

Abstract
BACKGROUND AND PURPOSE
Melanin-concentrating hormone receptor 1 (MCH1 receptor) antagonists are being considered as anti-obesity agents. The present study reports a new class of MCH1 receptor antagonists with an 8-methylquinoline scaffold. The molecular mechanism of MCH1 receptor blockade by these antagonists was examined.
EXPERIMENTAL APPROACH
The pharmacological properties of the 8-methylquinolines as exemplified by MQ1 were evaluated by use of multiple biophysical and cell-based functional assays.
KEY RESULTS
Multiple signalling pathways for Gαi and Gαq, and β-arrestin were inhibited by MQ1. Furthermore, MQ1 produced an insurmountable antagonism, causing a rightward shift of the curve for concentration-dependent binding of MCH along with a progressive reduction of the maximal response. The dissociation kinetics for MQ1 were determined from washout experiments as well as by affinity selection-MS. In short, MQ1 was shown to be a slowly dissociating reversible MCH1 receptor blocker with a low Koff value.
CONCLUSION AND IMPLICATIONS
This is the first time that a slowly dissociating negative allosteric modulator of the MCH1 receptor has been demonstrated to inhibit the numerous signalling pathways of this receptor. The characteristics of MQ1 are superior and distinct from previously reported MCH1 receptor antagonists, making members of this chemotype attractive as drug candidates.
Keywords: melanin-concentrating hormone receptor 1 (MCH1 receptor), antagonist, time-dependent inhibition, slow dissociation, negative allosteric modulator, obesity
Introduction
Obesity is defined as abnormal or excessive accumulation of fat that may impair health and nearly 400 million adults worldwide are obese, a number that has more than doubled since 1980 (Low et al., 2009). Obesity is a serious problem because it causes or aggravates various diseases, such as type 2 diabetes, hypertension, stroke, depression, sleep disorders and certain types of cancer, resulting in a huge socio-economic burden (Bray and Bellanger, 2006). Despite the intensive research efforts made by many pharmaceutical and biotechnology companies to develop anti-obesity drugs, only a few drugs are currently available to treat obesity and this remains a massive unmet medical need (Rodgers et al., 2012).
Melanin-concentrating hormone (MCH) is a disulfide-linked cyclic nonadecapeptide that is mainly expressed in the lateral hypothalamus and zona incerta, which are regions with extensive projections throughout the brain (Vaughan et al., 1989; Presse et al., 1990; Bittencourt et al., 1992; Saper et al., 2002). After the discovery of MCH, a number of studies demonstrated that it is involved in regulating food intake, energy homeostasis and weight gain (Qu et al., 1996; Della-Zuana et al., 2002; Ito et al., 2003; Pereira-da-Silva et al., 2003).
Two GPCRs for MCH have been reported so far. Melanin-concentrating hormone receptor 1 (MCH1 receptor), which was originally cloned and named SLC-1, has been characterized as a receptor with a high affinity for MCH by several groups (Chambers et al., 1999; Lembo et al., 1999; Shimomura et al., 1999; Saito, 2001). The MCH1 receptor is primarily expressed in the CNS, including the hippocampus, nucleus accumbens and hypothalamus (Pissios et al., 2006). It has been shown that the MCH1 receptor couples with multiple G-proteins, including Gαi and Gαq (Chambers et al., 1999; Lembo et al., 1999; Hawes et al., 2000; Pissios et al., 2006). Also there have been a number of reports suggesting a role for MCH1 receptors in feeding and energy expenditure. For example, MCH1 receptor-deficient mice are reported to be lean and resistant to dietary obesity due to their hyperactivity and increased energy expenditure (Chen et al., 2002; Marsh et al., 2002). In addition, administration of low molecular weight antagonists that block MCH1 receptors suppresses MCH-induced food intake (Takekawa et al., 2002) and reduces weight gain (Borowsky et al., 2002). These findings indicate that MCH1 receptor plays a key role in the regulation of feeding and energy expenditure, suggesting that an MCH1 receptor antagonist could be a promising anti-obesity agent. In contrast, little is known about the physiological role of MCH2 receptors (An et al., 2001; Hill et al., 2001; Mori et al., 2001; Rodriguez et al., 2001; Sailer et al., 2001).
So far, numerous pharmaceutical and biotechnology companies have discovered low molecular weight MCH1 receptor antagonists and extensive clinical trials have been conducted (McBriar, 2006; Luthin, 2007; Cheon, 2012). Several groups have reported on the in vitro characterization of some of these MCH1 receptor antagonists (Borowsky et al., 2002; Chaki et al., 2005; David et al., 2007), but in-depth studies on the molecular mechanism of MCH1 receptor blockade have not yet been conducted.
Here we describe a potent low molecular weight MCH1 receptor blocker that inhibits multiple signalling pathways. The antagonism induced by MQ1 (Kasai et al., 2012) is allosteric, time-dependent and reversible affording MQ1 distinctive advantages over previously reported antagonists (McBriar, 2006; Luthin, 2007; Cheon, 2012). MQ1 is a highly selective slowly dissociating negative allosteric modulator (NAM) of MCH1 receptors with a residence time (half-life) of 78 min (95% CI: 72–86 min).
Methods
Materials
MQ1 [4-(cyclopropylmethoxy)-N-(8-methyl-3-((1R)-1-(pyrrolidin-1-yl)ethyl)quinolin-7-yl)-benzamide] (Kasai et al., 2012), MQ2 [4-(4-hydroxybutoxy)-N-(8-methyl-3-((1R)-1-(pyrrolidin-1-yl)ethyl)quinolin-7-yl)benz-amide] and MQ3 [4-(cyclopropylmethoxy)-N-(8-methyl-3-(((1-oxidotetrahydro-2H-thiopyran-4-yl)amino)methyl)quinolin-7-yl)benzamide] (Figure 1) were synthesized by Takeda Pharmaceutical Company (Osaka, Japan). The synthesis of MQ2 and MQ3, as well as the sources of other materials used in this study, are summarized in the Supporting Information.
Figure 1.
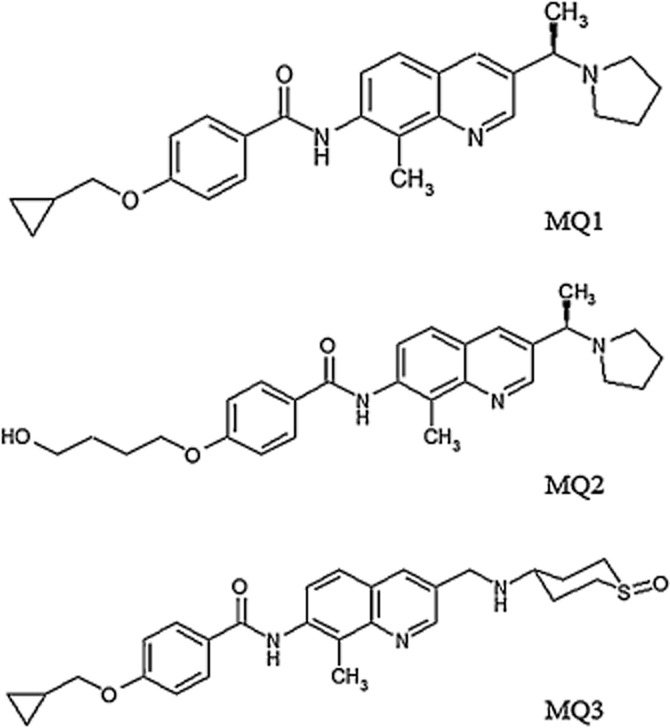
Chemical structures of MCH1 receptor antagonists.
Establishment of stable cell lines
The development of two stable cell lines expressing human MCH1 receptors, CHO (dhfr-)-hMCH1 receptor and CHO-K1-BAEA-hMCH1 receptor is described in the Supporting Information.
cAMP assay
The cAMP assay was carried out by using an AlphaScreen cAMP Assay Kit from PerkinElmer (Covina, CA, USA) according to the manufacturer's instructions with minor modifications (Supporting Information).
