

Shc proteins traditionally couple ligand-activated receptor tyrosine kinases to proximal effectors in signal transduction cascades. ShcD challenges this paradigm by activating the EGF receptor from within the cell, circumventing the need for extracellular stimulation.
Abstract
Proto-oncogenic Src homology and collagen (Shc) proteins have been considered archetypal adaptors of epidermal growth factor receptor (EGFR)–mediated signaling. We report that in addition to its role as an EGFR-binding partner and Grb2 platform, ShcD acts noncanonically to promote phosphorylation of select EGFR residues. Unexpectedly, Y1068, Y1148, and Y1173 are subject to ShcD-induced, cell-autonomous hyperphosphorylation in the absence of external stimuli. This response is not elicited by other Shc proteins and requires the intrinsic EGFR kinase, as well as the ShcD phosphotyrosine-binding (PTB) domain. Assessments of Erk, Akt, phospholipase C 1γ, and FAK pathways reveal no apparent distal signaling targets of ShcD. Nevertheless, the capacity of cultured cells to repopulate a wounded monolayer is markedly accelerated by ShcD in an EGFR kinase–dependent manner. Furthermore, detection of overexpressed ShcD coincident with EGFR phosphorylation in human gliomas suggests a clinical application for these findings. We thus demonstrate unique and relevant synergy between ShcD and EGFR that is unprecedented among signaling adaptors.
INTRODUCTION
The adaptor proteins of signal transduction provide vital links between receptor and effector components of a pathway, functioning to amplify or convert incoming signals or organize substrates in a cascade (Pawson, 1997). Src homology and collagen (Shc) family proteins are prototypes for this class of molecule, all sharing a common modular architecture consisting of an amino-terminal phosphotyrosine-binding (PTB) domain, a central collagen homology 1 (CH1) region rich in glycine, proline, and key tyrosine residues, and a carboxy-terminal Src homology 2 (SH2) domain. Both the PTB and SH2 domains can independently facilitate selective, motif-specific interaction with phosphotyrosine moieties in a variety of contexts, including activated receptor tyrosine kinases (RTKs) and integrins, as well as antigen, cytokine, and hormone receptors (Ravichandran, 2001; Wills and Jones, 2012). Subsequent phosphorylation of critical CH1-region tyrosine residues generates recognition motifs for proximal effectors like growth factor receptor–bound protein 2 (Grb2), rendering Shc a scaffold for development of the signaling complex (van der Geer et al., 1996a). The resulting cascade is ultimately capable of regulating many processes, including cellular differentiation, proliferation, and migration (Ravichandran, 2001; Wills and Jones, 2012). A subset of the Shc proteins also contains an extended amino-terminal CH2 region that is not directly implicated in canonical growth factor–mediated signaling (Wills and Jones, 2012).
Four mammalian Shc loci encoding eight alternatively spliced isoforms or start codon variants have been described, designated ShcA/Shc (Pelicci et al., 1992), ShcB/Sli, ShcC/Rai (O'Bryan et al., 1996; Pelicci et al., 1996), and the least-characterized homologue, ShcD or RaLP (Fagiani et al., 2007; Jones et al., 2007). Each protein has a distinct expression pattern through development and at maturity, with ShcA being nearly ubiquitous outside the nervous system, whereas ShcB and C are more restricted to the CNS (O'Bryan et al., 1996; Sakai et al., 2000). In the mouse embryo, ShcD localizes to CNS, muscle, and epithelia, as well as to multiple neural crest–derived tissues (Hawley et al., 2011). Accordingly, the adaptors make diverse physiological contributions and are differentially implicated in a variety of cancers, including those of breast, skin, and neuroendocrine tissues (Wills and Jones, 2012).
The first identified Shc binding partner was the epidermal growth factor receptor (EGFR; Pelicci et al., 1992), a member of the ErbB family of RTKs that is broadly expressed and responsive to several distinct ligands, including epidermal growth factor (EGF; Wells, 1999). The receptors are capable of activating at least five major pathways, including Ras/mitogen-activated protein kinase (MAPK; Burgess, 2008). In addition to their roles in development, regeneration, migration, and differentiation of a variety of cell types (Wells, 1999), the ErbB proteins are essential to the progression of many cancers and are established therapeutic targets (Cai et al., 2010). From the signaling perspective, ShcA has been deemed the principal EGFR adaptor (Soler et al., 1994), capable of mediating ligand-induced MAPK pathway activation (Gotoh et al., 1997). Studies using ShcA-deficient mouse embryo fibroblast cells nevertheless suggest that the adaptor's role may be subtle, as its genetic removal does not prevent MAPK activation downstream of EGF (Lai and Pawson, 2000). Instead, it reduces cellular sensitivity to low levels of ligand, supporting the hypothesis that ShcA refines the signaling process rather than controlling it outright (Lai and Pawson, 2000).
The recent discovery of ShcD raises questions about the novel contributions of this adaptor to signal transduction. ShcD has a demonstrated capacity to bind the EGFR (Smith et al., 2006) and respond to EGF signaling (Fagiani et al., 2007); however, the context and consequences of the interaction have yet to be elucidated. It has several unique structural features, including three nonconserved tyrosine residues subject to inducible phosphorylation, for a total of six in the CH1 region, at least one of which is an additional Grb2-docking site (Jones et al., 2007). It also contains a more extensive CH2 region and lacks the α-adaptin motif that is otherwise conserved across Shc proteins (Jones et al., 2007). Each permutation of the basic Shc architecture has the potential to drastically alter the signaling properties of the molecule, thus prompting us to characterize its associations and effects on EGFR.
