [DOI] [PubMed] [Google Scholar]
- Nag S, Takahashi JL, Kilty DW. Role of vascular endothelial growth factor in blood-brain barrier breakdown and angiogenesis in brain trauma. J Neuropathol Exp Neurol. 1997;56:912–921. doi: 10.1097/00005072-199708000-00009. [DOI] [PubMed] [Google Scholar]
- Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37:3–23. doi: 10.1111/j.1365-2990.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- Nagel S, Talbot NP, Mecinovic J, Smith TG, Buchan AM, Schofield CJ. Therapeutic manipulation of the HIF hydroxylases. Antioxid Redox Signal. 2010;12:481–501. doi: 10.1089/ars.2009.2711. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Mathieson G, Hüttner I. Blood-brain barrier opening to horseradish peroxidase in acute arterial hypertension. Acta Neuropathol. 1979;48:45–53. doi: 10.1007/BF00691790. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, et al. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol. 2007;27:687–694. doi: 10.1007/s10571-007-9195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Deli M, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, et al. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int. 2009;54:253–263. doi: 10.1016/j.neuint.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Berger P. Coordination of VEGF receptor trafficking and signaling by coreceptors. Exp Cell Res. 2013;319:1340–1347. doi: 10.1016/j.yexcr.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Nalivaevaa NN, Fisk L, Kochkina EG, Plesneva SA, Zhuravin IA, Babusikova E, et al. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann N Y Acad Sci. 2004;1035:21–33. doi: 10.1196/annals.1332.002. [DOI] [PubMed] [Google Scholar]
- Nico B, Ribatti D. Morphofunctional aspects of the blood-brain barrier. Curr Drug Metab. 2012;13:50–60. doi: 10.2174/138920012798356970. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, Churchill AJ, et al. Expression of pro-and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci. 2008;121:3487–3495. doi: 10.1242/jcs.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunshola OO. In vitro modeling of the blood-brain barrier: simplicity versus complexity. Curr Pharm Des. 2011;17:2755–2761. doi: 10.2174/138161211797440159. [DOI] [PubMed] [Google Scholar]
- Ogunshola OO, Al-Ahmad A. HIF-1 at the blood-brain barrier: a mediator of permeability? High Alt Med Biol. 2012;13:153–161. doi: 10.1089/ham.2012.1052. [DOI] [PubMed] [Google Scholar]
- Ogunshola OO, Antoniou X. Contribution of hypoxia to Alzheimer's disease: is HIF-1alpha a mediator of neurodegeneration? Cell Mol Life Sci. 2009;66:3555–3563. doi: 10.1007/s00018-009-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada T, Gu YH, Kanazawa M, Tsubota Y, Hawkins BT, Spatz M, et al. Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by beta(1)-integrins. J Cereb Blood Flow Metab. 2011;31:1972–1985. doi: 10.1038/jcbfm.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolinelli R, Corada M, Orsenigo F, Dejana E. The molecular basis of the blood brain barrier differentiation and maintenance. Is it still a mystery? Pharmacol Res. 2011;63:165–171. doi: 10.1016/j.phrs.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–784. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papavassiliou E, Gogate N, Proescholdt M, Heiss JD, Walbridge S, Edwards NA, et al. Vascular endothelial growth factor (vascular permeability factor) expression in injured rat brain. J Neurosci Res. 1997;49:451–460. doi: 10.1002/(sici)1097-4547(19970815)49:4<451::aid-jnr6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Paris D, Patel N, DelleDonne A, Quadros A, Smeed R, Mullan M. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett. 2004;366:80–85. doi: 10.1016/j.neulet.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Park K, Rosell A, Foerch C, Xing C, Kim WJ, Lee S, et al. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke. 2009;40:2836–2842. doi: 10.1161/STROKEAHA.109.554824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Kim NH, Jo I. Hypoxia and vascular endothelial growth factor acutely up-regulate angiopoietin-1 and Tie2 mRNA in bovine retinal pericytes. Microvasc Res. 2003;65:125–131. doi: 10.1016/s0026-2862(02)00035-3. [DOI] [PubMed] [Google Scholar]