Calcium flux assay
The calcium flux assay was performed by using a FLIPR Tetra (Molecular Devices, Sunnyvale, CA, USA) according to the manufacturer's instructions with minor modifications (Supporting Information).
PathHunter β-arrestin recruitment assay
The PathHunter β-arrestin recruitment assay was performed according to the manufacturer's instructions with minor modifications (Supporting Information).
[125I]-MCH-(4-19) binding assay
Preparation of human MCH1 receptor membranes, preparation of radiolabelled [125I]-MCH-(4-19), and the receptor-binding assay were all performed as described previously (Takekawa et al., 2002) with minor modifications (Supporting Information).
[125I]-MCH-(4-19) dissociation assay
The [125I]-MCH-(4-19) dissociation assay was performed as described in the Supporting Information.
Equilibrium binding and dissociation assay with affinity selection-MS
The equilibrium binding assay and dissociation assay with affinity selection-MS were performed by using a LC-MS system (API5000 LC/MS/MS system; AB SCIEX, Tokyo, Japan) (Supporting Information).
Mutation analysis
Site-directed mutagenesis and transient transfection were performed as described in the Supporting Information.
Data analysis and statisitics
Data analysis was performed with GraphPad Prism5 software (GraphPad, San Diego, CA). The values of EC50, IC50, and Emax were calculated by fitting the data to a sigmoidal dose-response equation. For analysis of dissociation kinetics, the dissociation rate constant (Koff) was calculated by fitting the data to the formula Y = Ae-kx, which is an exponential decay model. Unless otherwise stated, EC50, IC50, and Kd values are expressed as the mean with 95% confidence interval (CI) for four experiments (n = 4), each of which was performed more than twice. Unless otherwise stated, statistical significance was determined by using an Student's unpaired t-test. ANOVA with a Dunnett's test was used for the results presented in Tables 1992 and 2002. A value of P < 0.05 was taken as statistically significant.
Results
Antagonistic effect of MQ1 on multiple signalling pathways
It was previously reported that screening for MCH1 receptor antagonists and subsequent chemical modification of lead compounds resulted in the discovery of MQ1 (Figure 1) (Kasai et al., 2012). In the membrane binding assay using radiolabelled MCH-(4-19), which is a structural analogue of MCH with agonistic activity, EC50 1.6 nM (95% CI: 0.44–5.7 nM) in the cAMP assay, MQ1 inhibited the binding of [125I]-MCH-(4-19) to human MCH1 receptor membrane fractions with an IC50 value of 2.2 nM (95% CI: 1.8–2.7 nM) (Table 2001).
Table 1.
Affinity and potency for MCH and three antagonists in multiple assays
| Assay type | Kd value (95% CI) (nM) | MCH EC50 (95% CI) (nM) | MQ1 IC50 (95% CI) (nM) | MQ2 IC50 (95% CI) (nM) | MQ3 IC50 (95% CI) (nM) |
|---|---|---|---|---|---|
| [125I]-MCH-(4-19) binding assay (1 h of incubation) | – | – | 2.2 (1.8–2.7) | 28 (15–52) | 16 (10–25) |
| [125I]-MCH-(4-19) binding assay (at equilibria) | – | – | 0.32 (0.26–0.41) | 21 (11–41) | 11 (7.8–17) |
| Ki | 0.058 (0.031–0.085) | – | 0.16 | 11 | 5.6 |
| cAMP assay | – | 1.8 (0.81–4.3) | 5.7 (2.7–12) | 27 (12–58) | 45 (15–136) |
| Calcium flux assay | – | 0.70 (0.53–0.92) | 31 (18–51) | 230 (200–280) | 64 (54–75) |
| PathHunter B-arrestin assay | – | 2.5 (1.9–3.3) | 1.7 (1.4–2.0) | 53 (32–87) | 6.8 (4.6–10) |
The inhibitory effect (IC50) of MQ1, MQ2 and MQ3 was assessed with the [125I]-MCH-(4-19) binding assay with 1 and 8 h of incubation. The calcium flux assay, PathHunter β-arrestin recruitment assay and cAMP assay were performed in the presence of 2, 10 and 5 nM of MCH respectively. Values are means of three independent experiments and 95% confidence intervals (CI) conducted in duplicate (n = 2) for the [125I]-MCH-(4-19) binding assay and quadruplicate (n = 4) for the other assays.
Table 2.
Emax values of MCH at different concentrations of three antagonists in the PathHunter β-arrestin recruitment assay
| MQ1 | ||||
| Control (nM) | 1 | 10 | 100 | 1000 |
| 100 | 77 ± 1.2*** | 52 ± 1.0*** | 33 ± 1.0*** | 24 ± 1.1*** |
| MQ2 | ||||
| Control (nM) | 100 | 1000 | 3000 | 10 000 |
| 100 | 109 ± 4.6 | 95 ± 5.3 | 80 ± 5.5 | 44 ± 4.9*** |
| Peptide antagonist | ||||
| Control (nM) | 10 | 100 | 1000 | 3000 |
| 100 | 96 ± 1.9 | 98 ± 2.2 | 104 ± 3.2 | 92 ± 3.2 |
Emax values of MCH with MQ1, MQ2 and a peptide antagonist were assessed with the PathHunter β-arrestin recruitment assay. All values are expressed as mean ± SEM of four values from a representative experiment of three separate experiments. Statistical comparison was performed using anova with a Dunnett's test.
P < 0.001 vs. Emax control values).
Table 3.
Emax values of MCH at different concentrations of MQ1 and MQ2 in the cAMP assay
| MQ1 | |||||
| Control (nM) | 10 | 100 | 300 | 1000 | 3000 |
| 100 | 99 ± 1.8 | 98 ± 1.5 | 95 ± 2.6 | 92 ± 0.91** | 88 ± 0.91*** |
| MQ2 | |||||
| Control (nM) | 100 | 300 | 1000 | 3000 | |
| 100 | 96 ± 1.8 | 89 ± 1.9 | 85 ± 2.5** | 75 ± 0.81** |
Emax values of MCH with MQ1 and MQ2 were assessed with the cAMP assay. All values are expressed as mean ± SEM of four values from a representative experiment of three separate experiments. Statistical comparison was performed using anova with a Dunnett's test.
P < 0.01.
P < 0.001 vs. Emax control values.
We subsequently investigated the antagonistic effect of MQ1 in functional assays using cells that stably expressed human MCH1 receptor. It has already been indicated that the MCH1 receptor couples with multiple G-proteins, including Gαi, Gαo and Gαq (Hawes et al., 2000). We established a cAMP assay and a calcium flux assay in which MCH stimulated these G-protein signalling pathways with an EC50 value of 1.8 nM (95% CI: 0.81–4.3 nM) and 0.70 nM (95% CI: 0.53–0.92 nM) respectively. The IC50 value obtained for MQ1 in the cAMP assay and the calcium flux assay was 5.7 nM (95% CI: 2.7–12 nM) and 31 nM (95% CI: 18–51 nM) respectively (Table 2001).
While activated GPCRs transduce G-protein signals, they generally also activate β-arrestin-mediated signalling, which is thought to be involved in various physiological functions (Xiao et al., 2010). Therefore, we examined the antagonistic effect of MQ1 on β-arrestin-mediated signalling by establishing a PathHunter β-arrestin recruitment assay, in which the interaction of GPCR and β-arrestin was detected by using enzyme fragment complementation technology (Eglen, 2002). In this assay, MCH induced the recruitment of β-arrestin to the MCH1 receptor with an EC50 value of 2.5 nM (95% CI: 1.9–3.3 nM) and MQ1 showed antagonism with an IC50 value of 1.7 nM (95% CI: 1.4–2.0 nM) (Table 2001). These results demonstrated that MQ1 had the ability to inhibit multiple signalling pathways mediated by Gαi, Gαq and β-arrestin.