RESULTS
The ShcD PTB domain binds inducibly to EGFR Y1148 after EGF stimulation
Spot peptide array data reveal a putative interaction between ShcD and EGFR (Smith et al., 2006). To verify this interaction and subsequently map the association to the domains and motifs responsible, we performed binding studies in the COS-1 model cell line, selected for its robust endogenous expression of EGFR, ligand responsiveness, and amenability to manipulation. In these cells, overexpressed FLAG-tagged ShcD coprecipitates the EGFR after EGF stimulation (Figure 1A). We then performed parallel immunoprecipitation assays to compare the binding capacities of ShcA, B, C, and D to the EGFR and the Grb2 adaptor in the presence and absence of ligand (Figure 1B). Consistent with previous findings, we noted that upon EGF administration, ShcA and ShcB coprecipitated a phosphorylated protein of ∼170 kDa, presumed to be the EGFR. ShcA and ShcB also became phosphorylated under these stimulatory conditions, and their capacities to bind Grb2 increased markedly. Conversely, ShcC failed to bind EGFR or Grb2 and undergo phosphorylation (Figure 1B), confirming that it is not a physiological substrate of the EGFR, as previously shown (Pelicci et al., 2002). ShcD, however, was found to have a binding profile similar to that of ShcA and ShcB, in which EGF stimulates ShcD tyrosine phosphorylation and induces its interaction with EGFR and Grb2 (Figure 1B). To examine whether this interaction is mediated by the PTB and/or SH2 domain of ShcD, we next incubated the isolated glutathione S-transferase (GST)–conjugated PTB and SH2 domains with lysate from untreated or EGF-stimulated COS-1 cells. As evidenced by Figure 1C, the ShcD PTB domain, but not the SH2 domain, demonstrated robust affinity for EGFR in the presence of EGF, and this interaction was fully abrogated by the introduction of a point mutation (R315Q; PTB*) that disrupts the pTyr-binding capacity of the domain (Jones et al., 2007). Similarly, the ShcA PTB domain exhibited strong association with the tyrosine-phosphorylated EGFR. Finally, we sought to identify the EGFR tyrosine residue(s) that interact with the ShcD PTB domain and clarify whether the association is direct or mediated by additional factors. Four tyrosine residues of the EGFR cytoplasmic tail, consisting of sites Y1086, Y1114, Y1148, and Y1173, exist within the consensus NPXY motif recognized by Shc PTB domains (van der Geer et al., 1995, 1996b). To evaluate the affinity of ShcD for these isolated regions, we performed binding assays using synthetic, biotinylated EGFR peptides spanning these sites, in conjunction with purified GST fusion proteins. Direct interaction was seen between the phosphopeptide corresponding to EGFR phosphorylated on Y1148 and both the ShcD and ShcA PTB domains, and this association was abolished with the ShcD PTB* mutation (Figure 1D). Furthermore, substitution of either the asparagine or proline to alanine in the NPXY recognition sequence (NA and PA, respectively) or replacement of phosphotyrosine with tyrosine in the EGFR Y1148 peptide produced a marked reduction in ShcD-PTB coprecipitation (Figure 1D). Collectively these findings confirm an interaction between the ShcD PTB domain and phosphorylated tyrosine residue 1148 on the EGFR.
FIGURE 1:

The ShcD PTB domain binds to phosphorylated EGFR residue Y1148 after ligand stimulation. (A) COS-1 cells transfected with FLAG-tagged ShcD or vector were serum starved and EGF stimulated. FLAG immunoprecipitations were followed by detection of EGFR. (B) Cells expressing ShcA–D were similarly processed and analyzed for phosphotyrosine and Grb2 coprecipitation. (C) Purified GST-fusion proteins corresponding to the wild-type or disabled (*) ShcD PTB and SH2 domains and the ShcA PTB domain were incubated with glutathione beads and lysate from nontransfected, untreated, or EGF-stimulated cells. (D) The ShcD docking site on EGFR was identified by combining ShcD GST-fusion domains in C with synthetic, biotin-labeled EGFR peptides corresponding to known PTB consensus sites (NPXpY). In addition to phosphorylated and nonphosphorylated EGFR variants, we used the recognition-motif mutants APXpY (pY1148NA) and NAXpY (pY1148PA). IP, immunoprecipitation; IB, immunoblot; PD, pull down; WCL, whole-cell lysate.
ShcD promotes constitutive phosphorylation of the EGFR
Despite mounting biochemical evidence of an interaction between EGFR and ShcD, the relevance of this association to cellular dynamics and signal transduction has not yet been established. We thus intended to monitor temporal, ligand-induced changes in the subcellular localization of green fluorescent protein (GFP)–conjugated ShcD and the active EGFR to establish whether and how the two constituents associate under physiological conditions. The phosphorylation status of EGFR Tyr-1068, a residue that serves as both the Grb2-docking site (Batzer et al., 1994; Rojas et al., 1996) and an indicator of intrinsic EGFR kinase activity (Gan et al., 2010), was selected as a metric of receptor activation.
To monitor site-specific modifications, we identified an antibody selective for the EGFR pY1068 motif and confirmed that it did not cross-react with the inactive receptor or phosphorylated ShcD in Western blots or immunocytochemistry (Supplemental Figure S1). We then probed ShcD-GFP–transfected COS-1 cells to discern the patterns of phosphorylation and subcellular localization that occur before and after EGF stimulation. Unexpectedly, we detected EGFR Y1068 phosphorylation prior to ligand exposure in cells expressing ShcD but not in the surrounding nontransfectants, and this signal remained elevated for the duration of the experiment (Figure 2A). Conversely, in cells lacking ShcD, EGFR phosphorylation on Y1068 was evident 30 and 60 min after stimulation, and it returned to basal levels by 120 min posttreatment (Figure 2A).
FIGURE 2:

EGFR is constitutively phosphorylated in the presence of ShcD. (A) Localization and signaling dynamics of proteins of interest were monitored over an extended time course using confocal microscopy of ShcD-GFP/GFP–transfected, serum-starved cells pulsed with 50 ng/ml EGF and chased for up to 120 min before fixation. Receptor activation was monitored by staining with an antibody specific to the phosphorylated Y1068 EGFR residue. Also shown at select time points are localization plots for green and red channels and van Steensel x-translation to quantify relationship significance. Scale bar, 40 μm. (B) The phosphorylation response was further characterized by immunoprecipitating EGFR from cellular lysates and immunoblotting with a generic phosphotyrosine antibody. Cells were maintained in serum-deprived conditions (EGF–) or stimulated for 10 min (EGF+) before lysis. (C) Comparison of the capacity of overexpressed Shc proteins to induce EGFR phosphorylation during serum withdrawal. IP, immunoprecipitation; IB, immunoblot.
We also observed considerable colocalization between ShcD-GFP and EGFR pY1068 punctae at all time points assessed. The correlation is depicted graphically by plotting individual pixel intensities of the red (pEGFR) channel on the abscissa against those of the corresponding green (ShcD) signal on the ordinate axis to interpret the degree of pattern overlap (Figure 2A). The significance of this relationship was analyzed using van Steensel x-translation to generate comparisons between the native pEGFR distribution pattern and a scrambled ShcD signal displaced in the abscissa (van Steensel et al., 1996). Pearson's correlation coefficient (r) is shown for the original merged signals and after various degrees of translation (Figure 2A). Legitimately colocalized signals produce the highest r from the unaltered images (Δx = 0), whereas translations result in rapid loss of the relationship. The characteristic peaked plot was observed with the ShcD samples but not with those expressing GFP alone (Figure 2A). Quantification likewise confirmed that the agreement between EGFR pY1068 and ShcD signals was indeed significant (Supplemental Table S1).