- Patel HC, Boutin H, Allan SM. Interleukin-1 in the brain: mechanisms of action in acute neurodegeneration. Ann N Y Acad Sci. 2003;992:39–47. doi: 10.1111/j.1749-6632.2003.tb03136.x. [DOI] [PubMed] [Google Scholar]
- Peers C, Dallas ML, Boycott HE, Scragg JL, Pearson H, Boyle JP. Hypoxia and neurodegeneration. Ann N Y Acad Sci. 2009;1177:169–177. doi: 10.1111/j.1749-6632.2009.05026.x. [DOI] [PubMed] [Google Scholar]
- Plate KH. Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol. 1999a;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999b;58:654–666. doi: 10.1097/00005072-199906000-00010. [DOI] [PubMed] [Google Scholar]
- Plateel M, Teissier E, Cecchelli R. Hypoxia dramatically increases the nonspecific transport of blood-borne proteins to the brain. J Neurochem. 1997;68:874–877. doi: 10.1046/j.1471-4159.1997.68020874.x. [DOI] [PubMed] [Google Scholar]
- Pluta R. Role of ischemic blood-brain barrier on amyloid plaques development in Alzheimer's disease brain. Curr Neurovasc Res. 2007;4:121–129. doi: 10.2174/156720207780637207. [DOI] [PubMed] [Google Scholar]
- Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J, et al. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J Cereb Blood Flow Metab. 2002;22:520–525. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Rascher G, Fischmann A, Kroger S, Duffner F, Grote EH, Wolburg H. Extracellular matrix and the blood-brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin. Acta Neuropathol. 2002;104:85–91. doi: 10.1007/s00401-002-0524-x. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Siddiq A, Smirnova N, Karpisheva K, Haskew-Layton R, McConoughey S, et al. Harnessing hypoxic adaptation to prevent, treat, and repair stroke. J Mol Med. 2007;85:1331–1338. doi: 10.1007/s00109-007-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Siddiq A, Aminova L, Langley B, McConoughey S, Karpisheva K, et al. Small molecule activation of adaptive gene expression: tilorone or its analogs are novel potent activators of hypoxia inducible factor-1 that provide prophylaxis against stroke and spinal cord injury. Ann N Y Acad Sci. 2008;1147:383–394. doi: 10.1196/annals.1427.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raub TJ, Kuentzel SL, Sawada GA. Permeability of bovine brain microvessel endothelial cells in vitro: barrier tightening by a factor released from astroglioma cells. Exp Cell Res. 1992;199:330–340. doi: 10.1016/0014-4827(92)90442-b. [DOI] [PubMed] [Google Scholar]
- Reuss B, Dono R, Unsicker K. Functions of fibroblast growth factor (FGF)-2 and FGF-5 in astroglial differentiation and blood-brain barrier permeability: evidence from mouse mutants. J Neurosci. 2003;23:6404–6412. doi: 10.1523/JNEUROSCI.23-16-06404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rist RJ, Romero IA, Chan MW, Couraud PO, Roux F, Abbott NJ. F-actin cytoskeleton and sucrose permeability of immortalised rat brain microvascular endothelial cell monolayers: effects of cyclic AMP and astrocytic factors. Brain Res. 1997;768:10–18. doi: 10.1016/s0006-8993(97)00586-6. [DOI] [PubMed] [Google Scholar]
- Roberts J, Kahle MP, Bix GJ. Perlecan and the blood-brain barrier: beneficial proteolysis? Front Pharmacol. 2012;3:155. doi: 10.3389/fphar.2012.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, et al. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zlokovic BV. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:pii. doi: 10.1101/cshperspect.a011452. a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Sa-Pereira I, Brites D, Brito MA, Sá-Pereira I. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–347. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- Schmid-Brunclik N, Burgi-Taboada C, Antoniou X, Gassmann M, Ogunshola OO, Bürgi-Taboada C. Astrocyte responses to injury: VEGF simultaneously modulates cell death and proliferation. Am J Physiol Regul Integr Comp Physiol. 2008;295:R864–R873. doi: 10.1152/ajpregu.00536.2007. [DOI] [PubMed] [Google Scholar]
- Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–2557. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- Scholler K, Trinkl A, Klopotowski M, Thal SC, Plesnila N, Trabold R, et al. Characterization of microvascular basal lamina damage and blood-brain barrier dysfunction following subarachnoid hemorrhage in rats. Brain Res. 2007;1142:237–246. doi: 10.1016/j.brainres.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Schroeter ML, Mertsch K, Giese H, Muller S, Sporbert A, Hickel B, et al. Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett. 1999;449:241–244. doi: 10.1016/s0014-5793(99)00451-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease?: genes, proteins, and therapy. Perspective. 2001;81:741–767. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Li S, Chung SH, Zhu L, Stayt J, Su T, et al. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-beta1-induced permeability of centrally derived vascular endothelium. Eur J Cell Biol. 2011;90:323–332. doi: 10.1016/j.ejcb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemori Y, Katayama Y, Mori T, Maeda T, Kawamata T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir Suppl. 2006;96:130–133. doi: 10.1007/3-211-30714-1_29. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Abe MA, Maeda T, Ohtsuki S, Terasaki T, et al. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J Cell Physiol. 2011;226:255–266. doi: 10.1002/jcp.22337. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, et al. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem Res. 2012;37:401–409. doi: 10.1007/s11064-011-0626-8. [DOI] [PubMed] [Google Scholar]
- Siddharthan V, Kim YV, Liu S, Kim KS. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007;1147:39–50. doi: 10.1016/j.brainres.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Sharma G, Mishra V, Raghubir R. Hypoxia inducible factor-1: its potential role in cerebral ischemia. Cell Mol Neurobiol. 2012;32:491–507. doi: 10.1007/s10571-012-9803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sköld MK, Gertten CV, Sandberg-Nordqvist A-C, Mathiesen T, Holmin S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J Neurotrauma. 2002;19:353–367. doi: 10.1089/neu.2005.22.353. [DOI] [PubMed] [Google Scholar]
- Smirnova NA, Hushpulian DM, Speer RE, Gaisina IN, Ratan RR, Gazaryan IG. Catalytic mechanism and substrate specificity of HIF prolyl hydroxylases. Biochemistry. 2012;77:1108–1119. doi: 10.1134/S0006297912100033. [DOI] [PubMed] [Google Scholar]
- Sobue K, Yamamoto N, Yoneda K, Hodgson ME, Yamashiro K, Tsuruoka N, et al. Induction of blood-brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res. 1999;35:155–164. doi: 10.1016/s0168-0102(99)00079-6. [DOI] [PubMed] [Google Scholar]
- Song HS, Son MJ, Lee YM, Kim WJ, Lee S-W, Kim CW, et al. Oxygen tension regulates the maturation of the blood-brain barrier. Biochem Biophys Res Commun. 2002;290:325–331. doi: 10.1006/bbrc.2001.6205. [DOI] [PubMed] [Google Scholar]
- Sorond FA, Ratan RR. Ironing-out mechanisms of neuronal injury under hypoxic-ischemic conditions and potential role of iron chelators as neuroprotective agents. Antioxid Redox Signal. 2000;2:421–436. doi: 10.1089/15230860050192206. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Rooijen NV, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25:593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- Stanimirovic DB, Friedman A. Pathophysiology of the neurovascular unit: disease cause or consequence? J Cereb Blood Flow Metab. 2012;32:1207–1221. doi: 10.1038/jcbfm.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail – chick transplantation chimeras. Dev Biol. 1981;84:183–192. doi: 10.1016/0012-1606(81)90382-1. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem. 1996;271:13636–13641. doi: 10.1074/jbc.271.23.13636. [DOI] [PubMed] [Google Scholar]
- Sun M-C, Honey CR, Berk C, Wong NLM, Tsui JKC. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:9–20. doi: 10.3171/jns.2003.98.3.0565. [DOI] [PubMed] [Google Scholar]