Time dependence of inhibition by MQ1
The inhibitory effect of MQ1 was slightly weaker in the calcium flux assay than in the other assays. This discrepancy motivated us to investigate the possibility that inhibition by the compound was time dependent, because it was considered likely that the calcium flux assay was conducted at hemi-equilibrium, while the other three assays (the binding assay, cAMP assay and PathHunter β-arrestin recruitment assay) were performed with a long enough incubation time for equilibrium to be reached. Accordingly, the PathHunter β-arrestin recruitment assay was performed with various incubation times to examine whether MQ1 exhibited time-dependent inhibition. We found that the IC50 value decreased as the incubation time became longer. Without pre-incubation, the IC50 value was 14 nM (95% CI: 9.6–20 nM), whereas after pre-incubation for 30, 60 or 120 min, the value was significantly decreased to 4.1 nM (95% CI: 2.8–6.0 nM) (P < 0.01), 2.6 nM (95% CI: 1.9–3.5 nM) (P < 0.01) and 1.9 nM (95% CI: 1.3–2.6 nM) (P < 0.01) respectively (Figure 2A). In a similar manner, we also evaluated the time dependence of inhibition by MQ1 in the [125I]-MCH-(4-19) binding assay and observed that its inhibitory effect increased in a time-dependent manner. The IC50 value was 2.5 nM (95% CI: 1.6–4.0 nM) without pre-incubation, whereas after pre-incubation for 30, 60 or 120 min, the IC50 was 1.0 nM (95% CI: 0.45–2.3 nM), 0.57 nM (95% CI: 0.36–0.89 nM) (P < 0.01) and 0.39 nM (95% CI: 0.26–0.59 nM) (P < 0.01) respectively (Figure 2B). Taken together, these results suggested that MQ1 is an MCH1 receptor antagonist that shows time-dependent inhibition.
Figure 2.
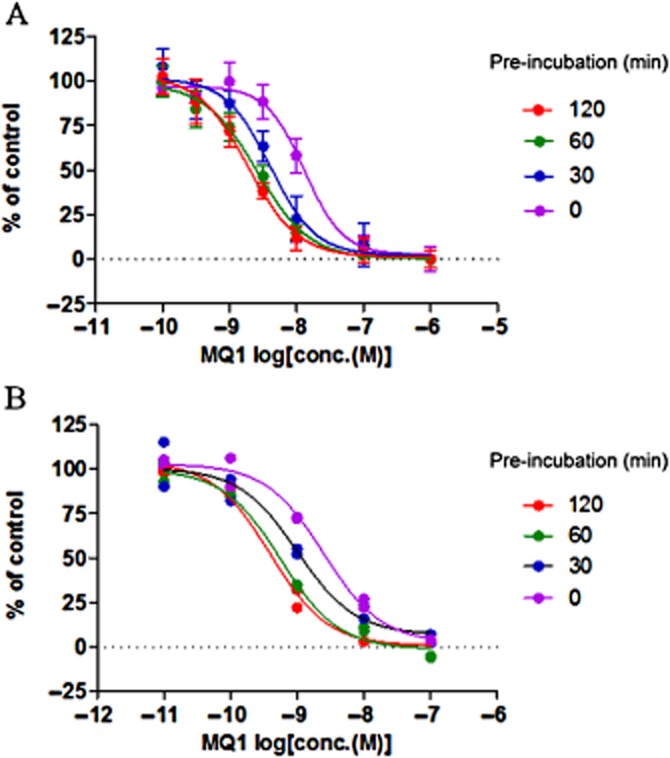
Time-dependent inhibition by MQ1. (A) Concentration-dependent inhibition by MQ1 was assessed in the PathHunter β-arrestin recruitment assay. CHO-K1-BAEA-hMCH1 receptor cells were pre-incubated with MQ1 for 0, 30, 60 or 120 min before incubation for 30 min with MCH (10 nM). Data points are the mean ± SD of four values from a representative experiment of three separate experiments. (B) Concentration-dependent inhibition by MQ1 was assessed in the [125I]-MCH-(4-19) binding assay. Human MCH1 receptor membrane fractions were pre-incubated with MQ1 for 0, 30, 60 or 120 min before incubation for 30 min with [125I]-MCH-(4-19) (50 pM). Each data point (n = 2) is plotted on the graph. Results are from representative experiments that were performed twice.
Reversible inhibition by MQ1
The time-dependence of inhibition by MQ1 (demonstrated above) raised the possibility that it might be an irreversible antagonist. Therefore, we investigated whether the inhibitory effect of MQ1 was based on covalent binding to MCH1 receptors. We developed an equilibrium binding assay using affinity selection-MS to test whether MQ1 that had bound to MCH1 receptors could be displaced by MQ2, a structurally related MCH1 receptor antagonist (Figure 1) with an IC50 value of 28 nM (95% CI: 15–52 nM) in the [125I-MCH-(4-19) binding assay (Table 2001, Supporting Information Figure S1b). MQ1 showed saturable binding to membrane fractions expressing MCH1 receptors in the absence of MQ2, whereas it was completely displaced by an excess of MQ2 (Figure 3). These findings indicate that the binding of MQ1 to MCH1 receptors is reversible.
Figure 3.
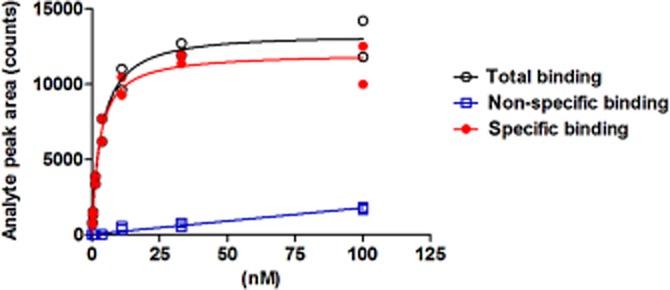
Saturation of the binding of MQ1 to MCH1 receptors assessed by affinity selection-MS. Human MCH1 receptor membrane fractions were incubated with various concentrations of MQ1 in the absence (total binding) or presence (non-specific) of MQ2 (30 μM) for 210 min at room temperature. Specific binding was determined as the difference between binding in the absence or presence of MQ2. The analyte peak area is displayed versus the concentration of MQ1. Each data point (n = 2) is plotted on the graph. Results are from representative experiments that were performed twice.
Inhibitory effect of MQ1 after washout
Time-dependent reversible inhibition is generally considered to be caused by slow dissociation of a compound from its receptor. To confirm that this applied to MQ1, we performed washout experiments using the PathHunter β-arrestin recruitment assay. Pretreatment with various concentrations of test compounds for 2 h inhibited MCH-induced recruitment of β-arrestin in a concentration-dependent manner (Figure 4). The inhibitory effect of MQ1 was still observed even after the cells were washed twice before addition of MCH (Figure 4A). In contrast, MQ2 did not show time-dependent inhibition in the [125I]-MCH-(4-19) binding assay (Supporting Information Figure S1b), and its binding was significantly reduced after the same washout procedure (Figure 4B). These results suggest that slow dissociation from the receptor contributed to the time-dependence of inhibition by MQ1.
Figure 4.