Whereas colocalization of Shc proteins with EGFR is a documented consequence of ligand exposure (Sato et al., 2000), it has not previously been described in the absence of stimulation, nor is it typically associated with increased receptor phosphorylation. To further investigate the intriguing observation that ShcD enhances EGFR Y1068 phosphorylation, we examined lysates from COS-1 cells expressing ShcD. Immunoprecipitation of the EGFR followed by Western blotting for generic phosphotyrosine residues confirmed that the receptor exhibits elevated levels of phosphorylation in ShcD-expressing cells after serum starvation and before ligand stimulation (Figure 2B). This trend was likewise observed in the whole-cell lysate, using both the EGFR pY1068 site-specific antibody and the phosphotyrosine antibody.
Finally, considering the amino acid sequence identity across both the PTB and SH2 domains of Shc proteins (Jones et al., 2007), we questioned whether the capacity of ShcD to facilitate ligand-free phosphorylation of EGFR extended to other members of the adaptor family. Figure 2C depicts immunoblotted lysate from transfected, serum-deprived cells expressing GFP-conjugated ShcA, B, C, or D or GFP alone. Although minimal, basal level tyrosine phosphorylation of the EGFR band (∼180 kDa) was detected in most lanes, marked up-regulation occurred only in the ShcD sample. This implies that ShcD possesses novel signaling characteristics not present in its homologues.
Multiple EGFR tyrosine residues are affected by ShcD
The technical limitations of antibody-based cellular analysis preclude comprehensive annotation and quantitation of the EGFR phosphorylation sites influenced by ShcD. Thus, to circumvent this constraint, we used a mass spectrometry (MS) technique that was previously optimized to analyze EGFR phosphorylation (Tong et al., 2009). It relies on overexpression of FLAG epitope–tagged EGFR in the HEK-293 cell line, which naturally expresses negligible levels of endogenous EGFR (Tong et al., 2008). We first sought to determine the suitability of this approach by assessing the level of EGFR-FLAG phosphorylation arising in HEK-293 cells upon transient transfection with ShcD-GFP (Figure 3A). This yielded an increase in EGFR phosphorylation similar to that observed in COS-1 cells (Supplemental Figure S2), validating the phenotype in an alternative experimental model and allowing us to proceed with MS.
FIGURE 3:

ShcD promotes phosphorylation of multiple EGFR residues. HEK-293 cells overexpressing FLAG-tagged EGFR were subjected to analysis by liquid chromatography–tandem mass spectrometry. (A) To confirm the observations made with COS-1 cells, ShcD was expressed ectopically in HEK-293-EGFR cells, followed by serum deprivation and IP/IB. (B) Tandem mass spectrum of the EGFR-derived phosphopeptide GSHQISLDNPDyQQDFFPK, where y denotes pTyr (at position 1148). The y and b series of ions are indicated along with the derived sequence of the peptide shown at the top. Additional information on this peptide is contained in Supplemental Table S1. (C) The XICs for this peptide with (left, m/z = 1158.51) and without (right, m/z = 1118.51) phosphorylation at 1148. (D) Schematic representation of EGFR residues subject to ShcD-induced phosphorylation.
For the purposes of MS analysis, EGFR-FLAG was transiently cotransfected along with untagged ShcD to afford control over receptor-adaptor stoichiometry and allow for FLAG-based purification of EGFR.
MS analysis revealed four EGFR pTyr sites in unstimulated cells cotransfected with ShcD and EGFR constructs (ShcD/EGFR) (Table 1 and Supplemental Table S2). A representative MS/MS spectrum of the EGFR pTyr peptide containing pY1148 is shown in Figure 3B. (Remaining peptide spectra are given in Supplemental Figure S3.) The mass-to-charge (m/z) difference between y7 and y8 indicates the pTyr residue (m/z = 253.0) of this peptide. To compare the relative abundance of EGFR phospho isoforms with or without phosphorylation at Y1148, we compared their extracted ion currents (XICs; Figure 3C). This indicated a greater amount of the pY1148 peptide in cells coexpressing ShcD and EGFR than in cells not expressing ectopic ShcD (Figure 3C, left, top vs. bottom). Accordingly, there was less of the nonphosphorylated version of the peptide recovered from cells coexpressing both proteins compared with cells expressing only ectopic EGFR (right, top vs. bottom). Based on these data, the estimated stoichiometry of phosphorylation at position Y1148 in ShcD/EGFR cells was calculated as ∼22% (Table 1), which was >10-fold higher than measured in control EGFR cells. Three of the four EGFR pTyr sites identified by MS analysis have elevated stoichiometry in ShcD/EGFR cells (Table 1 and Figure 3D), consistent with the Western blot result showing higher levels of EGFR phosphorylation in the ShcD/EGFR cells (Figure 3A). Therefore, the MS analysis confirmed that there are ShcD-dependent increases in the phosphorylation of multiple EGFR tyrosine residues.
TABLE 1:
Estimated stoichiometry of EGFR tyrosine phosphorylation in EGFR- or ShcD/EGFR-expressing cells.
| Estimated stoichiometry (%) | |||
|---|---|---|---|
| EGFR peptide sequencea | p-Tyr site | EGFR | ShcD/EGFR |
| (R)YSSDPTGALTEDSIDDTFLPVPEyINQSVPK(R) | 1068 | 4 | 9 |
| (K)RPAGSVQNPVyHNQPLNPAPSR(D) | 1086 | 5 | 5 |
| (K)GSHQISLDNPDyQQDFFPK(E) | 1148 | 2 | 22 |
| (K)GSTAENAEyLR(V) | 1173 | 12 | 32 |
aResidues in parentheses are deduced and shown to indicate the peptides as tryptic fragments. Phosphotyrosine is denoted by lowercase y.