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata F, Dohgu S, Matsumoto J, Takahashi H, Machida T, Wakigawa T, et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J Neuroinflammation. 2011;8:106. doi: 10.1186/1742-2094-8-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Wu P, Su J, Xiang J, Cai D, Dong Q. Effects of Aquaporin-4 on edema formation following intracerebral hemorrhage. Exp Neurol. 2010;223:485–495. doi: 10.1016/j.expneurol.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Nagy Z, Brightman MW. Tight junctions of brain endothelium in vitro are enhanced by astroglia. J Neurosci. 1987;7:3293–3299. doi: 10.1523/JNEUROSCI.07-10-03293.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Naito M, Mikami K, Tsuruo T. Enhanced expression by the brain matrix of P-glycoprotein in brain capillary endothelial cells. Cell Growth Differ. 1994;5:1145–1152. [PubMed] [Google Scholar]
- Taya K, Gulsen S, Okuno K, Prieto R, Marmarou CR, Marmarou A. Modulation of AQP4 expression by the selective V1a receptor antagonist, SR49059, decreases trauma-induced brain edema. Acta Neurochir Suppl. 2008;102:425–429. doi: 10.1007/978-3-211-85578-2_83. [DOI] [PubMed] [Google Scholar]
- Tejima E, Guo S, Murata Y, Arai K, Lok J, Van Leyen K, et al. Neuroprotective effects of overexpressing tissue inhibitor of metalloproteinase TIMP-1. J Neurotrauma. 2009;26:1935–1941. doi: 10.1089/neu.2009.0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Wyss-Coray T. A role for TGF-beta signaling in neurodegeneration: evidence from genetically engineered models. Curr Alzheimer Res. 2006;3:505–513. doi: 10.2174/156720506779025297. [DOI] [PubMed] [Google Scholar]
- Thanabalasundaram G, Pieper C, Lischper M, Galla H-J. Regulation of the blood-brain barrier integrity by pericytes via matrix metalloproteinases mediated activation of vascular endothelial growth factor in vitro. Brain Res. 2010;1347:1–10. doi: 10.1016/j.brainres.2010.05.096. [DOI] [PubMed] [Google Scholar]
- Thanabalasundaram G, Schneidewind J, Pieper C, Galla H-J. The impact of pericytes on the blood-brain barrier integrity depends critically on the pericyte differentiation stage. Int J Biochem Cell Biol. 2011;43:1284–1293. doi: 10.1016/j.biocel.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Tilling T, Korte D, Hoheisel D, Galla HJ. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem. 1998;71:1151–1157. doi: 10.1046/j.1471-4159.1998.71031151.x. [DOI] [PubMed] [Google Scholar]
- Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, et al. Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, et al. Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50:307–320. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- Turner DA, Adamson DC. Neuronal-astrocyte metabolic interactions. J Neuropathol Exp Neurol. 2011;70:167–176. doi: 10.1097/NEN.0b013e31820e1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Valable S, Montaner J, Bellail A, Berezowski V, Brillault J, Cecchelli R, et al. VEGF-induced BBB permeability is associated with an MMP-9 activity increase in cerebral ischemia: both effects decreased by Ang-1. J Cereb Blood Flow Metab. 2005;25:1491–1504. doi: 10.1038/sj.jcbfm.9600148. [DOI] [PubMed] [Google Scholar]
- Vangeison G, Rempe DA. The Janus-faced effects of hypoxia on astrocyte function. Neuroscientist. 2009;15:579–588. doi: 10.1177/1073858409332405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci. 2008;28:1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21. doi: 10.1002/(sici)1098-1136(199709)21:1<2::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Vilalta A, Sahuquillo J, Poca MA, De Los Rios J, Cuadrado E, Ortega-Aznar A, et al. Brain contusions induce a strong local overexpression of MMP-9. Results of a pilot study. Acta Neurochir Suppl. 2008;102:415–419. doi: 10.1007/978-3-211-85578-2_81. [DOI] [PubMed] [Google Scholar]
- Vogel C, Bauer A, Wiesnet M, Preissner KT, Schaper W, Marti HH, et al. Flt-1, but not Flk-1 mediates hyperpermeability through activation of the PI3-K / Akt pathway. J Cell Physiol. 2007;212:236–243. doi: 10.1002/jcp.21022. [DOI] [PubMed] [Google Scholar]