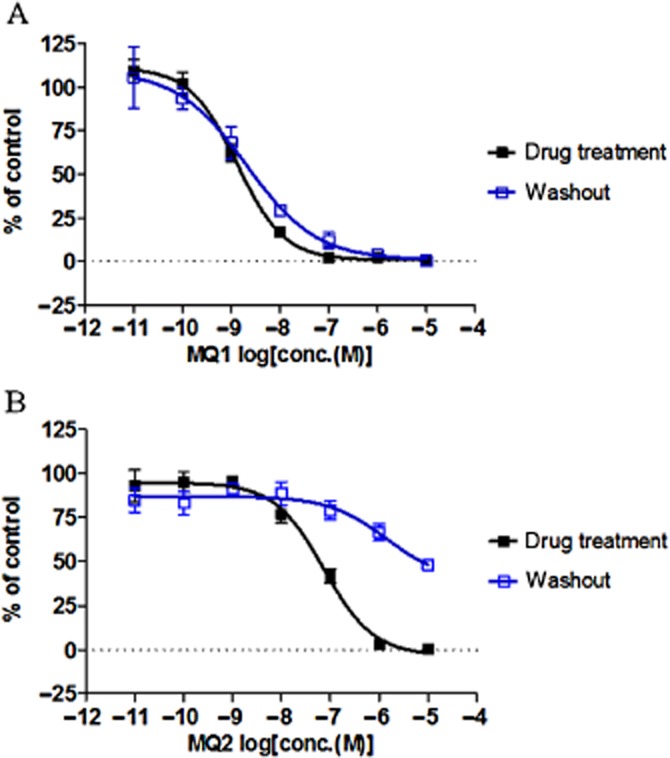
Inhibitory effect of two MCH1 receptor antagonists after washout. CHO-K1-BAEA-hMCH1 receptor cells were pretreated with various concentrations of (A) MQ1 or (B) MQ2 dissolved in Opti-MEM with 0.1% BSA for 2 h at 37°C under 5% CO2. Then, Opti-MEM medium containing the compounds was removed and the cells were washed twice with 50 μL of PBS. Next, the cells were stimulated with 25 μL of MCH (10 nM) for 2 h at 37°C under 5% CO2. All data points are the mean ± SD of four values from a representative experiment of two separate experiments.
Dissociation kinetics of MQ1
To better understand the dissociation kinetics of MQ1, we performed a dissociation assay based on the binding assay with affinity selection-MS. After MQ1 was pre-incubated with human MCH1 receptor membrane fractions, dissociation was assessed over time following the addition of 50 μM MQ3 (Figure 1), which is a structurally related MCH1 receptor antagonist with an IC50 value of 16 nM (95% CI: 10–25 nM) in the [125I]-MCH-(4-19) binding assay (Table 2001, Supporting Information Figure S1c). MQ3 was used because we had previously observed that it did not demonstrate time-dependent inhibition in the [125I]-MCH-(4-19) binding assay (Supporting Information Figure S1c). MQ1 slowly dissociated from the receptor (Figure 5), and the dissociation rate constant (Koff) was calculated to be 0.53 h−1 (95% CI: 0.48-0.58 h−1). In contrast, MQ2 was not expected to undergo slow dissociation based on the results of the washout experiments in Figure 4. As predicted, it showed relatively rapid dissociation from the receptor (Figure 5) and its Koff value was calculated to be 2.6 h−1 (95% CI: 2.2–3.0 h−1). These findings were consistent with the results of the washout experiments (Figure 4).
Figure 5.
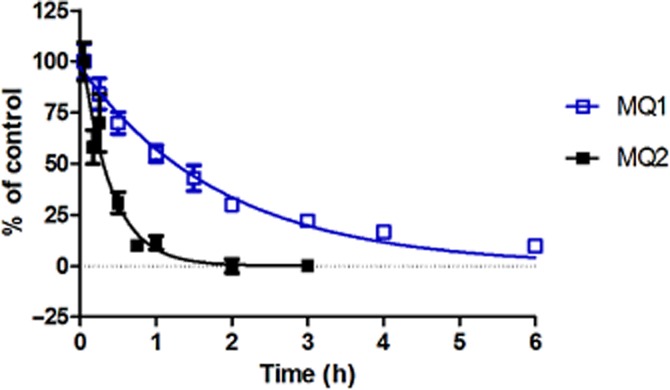
Dissociation kinetics of MCH1 receptor antagonists. Specific binding of MQ1 and MQ2 to human MCH1 receptor membrane fractions was measured over time as described in the Methods section. Initial binding of each compound and the plateau of specific binding were set as 100 and 0% respectively. Data points are the mean ± SD of six values from a representative experiment of two separate experiments.
Insurmountable antagonism induced by MQ1
To investigate the mechanism of the inhibition induced by MQ1, the effect of MQ1 on concentration-dependent binding of MCH was assessed in the PathHunter β-arrestin recruitment assay. To exclude the possibility that the results might be influenced by the slow dissociation of MQ1, we performed the experiments with an incubation time of 8 h, because we had already confirmed that the system reached equilibrium within 4 h as the EC50 values of MCH obtained at each concentration of MQ1 did not change between 4 and 8 h of incubation (Supporting Information Table S1). We also monitored the inhibitory effects of MQ1 over time and confirmed that the IC50 values remained the same after 4 h of incubation (Supporting Information Figure S2), in accordance with the results of Supporting Information Table S1. The addition of increasing concentrations of MQ1 caused both a rightward shift of the curve for concentration-dependent binding of MCH and a progressive reduction of the maximal response (Figure 6A, Table 1992). In addition, MQ2 showed a rapid dissociation from the MCH1 receptor (Figure 5) and also demonstrated insurmountable antagonism (Figure 6B, Table 1992). In contrast, a peptide antagonist (Gva-Cys-Met-Leu-Gly-Arg-Val-Tyr-Ava-Cys-NH2) that was expected to be competitive with MCH caused a parallel rightward shift without reducing the maximal response (Figure 6C, Table 1992). These results suggest that MQ1 and MQ2 act as NAMs, while the peptide antagonist was a competitive inhibitor.
Figure 6.
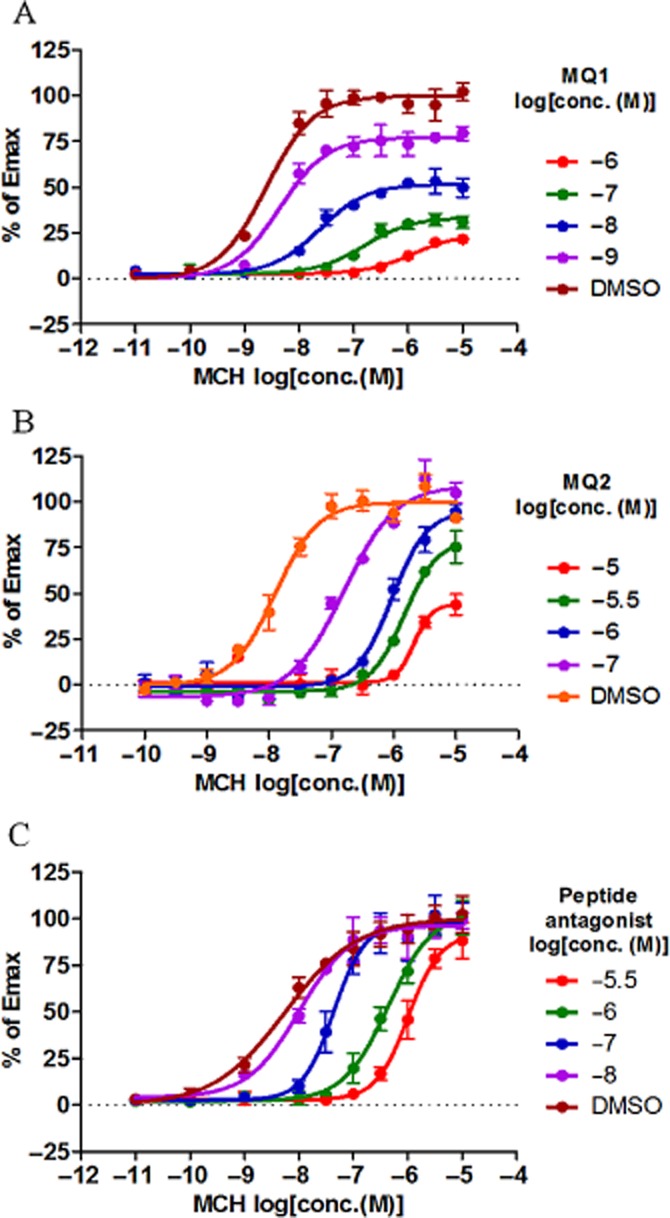
Effect of antagonists on concentration-dependent activity of MCH in the PathHunter β-arrestin recruitment assay. Concentration-dependent inhibition by (A) MQ1 and (B) MQ2 in the presence of increasing concentrations of MCH was measured in the PathHunter β-arrestin recruitment assay after incubation for 8 h. (C) Concentration-dependent inhibition by a peptide antagonist (Gva-Cys-Met-Leu-Gly-Arg-Val-Tyr-Ava-Cys-NH2) in the presence of increasing concentrations of MCH was measured in the PathHunter β-arrestin recruitment assay after overnight incubation. The magnitude of the response at each concentration was compared with the amplitude of the response to a maximally efficacious concentration of MCH (10 μM). Thus, results are expressed as % of Emax. All data points are the mean ± SD of four values from a representative experiment of three separate experiments.