ShcD requires the EGFR kinase to induce EGFR phosphorylation
Considering that Shc family proteins have no documented homology with tyrosine kinases, we were interested in identifying the enzyme(s) responsible for phosphorylating EGFR in the absence of external stimuli. The most obvious candidate was the intrinsic EGFR kinase itself, typically responsible for ligand-induced autophosphorylation of the receptor. To evaluate the contributions of this kinase to ShcD-induced phosphorylation, we used the selective EGFR tyrosine kinase inhibitor (TKI) AG 1478 (Tyrphostin) and first verified its capacity to counteract both basal and ligand-induced phosphorylation of the EGFR (Figure 4A). Subsequently, we applied the inhibitor to serum-deprived ShcD-expressing cells and recovered lysate for immunoblot analysis at five time points, ranging from pretreatment to 120-min incubation. Figure 4B reveals a steady decrease in the level of phosphorylation from 0 to 60 min, indicating that the effects of ShcD on the EGFR were reversed within 1 h of AG 1478 exposure. ShcD phosphorylation also declined rapidly upon treatment (Figure 4B), as revealed by immunoblotting with a phospho-Shc antibody that we previously validated for use with ShcD (Hawley et al., 2011).
FIGURE 4:

ShcD-induced EGFR phosphorylation requires the EGFR kinase and the ShcD PTB domain. (A) The capacity of 1 μM AG 1478 (Tyrphostin) vs. DMSO (vehicle) to prevent EGFR phosphorylation was verified using untransfected COS-1 cells in the presence and absence of 50 ng/ml EGF (10 min). (B) Subsequently, ShcD-overexpressing cells were serum starved and exposed to AG 1478 for varying amounts of time (in minutes) before lysis and immunoblotting. (C) The interplay between ShcD, the intrinsic EGFR kinase, and the cellular tyrosine phosphorylation profile was further analyzed via confocal microscopy of cells incubated with AG 1478 for 30–60 min and probed with anti-EGFR-pY1068 and anti-pTyr antibody. Scale bar, 40 μm. (D) Similarly, the SFK inhibitor PP2 (10 μM) was evaluated for its ability to counteract both ligand-induced (10 ng/ml, 10 min) and ShcD-induced EGFR phosphorylation. NTF, nontransfected cells. (E) To elucidate the molecular region of ShcD responsible for promoting EGFR phosphorylation, point-mutated variants of the protein containing disrupted PTB or SH2 domains (PTB*, SH2*) or the phosphorylation-defective tyrosine-to-phenylalanine substitutions (Y6F) were overexpressed in cells. Lysates from serum-deprived samples were immunoblotted for pTyr. IB, immunoblot.
To further investigate the extent of Tyrphostin-mediated inhibition, we used confocal microscopy to discern specific and general tyrosine phosphorylation patterns produced by cells before and after treatment with AG 1478 (Figure 4C). Predictably, the inhibitor ablated EGFR Y1068 phosphorylation (Figure 4C, top). Staining with a general antiphosphotyrosine antibody also revealed a drastic decrease in the overall extent of tyrosine phosphorylation in the cell (Figure 4C, bottom), arguing that EGFR is the major species subject to ShcD-induced phospho up-regulation and/or that any resulting proximal tyrosine-based signaling events are prevented by inactivating the EGFR kinase.
Although these data provide compelling evidence that the EGFR kinase is required for ShcD-induced phosphorylation, they do not eliminate the possibility of other collaborating factors. A prime candidate is the nonreceptor kinase c-Src, as it phosphorylates the activation lip of the EGFR kinase at residue Y845 and is believed to stabilize the enzyme, thereby potentiating catalysis (Biscardi et al., 1999). Accordingly, synergy between the EGFR and c-Src has been found to enhance tumorigenic cellular characteristics (Maa et al., 1995). To address the possibility that ShcD affects the EGFR via c-Src, we used the Src family kinase (SFK) inhibitor PP2. As depicted in Figure 4D, 30-min pretreatment with PP2 reduced the level of EGFR tyrosine phosphorylation elicited by EGF applied to nontransfected cells, whereas 90-min incubation with the TKI did not appear to counteract ShcD-induced phosphorylation. We therefore conclude that ShcD facilitates EGFR phosphorylation by influencing the intrinsic EGFR kinase in a c-Src independent manner.
Having determined the kinase responsible for phosphorylation, we next sought to identify the regions of ShcD that give rise to this phenotype. We focused on modules and residues of known and implied function, using mutant versions of the molecule that disrupt phosphotyrosine binding (PTB* and SH2*) or prevent phosphorylation of the six tyrosines in the CH1 region that coordinate proximal signaling (Tyr6Phe, or Y6F; Jones et al., 2007). Of these modifications, only the R315Q substitution in the PTB domain reduced the level of ShcD-induced EGFR phosphorylation (Figure 4E). This further emphasizes the importance of the PTB domain in mediating various aspects of the cellular ShcD-EGFR dynamic.
ShcD fails to influence major signaling nodes through EGFR
Shc proteins are classically characterized as mediators of EGF-induced MAPK signaling due to their ability to recruit Grb2 to the burgeoning signaling complex. It has become clear, however, that isoform-, homologue-, and context-specific differences exist in their signaling potentials (Wills and Jones, 2012). To elucidate the particular cascades affected by ShcD during serum starvation and after 50 ng/ml EGF exposure, we examined the phosphorylation status of a number of prominent signaling effectors. Although we found that phosphorylation increased on Akt, extracellular-signal-regulated kinase (Erk; MAPK), p38 (MAPK), and phospholipase C 1γ (PLC1γ) after ligand stimulation, the response did not appear to be further modulated by ShcD (Figure 5A). Indeed, the level of induction observed 10 min post EGF treatment was comparable between cells expressing ShcD-GFP or GFP alone (Figure 5A). Before stimulation, the phosphorylation profiles of ShcD- and GFP-expressing populations were likewise similar.