- Walshe TE, Saint-Geniez M, Maharaj AS, Sekiyama E, Maldonado AE, D'Amore PA. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE. 2009;4:e5149. doi: 10.1371/journal.pone.0005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Hui YN, Guo B, Ma JX. Strengthening tight junctions of retinal microvascular endothelial cells by pericytes under normoxia and hypoxia involving angiopoietin-1 signal way. Eye. 2007;21:1501–1510. doi: 10.1038/sj.eye.6702716. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience. 2009;159:1055–1069. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, et al. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45:325–337. doi: 10.1002/glia.10333. [DOI] [PubMed] [Google Scholar]
- Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt KA, Mark KS, Sandoval KE, Davis TP. Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvasc Res. 2008;75:91–96. doi: 10.1016/j.mvr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009a;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist. 2009b;15:180–193. doi: 10.1177/1073858408329509. [DOI] [PubMed] [Google Scholar]
- Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Xia Y, Choi H-K, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem. 2012;49C:24–40. doi: 10.1016/j.ejmech.2012.01.033. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Tagami M, Takenaga F, Yamori Y, Itoh S. Hypoxia-induced changes in tight junction permeability of brain capillary endothelial cells are associated with IL-1beta and nitric oxide. Neurobiol Dis. 2004;17:491–499. doi: 10.1016/j.nbd.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Yang S-P, Bae D-G, Kang HJ, Gwag BJ, Gho YS, Chae C-B. Co-accumulation of vascular endothelial growth factor with beta-amyloid in the brain of patients with Alzheimer's disease. Neurobiol Aging. 2004;25:283–290. doi: 10.1016/S0197-4580(03)00111-8. [DOI] [PubMed] [Google Scholar]
- Yeh W-L, Lu D-Y, Lin C-J, Liou H-C, Fu W-M. Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1alpha accumulation and VEGF expression. Mol Pharmacol. 2007;72:440–449. doi: 10.1124/mol.107.036418. [DOI] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Zehendner CM, Librizzi L, de Curtis M, Kuhlmann CR, Luhmann HJ. Caspase-3 contributes to ZO-1 and Cl-5 tight-junction disruption in rapid anoxic neurovascular unit damage. PLoS ONE. 2011;6:e16760. doi: 10.1371/journal.pone.0016760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng XN, Xie LL, Liang R, Sun XL, Fan Y, Hu G. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Ther. 2012;18:388–394. doi: 10.1111/j.1755-5949.2012.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Petrovic JM, Callaghan D, Jones A, Cui H, Howlett C, et al. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1beta (IL-1beta) in astrocyte cultures. J Neuroimmunol. 2006;174:63–73. doi: 10.1016/j.jneuroim.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Zhang X, Cheng M, Chintala SK. Optic nerve ligation leads to astrocyte-associated matrix metalloproteinase-9 induction in the mouse retina. Neurosci Lett. 2004;356:140–144. doi: 10.1016/j.neulet.2003.10.084. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao F-F, et al. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Croll SD, Chopp M. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience. 2002;113:683–687. doi: 10.1016/s0306-4522(02)00175-6. [DOI] [PubMed] [Google Scholar]
- Zhao B-QQ, Wang S, Kim H-YY, Storrie H, Rosen BR, Mooney DJ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zhou L, Chan KH, Chu LW, Kwan JSC, Song YQ, Chen LH, et al. Plasma amyloid-β oligomers level is a biomarker for Alzheimer's disease diagnosis. Biochem Biophys Res. 2012;423:697–702. doi: 10.1016/j.bbrc.2012.06.017. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- Zozulya A, Weidenfeller C, Galla HJ. Pericyte-endothelial cell interaction increases MMP-9 secretion at the blood-brain barrier in vitro. Brain Res. 2008;1189:1–11. doi: 10.1016/j.brainres.2007.10.099. [DOI] [PubMed] [Google Scholar]