To confirm that the insurmountable antagonism demonstrated by MQ1 and MQ2 was not an artefact specific to the PathHunter β-arrestin recruitment assay, we performed a similar experiment using the cAMP assay. We again observed a rightward shift of the curve for concentration-dependent binding of MCH and suppression of the maximal response with increasing concentrations of MQ1 and MQ2 (Figure 7, Table 2002), consistent with the results obtained in the PathHunter β-arrestin recruitment assay. Thus, the results of the cAMP assay further supported the possibility that MQ1 and MQ2 are NAMs.
Figure 7.
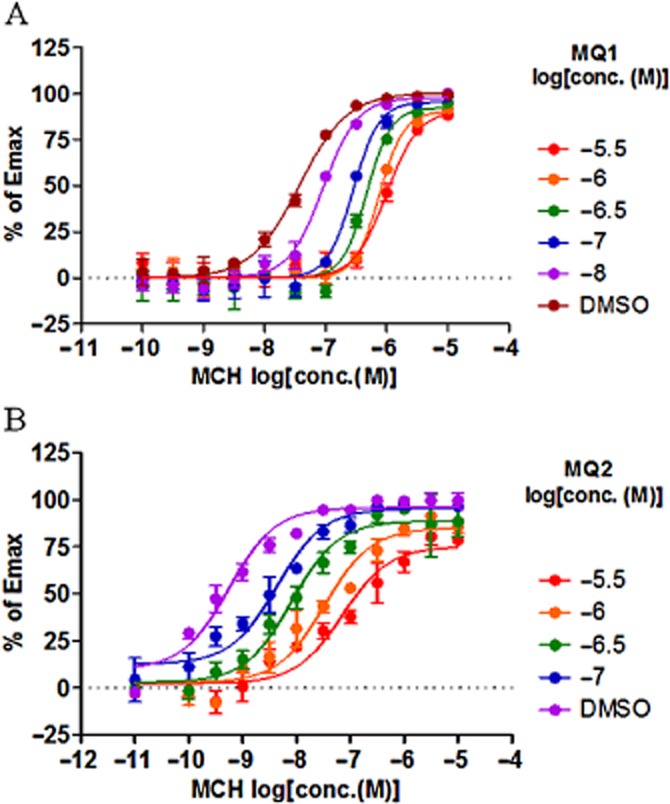
Effect of antagonists on concentration-dependent activity of MCH in the cAMP assay. Concentration-dependent antagonism by (A) MQ1 and (B) MQ2 in the presence of increasing concentrations of MCH was measured in the cAMP assay. Activity is plotted as percentage of maximal MCH response. Data points are the mean ± SD of four values from a representative experiment of three separate experiments.
Effect of MQ1 on the dissociation of [125I]-MCH-(4-19) from MCH1 receptors
One characteristic of allosteric modulators is their ability to alter the binding kinetics of orthosteric ligands (Christopoulos and Kenakin, 2002). Accordingly, we examined the dissociation kinetics of [125I]-MCH-(4-19) from MCH1 receptors to obtain further evidence about the allosteric interaction of MQ1. The dissociation of [125I]-MCH-(4-19) was detected after the addition of a substantial excess of unlabelled MCH-(4-19) to prevent the association of [125I-MCH-(4-19) with the receptor. The dissociation of [125I-MCH-(4-19) from MCH1 receptors occurred with a Koff value of 0.13 min−1 (95% CI: 0.070–0.19 min−1) (Figure 8). While MCH-(1-19) [which was expected to compete with MCH-(4-19)] did not have any effect on the dissociation rate of [125I]-MCH-(4-19), MQ1 significantly decreased the Koff value to 0.011 min−1 (95% CI: 0.0044–0.018 min−1) (P < 0.01) (Figure 8). We confirmed that MQ2 also significantly decreased the Koff value to 0.026 min−1 (95% CI: 0.0013–0.040 min−1) (P < 0.01). This demonstrates that MQ1 and MQ2 alter the dissociation kinetics of [125I]-MCH-(4-19) and clearly show that these compounds have an allosteric interaction with MCH1 receptors.
Figure 8.
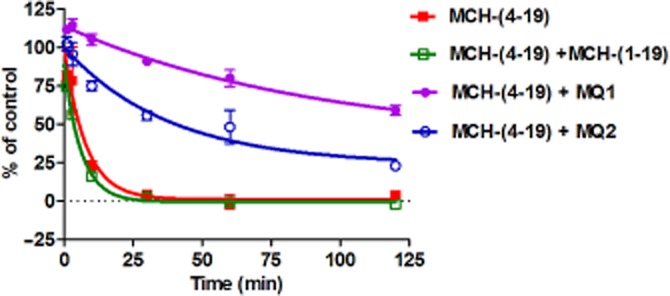
Effect of MQ1 on the dissociation of [125I]-MCH from MCH1 receptors. Human MCH1 receptor membrane fractions were incubated with 50 pM of [125I]-MCH-(4-19) for 2 h before adding an excess (1 μM) of unlabelled MCH-(4-19) to initiate dissociation of [125I]-MCH in the absence or presence of MQ1, MQ2 and MCH-(1-19). Each condition contains 1% DMDO. Initial binding and the plateau of specific binding were set at 100 and 0% respectively. Data points are the mean ± SD of three values from a representative experiment of three separate experiments.
Site-directed mutagenesis
To further confirm that MQ1 binds to a different site from that of MCH, we performed mutation analysis using a conventional alanine scan on transmembrane (TM) helices 3, 5 and 6 based on the information that the corresponding TM helices of the crystal structure of corticotrophin-releasing factor receptor 1 forms an allosteric pocket (Hollenstein et al., 2013). Eighteen residues predicted to be within this region were separately mutated to alanine or valine, and potency of MCH and MQ1 at each of these mutants was investigated in the PathHunter β-arrestin recruitment assay following transient transfection. Substitution of most of these individual residues did not show a significant change in the pEC50 or pIC50 value compared with the wild type (P > 0.05, Supporting Information Table S2) whereas the mutants A136V and H147A interestingly showed significant increases in the pIC50 value (Supporting Information Table S2). Furthermore, mutation of threonine 209 and glutamine 276, which are considered to be involved in the ligand binding (Macdonald et al., 2000), displayed significant decreases in the pEC50 value of MCH but showed no statistical changes in the pIC50 value of MQ1. These data further support the possibility that the binding mode of MQ1 is different from that of MCH.
Selectivity of MQ1 for MCH1 receptors
To assess the selectivity of MQ1 for MCH1 receptors, the effect of this compound on other drug targets was experimentally examined at Ricerca Biosciences (Concord, OH, USA) using equilibrium binding assays and enzyme activity assays. MQ1 (1 μM) did not show a strong effect on over 100 targets, including various GPCRs, enzymes and ion channels (data not shown). In particular, it had no significant effect on MCH2 receptors, somatostatin receptor 1 and μ-opioid receptor, all of which exhibit a high degree of homology with MCH1 receptors. These findings demonstrate that MQ1 is a highly selective NAM for MCH1 receptors.
Discussion
The results presented here demonstrate that MQ1 is an antagonist, which inhibits multiple signalling pathways of MCH1 receptors. In addition, MQ1 is a reversible antagonist that dissociates slowly from the receptor. Furthermore, it was demonstrated that MQ1 is a NAM of the MCH1 receptor.