FIGURE 5:
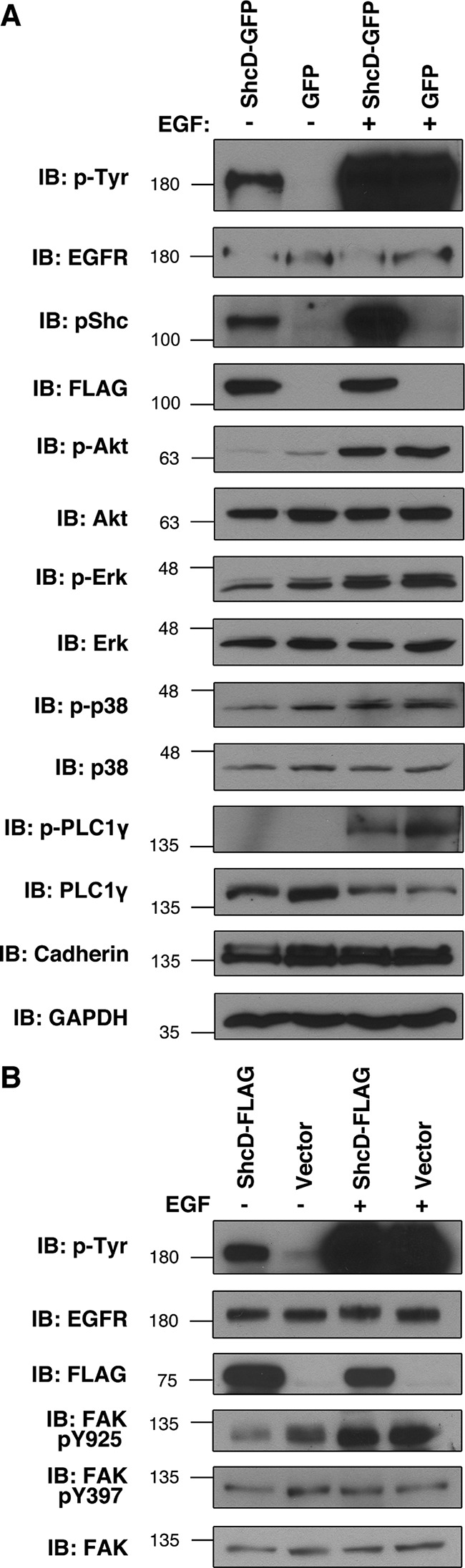
Proximal signaling through major hubs is not affected by ShcD expression. (A) The consequence of ShcD-GFP on signaling events occurring before and after stimulation with 50 ng/ml EGF (10 min) was monitored by immunoblotting lysates for common downstream effectors to compare total protein levels with the phosphorylated fraction. (B) Similarly, cells transfected with ShcD-FLAG were assayed for phosphorylation of FAK on two distinct residues.
Factors involved in cytoskeletal organization, cell adhesion, and motility can also be affected by Shc proteins (Wills and Jones, 2012). To survey the biochemical impact of ShcD on such components, we selected FAK as a central actin-mediating hub and EGFR effector and monitored its phosphorylation at two distinct motifs. The autophosphorylation residue Y397, which serves as a binding site for proteins, including Shc and Src (Mitra et al., 2005), was unaffected by the conditions of our analysis (Figure 5B). Y925, which is phosphorylated by Src and subsequently coordinates Grb2 (Mitra et al., 2005), was responsive to EGF stimulus but not modulated by ShcD expression. Thus the nature of the signaling cascade downstream of ShcD- activated EGFR remains speculative.
EGFR–ShcD synergy promotes monolayer reconstitution
Because biochemical analyses of proximal effectors failed to identify a clear target of constitutive, ShcD-induced EGFR phosphorylation, we elected to examine key cellular processes to determine whether the molecular perturbation had any phenotypic effect. Specifically, ShcD has been implicated as a mechanistic factor in melanoma metastasis, being indispensable for cellular migration (Fagiani et al., 2007). We thus questioned whether ShcD would have a similar effect in COS-1 cells and, if so, whether the enhanced migration could be attributed to the influence of ShcD over the EGFR kinase. To address each of these perspectives, we established confluent cultures of ShcD-GFP– or GFP-transfected cells, serum starved and treated them with the EGFR TKI AG 1478 or vehicle for 6 h, and introduced a narrow wound to the monolayer. The capacity of cells in each treatment to fill the void was monitored after wound infliction (T = 0 h) and again at 18 and 48 h and analyzed using TScratch software (Gebäck et al., 2009) to quantify the change in area. The results in Figure 6A depict accelerated wound closure in ShcD-expressing cultures treated with dimethyl sulfoxide (DMSO) as compared with those exposed to AG 1478 or expressing GFP alone. One-way analysis of variance (ANOVA) and Bonferroni's multiple comparison test performed on the average change in area across three biological replicates per condition confirmed a significant difference (p = 0.0038) between the ShcD plus DMSO samples and each of the other two treatments (Figure 6B).
FIGURE 6:

ShcD promotes EGFR-dependent cellular migration. (A) To determine the physiological consequences of ShcD-induced EGFR phosphorylation, confluent monolayers of transfected cells were serum starved and treated with DMSO (vehicle) or 1 μM AG 1478 6 h before inflicting scratch wounds. Matched regions of interest were imaged 0, 18, and 48 h after the procedure. Scale bar, 140 μm. (B) Wound closure was measured by T-Scratch software and expressed as Δ area. Averages of three biological replicates per condition are shown and were compared by one-way ANOVA (p = 0.0038), followed by Bonferroni's multiple comparison test. *Significant difference. Error bars denote SEM. (C) Levels of cellular apoptosis inferred from PARP cleavage patterns in suspension and adherent cells after serum deprivation and 10 min of EGF stimulation.
We were then curious to determine whether ShcD overexpression conferred resistance to apoptosis during serum starvation, as enhanced longevity could facilitate repopulation of the scratch wound. The enzyme poly(ADP ribose) polymerase (PARP) of the DNA repair response is a major target of caspase-3, which cleaves the protein at the recognition sequence DEVD-216 G-217 at the onset of apoptosis (Nicholson et al., 1995). Detection of the resulting 89- and 24-kDa fragments is thus indicative of programmed cell death. After overnight serum deprivation of ShcD- or GFP-transfected cells, we collected suspension cells and analyzed their PARP profiles in parallel with those of adherent cells. Immunoblotting with an antibody that recognizes full-length and cleaved PARP revealed that the majority of the protein had been processed by caspase 3 in suspension cells (Figure 6C). Levels of this 89-kDa form were equivalent between ShcD- and GFP-expressing populations, suggesting that both cultures were equally affected by apoptosis. Conversely, levels of the apoptotic form of PARP were minimal across both ShcD- and GFP-transfected adherent cells, whereas the full-length version was abundant, indicating a similar survival index between both treatments (Figure 6C). We therefore conclude that ShcD promotes scratch-wound repopulation without affecting cell viability.
Elevated ShcD expression and enhanced EGFR phosphorylation are detected within primary human brain tumors
To understand the potential physiological relevance of ShcD-induced EGFR activation, we sought to identify natural circumstances that promote ShcD up-regulation and involve hyperactive EGFR. The nervous system is a major site of ShcD expression (Jones et al., 2007; Hawley et al., 2011), prompting us to investigate whether oncogenic transformations in the brain are associated with changes in abundance of the ShcD adaptor. To address this question, we performed quantitative PCR (qPCR) on a selection of biopsy cores (n = 14) representing grade I–IV astrocytomas. These were analyzed in parallel with benign tissues recovered from lobectomy surgeries to determine the relative expression level of ShcD in malignant samples. Comparison of the normalized threshold cycle numbers (ΔCT) of healthy and diseased tissue revealed a significant difference between the two groups (p = 0.0020; Figure 7A). Astoundingly, this corresponds to a mean 41-fold ShcD induction in tumors relative to benign brain, with considerable variability between individual patients (SEM = 11.92; Figure 7B).