Inhibition of multiple signalling pathways
MQ1 inhibited Gαi and β-arrestin signalling with IC50 values of 5.7 nM (95% CI: 2.7–12 nM) and 1.7 nM (95% CI: 1.4–2.0 nM), respectively, consistent with its IC50 value of 2.2 nM (95% CI: 1.8–2.7 nM) in the [125I]-MCH-(4-19) binding assay. Compared with the results from these three assays, MQ1 had a slightly larger IC50 of 31 nM (95% CI: 18–51 nM) in the calcium flux assay, presumably because that assay was conducted at hemi-equilibrium. This hypothesis was supported by the finding that pre-incubation for 60 min increased the potency of MQ1 to 5.2 nM (95% CI: 2.1–13 nM) in the calcium flux assay (data not shown), which was similar to the results of the other assays. Therefore, when tested at equilibrium, MQ1 exhibited equal inhibitory activity in all of the cell-based functional assays that we performed, demonstrating its ability to inhibit multiple signalling pathways.
So far, several MCH1 receptor antagonists have been reported to inhibit Gαq and/or Gαi signalling (Borowsky et al., 2002; Takekawa et al., 2002; David et al., 2007). However, to the best of our knowledge, there has been no report about an MCH1 receptor antagonist with the ability to inhibit β-arrestin signalling. In addition to playing a key role in internalization and subsequent desensitization of receptors, β-arrestin has been found to be involved in more diverse signalling processes than was previously appreciated (Xiao et al., 2010). This study shows that MQ1 inhibits β-arrestin signalling, although it is not clear whether the compound inhibits β-arrestin signalling directly or blocks G-protein coupling, and as a result inhibits β-arrestin binding. Considering that the pharmacologically relevant signalling pathway of MCH1 receptors is not clearly understood and that it even remains uncertain whether or not MCH1 receptors activate multiple signalling pathways in vivo, inhibition of all the signalling pathways (including the β-arrestin pathway) that have been detected in recombinant overexpression systems could be important for a compound to exhibit the expected pharmacological effects in vivo. Hence, the finding that MQ1 can inhibit multiple signalling pathways might increase the likelihood of it exhibiting efficacy in vivo. Moreover, the importance of understanding the relationship between different signalling pathways and their physiological consequences in drug discovery programmes is becoming more strongly appreciated as recent studies have identified a wide variety of compounds that have differential effects on various signalling pathways (Violin and Lefkowitz, 2007; Drake et al., 2008; Gesty-Palmer et al., 2009; Kenakin, 2009; Reiter et al., 2012). Thus, our efforts to determine the potency of MQ1 for each signalling pathway could be important with regard to elucidating the physiologically relevant signalling pathway for MCH1 receptors in the drug discovery process.
Slow dissociation
We showed that the inhibitory effect of MQ1 increased over time in both the cell-based PathHunter β-arrestin recruitment assay (Figure 2A) and the cell-free [125I]-MCH-(4-19) binding assay (Figure 2B). Although it has to be emphasized that the system may not have reached re-equilibrium among the three molecules (MCH, MQ1 and MCH1 receptor), the changes in the IC50 values with different pre-incubation times suggest that MQ1 is a time-dependent inhibitor. In addition, the inhibitory effect of MQ1 was still observed after washout (Figure 4). These results suggest that MQ1 undergoes slow dissociation from the MCH1 receptor. It is generally considered that slow dissociation of an antagonist contributes to extending the receptor residence time and prolongs its effects, resulting in maximal antagonist activity in vivo (Copeland et al., 2006; Brinkerhoff et al., 2008; Copeland, 2010). For example, it has been reported that candesartan, a slowly dissociating angiotensin AT1 receptor antagonist, has a stronger antihypertensive effect than a more rapidly dissociating antagonist (Hansson, 2001; Van Liefde and Vauquelin, 2009), while the pharmacodynamics of the μ-opioid receptor antagonist buprenorphine have been attributed to its slow dissociation from the receptor (Yassen et al., 2006). Therefore, detailed evaluation of slow dissociation kinetics in order to accurately understand structure–activity relationships (SAR) may be important for maximizing the in vivo efficacy of compounds during the chemical optimization process. This concept motivated us to apply the affinity selection-MS-based equilibrium binding assay as a dissociation assay that enabled us to directly determine the Koff values of the test compounds. We found that MQ1 dissociated from the receptor with a Koff value of 0.53 h−1 (95% CI: 0.48–0.58 h−1) and its dissociation was five times slower than that of MQ2, in good agreement with the results of the washout experiments. It is noteworthy that subtle differences of the chemical structure resulted in such a significant difference of dissociation kinetics. This finding highlights the importance of understanding SAR as part of a drug discovery program. This kinetic analysis method using affinity selection-MS that we have devised should also be applicable to the development of other compounds with slow dissociation kinetics. Although a large number of MCH1 receptor antagonists have already been identified by various pharmaceutical companies, this is the first report, to our knowledge, that provides clear evidence of a compound that slowly dissociates from the receptor. Whereas some targets are reported to cause adverse side effects due to prolonged occupancy (Copeland et al., 2006; Bryant et al., 2008; Tummino and Copeland, 2008), it is thought that the unique property of MQ1 would be beneficial with respect to prolongation of pharmacodynamic efficacy in vivo.
Negative allosteric modulation
While studying the mode of the inhibition induced by MQ1, we observed insurmountable antagonism. In general, the following molecular mechanisms have been suggested to contribute to insurmountable antagonism: assessment of assay results at hemi-equilibrium, irreversible non-competitive inhibition, cytotoxicity of the test compound and negative allosteric modulation of the receptor (Ojima et al., 2011).
Firstly, to exclude the possibility that the assay was conducted at hemi-equilibrium, we incubated the cells with MQ1 and MCH for 8 h in our experiments. We did so because we considered that the system would reach equilibrium within this period, based on the results of previous experiments with different pre-incubation times (Figure 2). This hypothesis was supported by our observations that the potency of MCH obtained at each concentration of MQ1 did not change between 4 and 8 h of incubation (Supporting Information Table S1), indicating that the system reached equilibrium within 4 h. Therefore, we considered that the insurmountable antagonism exhibited by MQ1 was not due to performing the assay at hemi-equilibrium.
Secondly, we investigated the possibility that MQ1 was an irreversible non-competitive antagonist by using an equilibrium binding assay with affinity selection-MS. We found that bound MQ1 was displaced from the membrane fraction by the structurally related MQ2, suggesting that the binding of MQ1 was reversible (Figure 3).
Thirdly, to examine whether the reduction of the maximal response was due to cytotoxicity of MQ1, we evaluated cell viability after 8 h of incubation with the compound by using a CellTiter-Glo Luminescent Cell Viability assay (Promega, Tokyo, Japan). The results confirmed that there was no change in cell viability, indicating that MQ1 was not cytotoxic in this assay (data not shown).
Taken together, these findings suggested that MQ1 could be a NAM that reduces the efficiency of the receptor, resulting in insurmountable antagonism. To confirm that MQ1 had an allosteric interaction with the MCH1 receptor, we performed kinetic studies using radiolabelled MCH. The rate constants that govern the association (Kon) and dissociation (Koff) of ligands are sensitive indicators of the interaction of each ligand with a particular receptor conformation. Therefore, a change in receptor conformation induced by an allosteric modulator would theoretically be expected to lead to changes in orthosteric ligand association and/or dissociation properties (Christopoulos and Kenakin, 2002). Our kinetic studies revealed that MQ1 altered the rate of dissociation, whereas a competitive peptide agonist had no such effect, a finding that confirmed the allosteric interaction of MQ1 with MCH1 receptors. Interestingly, the dissociation rate of MCH from the receptor was reduced by MQ1 (Figure 8). Several NAMs have already been reported to display this property, for example, Ellis and Seidenberg showed that some NAMs for muscarinic acetylcholine receptors decreased the dissociation rate of orthosteric radioligands while still reducing binding affinity (Ellis and Seidenberg, 1992). Similar to these compounds, MQ1 is likely to be a NAM that induces a change in the receptor conformation, which results in the slowing of both dissociation and association.
We also performed mutation analysis to predict the binding site of MQ1 using the PathHunter β-arrestin recruitment assay. One of the advantages of using this assay is that the expression level of transfected receptors does not affect the potency of MCH, as there is a linear relationship between occupancy and effect in the PathHunter β-arrestin recruitment assay (Nickolls et al., 2011). To select residues for substitution, we used information obtained from the corticotropin-releasing factor receptor 1 crystal structure because it is the only crystal structure of a GPCR available in complex with a NAM, and because it clearly shows the allosteric site (Hollenstein et al., 2013). The potency of MCH at most of the mutant receptors is unaltered, suggesting that these mutations do not affect the overall receptor conformation and MCH binding to the MCH1 receptor, whereas alanine 136 and histidine 147, which are predicted to be within TM helix 3, might be involved in the binding of MQ1. These data further support the hypothesis that MQ1 allosterically binds to the MCH1 receptor when compared to MCH.
Finally, we demonstrated that MQ1 is a highly selective NAM for MCH1 receptors, because it had no obvious effect on other molecular targets with a high level of homology, including GPCRs. Allosteric modulators are generally considered to display considerable selectivity, presumably because many receptors exhibit greater divergence of sequence homology at allosteric sites compared with orthosteric sites (Christopoulos and Kenakin, 2002; Kenakin and Miller, 2010). Thus, the very high selectivity of MQ1 might be due to its binding to an allosteric site on the MCH1 receptor.
To date, a competitive antagonist (Borowsky et al., 2002), orthosteric insurmountable antagonist (David et al., 2007) and non-competitive antagonists (Chaki et al., 2005) for MCH1 receptors have been reported. However, to the best of our knowledge, there has been no report about a low molecular weight compound that clearly exhibits an allosteric interaction with the MCH1 receptor. The findings of this study are of importance because this was the first demonstration that there is an allosteric site of the MCH1 receptor to which a low molecular weight compound can bind. In addition, the allosteric site to which MQ1 binds could be useful in the drug discovery process for MCH1 receptor blockers, because this compound exerts preferable antagonistic effects, such as inhibition of multiple signalling pathways, slow dissociation from the receptor and high selectivity. Thus, we expect that our findings will help to accelerate the discovery of MCH1 receptor blockers that can be developed as anti-obesity agents.
Acknowledgments
The authors would like to express their gratitude to K Takami, T Okawa and T Murata for compound preparation. We also thank I Miyahisa, T Sameshima, M Hixon, K Okada, Y Hirozane and Y Shimizu for helpful discussion of data analysis. We would also like to acknowledge the support and encouragement of Y Nagisa, J Matsui and N Tarui when implementing this study.
Abbreviations
- MCH1 receptor
melanin-concentrating hormone receptor 1
- MQ1
4-(cyclopropylmethoxy)-N-(8-methyl-3-((1R)-1-(pyrrolidin-1-yl)ethyl)quinolin-7-yl)-benzamide
- NAM
negative allosteric modulator
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12529
Figure S1 Inhibitory effect of three MCH1 receptor antagonists. Concentration-dependent inhibition by MQ1 (A), MQ2 (B) and MQ3 (C) was assessed with the [125I]-MCH-(4-19) binding assay with 1 h □ and 8 h ▀ of incubation. Each data point (n=2) is plotted on the graph. Results are from representative experiments that were performed twice.
Figure S2 Time course of inhibitory effect (pIC50) of MQ1. CHO-K1-BAEA-hMCH1 receptor cells were incubated with MQ1 and 10 or 100 nM MCH for 1, 2, 4, 8, 24 and 32 h of incubation. Each data point represents pIC50 values of two independent experiments performed in quadruplicate (n=4).
Table S1 pEC50 values of MCH at different concentration of MQ1 with different incubation time. EC50 values of MCH at different concentration of MQ1 were assessed with the Path-Hunter β-arrestin recruitment assay after incubation for 4 and 8 h. All data are represented as mean ± SEM of four values from a representative experiment of three separate experiments. Statistical comparison of pEC50 values was performed using an unpaired t-test and no values were determined different (P > 0.05).
Table S2 Effects of mutations on potency for MCH and MQ1. Potency of MCH and the inhibitory effect of MQ1 for MCH1 receptor mutants were assessed with the Path-Hunter β-arrestin recruitment assay. Values are pEC50 and pIC50 means ± SEM of two independent experiments conducted in quadruplicate (n=4). Mutant values were compared with the wild type using an unpaired β-test (*P < 0.05).
References
- An S, Cutler G, Zhao JJ, Huang SG, Tian H, Li W, et al. Identification and characterization of a melanin-concentrating hormone receptor. Proc Natl Acad Sci U S A. 2001;98:7576–7581. doi: 10.1073/pnas.131200698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, et al. The melanin-concentrating hormone system of the rat brain: an immuno-and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, et al. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- Bray GA, Bellanger T. Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine. 2006;29:109–117. doi: 10.1385/ENDO:29:1:109. [DOI] [PubMed] [Google Scholar]
- Brinkerhoff CJ, Choi JS, Linderman JJ. Diffusion-limited reactions in G-protein activation: unexpected consequences of antagonist and agonist competition. J Theor Biol. 2008;251:561–569. doi: 10.1016/j.jtbi.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J, Post JM, Alexander S, Wang YX, Kent L, Schirm S, et al. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (I): in vitro effects on platelets. Thromb Res. 2008;122:523–532. doi: 10.1016/j.thromres.2008.03.026. [DOI] [PubMed] [Google Scholar]
- Chaki S, Funakoshi T, Hirota-Okuno S, Nishiguchi M, Shimazaki T, Iijima M, et al. Anxiolytic-and antidepressant-like profile of ATC0065 and ATC0175: nonpeptidic and orally active melanin-concentrating hormone receptor 1 antagonists. J Pharmacol Exp Ther. 2005;313:831–839. doi: 10.1124/jpet.104.081711. [DOI] [PubMed] [Google Scholar]
- Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, et al. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature. 1999;400:261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hu C, Hsu CK, Zhang Q, Bi C, Asnicar M, et al. Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology. 2002;143:2469–2477. doi: 10.1210/endo.143.7.8903. [DOI] [PubMed] [Google Scholar]
- Cheon HG. Antiobesity effects of melanin-concentrating hormone receptor 1 (MCH-R1) antagonists. Handb Exp Pharmacol. 2012;209:383–403. doi: 10.1007/978-3-642-24716-3_18. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- David DJ, Klemenhagen KC, Holick KA, Saxe MD, Mendez I, Santarelli L, et al. Efficacy of the MCHR1 antagonist N-[3-(1-{[4-(3,4-difluorophenoxy)phenyl]methyl}(4-piperidyl))-4-methylphenyl]-2-methylpropanamide (SNAP 94847) in mouse models of anxiety and depression following acute and chronic administration is independent of hippocampal neurogenesis. J Pharmacol Exp Ther. 2007;321:237–248. doi: 10.1124/jpet.106.109678. [DOI] [PubMed] [Google Scholar]
- Della-Zuana O, Presse F, Ortola C, Duhault J, Nahon JL, Levens N. Acute and chronic administration of melanin-concentrating hormone enhances food intake and body weight in Wistar and Sprague-Dawley rats. Int J Obes Relat Metab Disord. 2002;26:1289–1295. doi: 10.1038/sj.ijo.0802079. [DOI] [PubMed] [Google Scholar]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- Eglen RM. Enzyme fragment complementation: a flexible high throughput screening assay technology. Assay Drug Dev Technol. 2002;1:97–104. doi: 10.1089/154065802761001356. [DOI] [PubMed] [Google Scholar]
- Ellis J, Seidenberg M. Two allosteric modulators interact at a common site on cardiac muscarinic receptors. Mol Pharmacol. 1992;42:638–641. [PubMed] [Google Scholar]
- Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, et al. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1ra1. doi: 10.1126/scitranslmed.3000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson L. The relationship between dose and antihypertensive effect for different AT1-receptor blockers. Blood Press Suppl. 2001;3:33–39. doi: 10.1080/08037050152518348. [DOI] [PubMed] [Google Scholar]
- Hawes BE, Kil E, Green B, O'Neill K, Fried S, Graziano MP. The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology. 2000;141:4524–4532. doi: 10.1210/endo.141.12.7833. [DOI] [PubMed] [Google Scholar]
- Hill J, Duckworth M, Murdock P, Rennie G, Sabido-David C, Ames RS, et al. Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. J Biol Chem. 2001;276:20125–20129. doi: 10.1074/jbc.M102068200. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Ito M, Gomori A, Ishihara A, Oda Z, Mashiko S, Matsushita H, et al. Characterization of MCH-mediated obesity in mice. Am J Physiol Endocrinol Metab. 2003;284:E940–E945. doi: 10.1152/ajpendo.00529.2002. [DOI] [PubMed] [Google Scholar]
- Kasai S, Kamata M, Masada S, Kunitomo J, Kamaura M, Okawa T, et al. Synthesis, structure-activity relationship, and pharmacological studies of novel melanin-concentrating hormone receptor 1 antagonists 3-aminomethylquinolines: reducing human ether-a-go-go-related gene (hERG) associated liabilities. J Med Chem. 2012;55:4336–4351. doi: 10.1021/jm300167z. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Biased agonism. F1000 Biol Rep. 2009;1:87. doi: 10.3410/B1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo PM, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, et al. The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nat Cell Biol. 1999;1:267–271. doi: 10.1038/12978. [DOI] [PubMed] [Google Scholar]
- Low S, Chin MC, Deurenberg-Yap M. Review on epidemic of obesity. Ann Acad Med Singapore. 2009;38:57–59. [PubMed] [Google Scholar]
- Luthin DR. Anti-obesity effects of small molecule melanin-concentrating hormone receptor 1 (MCHR1) antagonists. Life Sci. 2007;81:423–440. doi: 10.1016/j.lfs.2007.05.029. [DOI] [PubMed] [Google Scholar]
- McBriar MD. Recent advances in the discovery of melanin-concentrating hormone receptor antagonists. Curr Opin Drug Discov Devel. 2006;9:496–508. [PubMed] [Google Scholar]
- Macdonald D, Murgolo N, Zhang R, Durkin JP, Yao X, Strader CD, et al. Molecular characterization of the melanin-concentrating hormone/receptor complex: identification of critical residues involved in binding and activation. Mol Pharmacol. 2000;58:217–225. doi: 10.1124/mol.58.1.217. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Harada M, Terao Y, Sugo T, Watanabe T, Shimomura Y, et al. Cloning of a novel G protein-coupled receptor, SLT, a subtype of the melanin-concentrating hormone receptor. Biochem Biophys Res Commun. 2001;283:1013–1018. doi: 10.1006/bbrc.2001.4893. [DOI] [PubMed] [Google Scholar]
- Nickolls SA, Waterfield A, Williams RE, Kinloch RA. Understanding the effect of different assay formats on agonist parameters: a study using the micro-opioid receptor. J Biomol Screen. 2011;16:706–716. doi: 10.1177/1087057111406548. [DOI] [PubMed] [Google Scholar]
- Ojima M, Igata H, Tanaka M, Sakamoto H, Kuroita T, Kohara Y, et al. In vitro antagonistic properties of a new angiotensin type 1 receptor blocker, azilsartan, in receptor binding and function studies. J Pharmacol Exp Ther. 2011;336:801–808. doi: 10.1124/jpet.110.176636. [DOI] [PubMed] [Google Scholar]
- Pereira-da-Silva M, Torsoni MA, Nourani HV, Augusto VD, Souza CT, Gasparetti AL, et al. Hypothalamic melanin-concentrating hormone is induced by cold exposure and participates in the control of energy expenditure in rats. Endocrinology. 2003;144:4831–4840. doi: 10.1210/en.2003-0243. [DOI] [PubMed] [Google Scholar]
- Pissios P, Bradley RL, Maratos-Flier E. Expanding the scales: the multiple roles of MCH in regulating energy balance and other biological functions. Endocr Rev. 2006;27:606–620. doi: 10.1210/er.2006-0021. [DOI] [PubMed] [Google Scholar]
- Presse F, Nahon JL, Fischer WH, Vale W. Structure of the human melanin concentrating hormone mRNA. Mol Endocrinol. 1990;4:632–637. doi: 10.1210/mend-4-4-632. [DOI] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, et al. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers RJ, Tschop MH, Wilding JP. Anti-obesity drugs: past, present and future. Dis Model Mech. 2012;5:621–626. doi: 10.1242/dmm.009621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Beauverger P, Naime I, Rique H, Ouvry C, Souchaud S, et al. Cloning and molecular characterization of the novel human melanin-concentrating hormone receptor MCH2. Mol Pharmacol. 2001;60:632–639. [PubMed] [Google Scholar]
- Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, et al. Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc Natl Acad Sci U S A. 2001;98:7564–7569. doi: 10.1073/pnas.121170598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y. [Searching for neurotransmitters as cognate ligands of orphan G protein-coupled receptor: finding receptor for melanin-concentrating hormone] Nihon Shinkei Seishin Yakurigaku Zasshi. 2001;21:77–82. [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Mori M, Sugo T, Ishibashi Y, Abe M, Kurokawa T, et al. Isolation and identification of melanin-concentrating hormone as the endogenous ligand of the SLC-1 receptor. Biochem Biophys Res Commun. 1999;261:622–626. doi: 10.1006/bbrc.1999.1104. [DOI] [PubMed] [Google Scholar]
- Takekawa S, Asami A, Ishihara Y, Terauchi J, Kato K, Shimomura Y, et al. T-226296: a novel, orally active and selective melanin-concentrating hormone receptor antagonist. Eur J Pharmacol. 2002;438:129–135. doi: 10.1016/s0014-2999(02)01314-6. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Van Liefde I, Vauquelin G. Sartan-AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol Cell Endocrinol. 2009;302:237–243. doi: 10.1016/j.mce.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Vaughan JM, Fischer WH, Hoeger C, Rivier J, Vale W. Characterization of melanin-concentrating hormone from rat hypothalamus. Endocrinology. 1989;125:1660–1665. doi: 10.1210/endo-125-3-1660. [DOI] [PubMed] [Google Scholar]
- Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villen J, et al. Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR) Proc Natl Acad Sci U S A. 2010;107:15299–15304. doi: 10.1073/pnas.1008461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassen A, Olofsen E, Romberg R, Sarton E, Danhof M, Dahan A. Mechanism-based pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of buprenorphine in healthy volunteers. Anesthesiology. 2006;104:1232–1242. doi: 10.1097/00000542-200606000-00019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Inhibitory effect of three MCH1 receptor antagonists. Concentration-dependent inhibition by MQ1 (A), MQ2 (B) and MQ3 (C) was assessed with the [125I]-MCH-(4-19) binding assay with 1 h □ and 8 h ▀ of incubation. Each data point (n=2) is plotted on the graph. Results are from representative experiments that were performed twice.
Figure S2 Time course of inhibitory effect (pIC50) of MQ1. CHO-K1-BAEA-hMCH1 receptor cells were incubated with MQ1 and 10 or 100 nM MCH for 1, 2, 4, 8, 24 and 32 h of incubation. Each data point represents pIC50 values of two independent experiments performed in quadruplicate (n=4).
Table S1 pEC50 values of MCH at different concentration of MQ1 with different incubation time. EC50 values of MCH at different concentration of MQ1 were assessed with the Path-Hunter β-arrestin recruitment assay after incubation for 4 and 8 h. All data are represented as mean ± SEM of four values from a representative experiment of three separate experiments. Statistical comparison of pEC50 values was performed using an unpaired t-test and no values were determined different (P > 0.05).
Table S2 Effects of mutations on potency for MCH and MQ1. Potency of MCH and the inhibitory effect of MQ1 for MCH1 receptor mutants were assessed with the Path-Hunter β-arrestin recruitment assay. Values are pEC50 and pIC50 means ± SEM of two independent experiments conducted in quadruplicate (n=4). Mutant values were compared with the wild type using an unpaired β-test (*P < 0.05).