FIGURE 7:

Elevated ShcD expression and EGFR phosphorylation are characteristic of astrocytoma tumors. (A) qPCR was performed to compare ShcD mRNA levels in benign and malignant brain tissue. A significant difference (p = 0.0020) in ΔCT (threshold cycle value normalized to HPRT reference) exists between the groups. Error bars denote SEM. (B) ShcD fold induction of individual patient samples relative to normal tissue (mean, 41.29; SEM, 11.92). (C) Corresponding tissue sections were stained for the presence of EGFR pY1068 (green), tubulin (red), and DAPI (blue). (I, II) Astrocytomas, (III) benign tissue, (IV) no primary antibody control. Scale bar, 40 μm.
Matched paraffin-embedded sections were subsequently stained to detect EGFR pY1068 (green) in the context of generic cytological markers tubulin (red) and 4′,6-diamidino-2-phenylindole (DAPI; blue; Figure 7C). Although not quantitative, the resulting fluorescence immunohistochemical micrographs clearly reveal enhanced pEGFR signal in the gliomas (Figure 7C, I and II) when compared with benign tissue (Figure 7C, III) or a negative control lacking primary antibody (Figure 7C, IV). Both of the malignant samples depicted in Figure 7C showed robust ShcD up-regulation by qPCR, suggesting that elevated ShcD and EGFR phosphorylation coexist under pathological circumstances.
DISCUSSION
Through a combination of biochemical, cytological, and biophysical techniques, we have demonstrated that ShcD unexpectedly enhances EGFR autophosphorylation in the absence of external stimuli. This response is unprecedented among Shc proteins and signaling scaffolds in general; to date, the only cytoplasmic, nonkinase components identified as enhancers of EGFR phosphorylation have been members of the cytohesin family of guanine nucleotide exchange factors (GEFs; Bill et al., 2010). Our data portray ShcD as a direct, PTB-dependent binding partner of EGFR residue pY1148 that constitutively colocalizes with the active receptor and elicits ligand-independent phosphorylation of three EGFR residues via an unidentified mechanism that requires both the ShcD PTB domain and the intrinsic EGFR kinase. Although ShcD does not appear to influence signaling through common proximal effectors, the adaptor, in conjunction with the EGFR kinase, accelerates cell monolayer reconstitution after scratch injury. Of note, preliminary profiling of glioma mRNA suggests that brain tumors can be subject to drastic ShcD overexpression, implying the potential translational significance of our cellular observations.
At the molecular level, we report that ShcD has a more limited repertoire of EGFR recognition motifs than does ShcA. The latter is known to use either its PTB or SH2 domain to bind one of three distinct EGFR residues (Batzer et al., 1994; Sakaguchi et al., 1998), whereas ShcD predominantly interacts with EGFR pY1148 in a PTB-mediated manner. This requires both the correct consensus sequence (NPXY) and the phosphorylated moiety. It remains to be seen whether the absence of SH2-domain engagement alters the proximal output of ShcD-mediated cascades. Although biochemistry argues that ligand stimulation greatly enhances the interaction between EGFR and ShcD, their extensive colocalization before EGF treatment suggests that they weakly associate or exist as indirect partners in a pre-signaling complex.
The most striking feature of these pre-stimulus islands is the preponderance of phosphorylated EGFR exclusively in the presence of ShcD. Indeed, overexpression of the adaptor promotes constitutive phosphorylation of EGFR Y1068, Y1148, and Y1173 in the absence of extrinsic signals. Quantitative MS further determined that the induction level differs by residue, with the most dramatic response being the 11-fold increase over basal phosphorylation that occurs at Y1148. At the two additional sites, ShcD produces an approximate twofold to threefold phospho up-regulation. It is interesting to note that Y1148 is the sole ShcD binding site identified by our investigations. Compelling evidence that ShcA, B, and C do not affect basal EGFR phosphorylation positions ShcD as a unique member of this adaptor family.
Of importance, microscopy has established that the capacity of ShcD to facilitate EGFR phosphorylation is a cell-autonomous effect, since cocultured nontransfected cells are not subject to this phenomenon. This precludes the possibility that ShcD initiates an intrinsic autocrine or paracrine stimulus loop and instead invokes an intracellular mechanism to account for the changes in phosphotyrosine equilibrium. This ligand-free phosphorylation requires, at minimum, the ShcD PTB domain and the intrinsic EGFR kinase. Inhibition of the latter, via the selective TKI AG 1478, results in rapid loss of phosphorylation on both EGFR and ShcD, suggesting that the enzyme must be constitutively functional to sustain the phosphorylation that we observe. Considering that EGFR activation by Shc protein is a novel phenomenon, no immediate mechanisms can be inferred from behavior of the ShcD homologues. However, emerging models describe inside-out signaling and endogenous EGFR activation as novel alternatives to conventional ligand-induced transduction. The traditional perspective that the receptor is exclusively responsive to EGF or oncogenic mutation is being challenged by findings that demonstrate its sensitivity to allosteric regulation by the intracellular juxtamembrane region (Thiel and Carpenter, 2007; Macdonald-Obermann and Pike, 2009), reactive oxygen species (Goldkorn et al., 1998), and the cytohesin GEFs (Bill et al., 2010).
Considering the architecture of the adaptor molecule, it is reasonable to hypothesize that ShcD influences the enzymatic activity of EGFR by coordinating a pTyr residue, thereby protecting the site from dephosphorylation. However, there is no precedent for adaptor proteins to bind the catalytically important EGFR pY845 residue (Schulze et al., 2005), and protein recognition events at the interaction motifs of the EGFR carboxy terminus are not known to influence enzyme activity. Association-mediated protection may nevertheless help to explain the dramatic increase in phosphorylation on Y1148, the ShcD binding site. Perhaps colocalization of the adaptor with the receptor, as observed before and after ligand stimulation, shields this residue from phosphatases and thus prevents dephosphorylation. This concept of interaction-based phosphopreservation is not novel; indeed, the duration of ShcA attachment to an upstream binding partner is speculated to be an important parameter in determining phosphatase access (Suenaga et al., 2009).
Although details of the mechanism by which ShcD influences EGFR have yet to be determined, downstream consequences of this response are of prime interest. Prior investigations found that constitutive EGFR phosphorylation, even at a level below the ligand induction threshold, promotes cellular oncogenic potential (Huang et al., 1997). It has also been shown that silencing ShcD in metastatic melanomas reduces their tumorigenic capacity in nude mice (Fagiani et al., 2007). Nevertheless, reports have been inconsistent about the specific contributions that ShcD makes to major transduction pathways (Fagiani et al., 2007; Turco et al., 2012). Our discovery that ShcD does not affect EGFR signaling through the Akt, Erk, p38, PLC1γ, or FAK pathways in COS-1 cells, despite promoting receptor phosphorylation, may suggest that the cells were not captured at an informative time point after protein expression and serum starvation. The fact that ShcD transfectants sustain low levels of EGFR phosphorylation indefinitely raises questions of the subtle, long-term changes to signaling potential that may arise, and the best approaches to analyze such progression. Conversely, observations at the cellular level have been more definitive, revealing that ShcD and the EGFR kinase collaborate to facilitate cell monolayer wound closure in the absence of external stimuli. Metastatic melanoma cells likewise engage ShcD in MAPK-independent chemotactic cellular migration, prompting speculation that ShcD regulates an alternative, unidentified migratory pathway distinct from the Erk cascade (Fagiani et al., 2007).
Intriguingly, we found that the phenomenon of ShcD overexpression is not exclusive to melanomas but is also detected in gliomas. Despite reports of the low abundance of this protein in healthy tissues (Fagiani et al., 2007), it is now evident that ShcD overexpression occurs naturally under select circumstances and should be considered a pathologically relevant phenomenon. The clinical significance of EGFR deregulation is well documented in cancers of the nervous system, where overexpression and mutation of the receptor are common in high-grade tumors and indicative of poor survival (Shinojima et al., 2003). Accordingly, our immunohistochemical staining confirms that EGFR Y1068 hyperphosphorylation is characteristic of astrocytomas and occurs in tissues that likewise up-regulate ShcD. It is unclear whether ShcD plays a causal role in activating EGFR in this context, although it is noteworthy that the most common oncogenic mutation, ΔEGFR/EGFRvIII, manifests a cellular phenotype similar to that of overexpressed ShcD. Indeed, ΔEGFR sustains a low level of ligand-independent phosphorylation of residues Y1068, Y1148, and Y1173, contributing to the establishment, growth, maintenance, angiogenic potential, and overall tumorigenicity of gliomas (Huang et al., 1997; Mukasa et al., 2010; Bonavia et al., 2011).
To our knowledge, ShcD is the first noncatalytic protein found to participate in regulating the EGFR phosphotyrosine equilibrium, rendering it a novel and enigmatic component of the signal transduction machinery. Whether this phenomenon is exclusive to ShcD or exists as an unexplored paradigm in adaptor protein biology remains to be seen.
MATERIALS AND METHODS
Plasmids, antibodies, growth factors, and inhibitors
Previously, cDNA encoding full-length wild-type human ShcD (BC033907), as well as point-mutated and truncated versions of the protein, were introduced to the pcDNA3 vector (Invitrogen, Burlington, Canada) with a triple, carboxy-terminal FLAG epitope (Jones et al., 2007). These constructs, including ShcD PTB* (R315Q), SH2* (R548K), and Y6F (Y374F, Y375F, Y403F, Y413F, Y424F, Y465F; Jones et al., 2007), were subcloned into the pEGFP C2 vector (Clontech, Mountain View, CA) to generate amino-terminally tagged GFP-ShcD. Isolated PTB and SH2 domains (residues 180–351 and 524–603, respectively) were previously cloned into pGEX-4T-1 to generate GST-fused proteins (Jones et al., 2007).
Working aliquots of EGF (PeproTech, Dollard des Ormeaux, Canada) were prepared in sterile, molecular-grade water to concentrations of 50 ng/μl. EGFR kinase inhibitor AG 1478 (Tyrphostin) was acquired from Cell Signaling (Danvers, MA; product 9842) and reconstituted in DMSO to 1 mM stock. PP2 SFK Inhibitor (Tocris Biosciences, Bristol, United Kingdom) was dissolved in DMSO to a working concentration of 10 mM.
Primary antibodies were used for immunoprecipitation (IP), immunoblot, and immunofluorescence to detect total protein or phospho-specific motifs: anti-Akt (C67E7; Cell Signaling), anti-pAkt (S473; D9E; Cell Signaling), anti–pan-cadherin (ab16505; Abcam, Cambridge, MA), anti-EGFR (sc-03; Santa Cruz Biotechnology, Dallas, TX), anti–EGFR pY1068 (D7A5; Cell Signaling), anti-Erk (1/2; 137F2; Cell Signaling), anti-pErk (1/2; T202/Y204; D13.14.4E; Cell Signaling), anti-FAK (3285; Cell Signaling), anti–pFAK Y925 (3284; Cell Signaling), anti–pFAK Y397 (3283; Cell Signaling), anti-FLAG (M2; Sigma-Aldrich, Oakville, Canada), anti–glyceraldehyde-3-phosphate dehydrogenase (1D4; ABM, Richmond, Canada), anti-Grb2 (81/GRB2; BD Bioscience, Mississauga, Canada), Anti-p38 (ab7952; Abcam), anti-pp38 (T180/Y182; E229; Abcam), anti-PARP (46D11; Cell Signaling), anti-PLC1γ (D9H10; Cell Signaling), anti-pPLC1γ (Y783; 2821; Cell Signaling), anti-pShc (Y239/Y240; 2434; Cell Signaling), anti-tubulin (DM1A; Millipore, Billerica, MA), and anti-pTyr (4G10; Upstate Biotechnology, Billerica, MA). Horseradish peroxidase–conjugated goat anti-mouse and goat anti-rabbit secondary antibodies (Bio-Rad, Mississauga, Canada) were used for immunoblot detection, and goat anti-mouse or goat anti-rabbit Alexa Fluor 594 (Invitrogen) were used for immunostaining.
Cell culture, transfection, stimulation, and inhibition
COS-1 cells were maintained at 37ºC and 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). In preparation for lysis and protein recovery, cells were grown in 100 mm × 20 mm cell culture dishes with 10 ml of media; for immunofluorescence analysis they were seeded on sterile glass coverslips in 35-mm wells with 2 ml of media. Plasmid transfection was performed ∼24 h after cell passaging by introducing 2.5 or 5 μg of vector DNA in 5 or 25 μl of polyethyleneimine and 250 or 750 μl of FBS-free medium to 35- and 100-mm plates, respectively. Cells were left for a minimum of 24 h before being primed for treatment.
At 15–18 h before EGF stimulation, medium was removed from cells and replaced with serum-free DMEM containing 1% P/S. EGF was diluted in fresh DMEM to a final concentration of 50 ng/ml and applied to cells. The 100-mm dishes for protein analysis were incubated directly at 37ºC for the indicated time interval. Glass slides were pulsed with EGF at 4ºC for 30 min, washed once with 1× phosphate-buffered saline (PBS), and chased at 37ºC in fresh DMEM before fixation.
Kinase inhibitors were likewise applied after overnight serum starvation. For the purposes of system validation, nontransfected cells were simultaneously exposed to 1 μM AG 1478 (or DMSO) and 50 ng/ml EGF for 30 min before lysis. ShcD-transfected populations were treated with 1 μM AG 1478 in the absence of EGF and lysed at specific time points for immunoblot analysis. Immunocytochemistry samples were incubated with the inhibitor for 30–60 min before fixation and staining.
Transfected, unstimulated cells were incubated with 10 μM PP2 for 90 min before lysis. Nontransfected counterparts were preincubated with PP2 for 30 min and treated with 10 ng/ml EGF for an additional 10 min.
Cell lysis, immunoprecipitation, GST pull down, and peptide mapping
Immediately after stimulation, cells were placed on ice and washed once with cold 1× PBS, then lysed in 200–300 μl of PLC buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, 10 mM sodium pyrophosphate, 100 mM sodium fluoride) freshly supplemented with protease inhibitors (1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) to generate PLC Plus buffer, and centrifuged for 10 min at 13,000 rpm and 4ºC to recover the cleared supernatant.
To immunoprecipitate select proteins, fractions of total lysate (150–200 μl) were nutated overnight at 4ºC in an 800-μl volume of primary antibody, 10% anti-mouse bead-conjugated secondary antibody (100 μl), and PLC Plus buffer. Alternatively, GST-fused bait proteins (ShcD PTB and SH2 domains, ShcA PTB domain) were purified from BL21 Escherichia coli cells and incubated with whole-cell lysate, as described for immunoprecipitation. IPs were centrifuged for 1–2 min at 3000 rpm to pellet beads and aspirated to remove supernatant. Three rounds of washing with 500 μl of PLC buffer, centrifugation, and aspiration were performed. Complete removal of supernatant from pellet was achieved after the final wash. A 20-μl amount of 2× SDS protein sample buffer was added to the bead pellet and incubated at 100ºC for 5–10 min immediately before gel loading.
Peptide mapping was performed by incubating purified ShcD and ShcA GST-fusion proteins with synthetic, biotinylated EGFR peptides corresponding to NPXY motifs. A 5-μg amount of each GST-fusion protein was combined with 10 μl of the 10 mM peptide preparation in 750 μl of PLC Plus and 30 μl of 50% streptavidin agarose beads, then processed and analyzed as for immunoprecipitation.
Immunoblotting
Whole-cell lysates (WCLs) were prepared for electrophoresis by combining them 1:1 with 2× SDS buffer and boiling at 100ºC for 5–10 min.
IP and WCL samples were electrophoresed on 8–10% SDS–polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Millipore) using a semidry transfer unit (Bio-Rad). Membranes were blocked in 5% BSA in Tris-buffered saline/Tween-20 (TBST) for at least 1 h before antibody incubation. Blots were washed three times for 8–10 min each in TBST after treatment with primary (16–72 h, 4ºC with nutation) and secondary (1 h, room temperature, with rocking) antibodies and visualized with ECL chemiluminescence detection reagent (Thermo Scientific, Ottawa, Canada) and film.
Immunofluorescence staining, confocal microscopy, and colocalization analysis
At indicated time points poststimulation, glass coverslips were removed from media, washed twice in 1 ml of 1× PBS, and fixed for 10 min in 4% paraformaldehyde in PBS. Cells were then permeabilized (0.2% Triton X-100 in PBS, 5 min), rinsed in PBS, blocked in PBS containing 5% BSA for 1 h with gentle rocking, PBS washed, incubated for 1 h with primary antibody, rinsed with PBS, incubated in a sealed, light-impermeable chamber with secondary antibody, rinsed with PBS, and mounted on glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen). All PBS rinses were performed three times with 1 ml/well PBS, unless otherwise noted.
Samples were imaged at 40 or 63× magnification on the Leica DM6000 multiphoton confocal laser scanning microscope equipped with a TSC SP5 scanner at a resolution of 1024 × 1024 dpi and a pinhole of Airy 1. Cells were scanned sequentially with 405-nm diode, 488-nm argon, and 594-nm HeNe lasers to avoid channel bleedthrough, and emission data were collected a minimum of 20 nm from the excitation wavelength to eliminate laser reflection. Images were colorized and composited in ImageJ software (National Institutes of Heath, Bethesda, MD) and assembled using Photoshop CS5 (Adobe).
ImageJ microscopy plug-ins were used to quantify the extent of colocalized signal in the green and red channels. Manders colocalization analyses (Manders et al., 1993) were performed on representative images and depicted graphically by plotting individual pixel intensities of each channel on the x- and y-axes. A van Steensel x-translation was used to generate comparisons between the observed pEGFR distribution pattern and a scrambled ShcD signal displaced on the abscissa (van Steensel et al., 1996). Pearson's r was calculated for the original overlaid signals and at various degrees of translation. Quantitatively, correlation is considered significant when r generated from the actual observations, robs, exceeds the coefficient produced from multiple, randomized green-channel images, rrand, at least 95% of the time. Forty-one x-translation iterations were performed.
Mass spectrometry sample preparation and data analysis
The pcDNA3 (control vector) and pcDNA.EGFR-FLAG (Tong et al., 2009) or ShcD and pcDNA.EGFR-FLAG DNA constructs were cotransfected into HEK-293 cells. The FLAG-tagged EGFR proteins were immunoprecipitated by anti-FLAF beads, eluted with 0.15% trifluoroacetic acid, and digested with trypsin. The tryptic peptides were then analyzed by a Q Exactive linear quadrupole Orbitrap analyzer (Thermo Scientific; Tong et al., 2009).
MS data were analyzed using SEQUEST (ThermoFinnigan, San Jose, CA) and X! Tandem (The Global Proteome Machine Organization). Scaffold, version Scaffold-3.6 (Proteome Software, Portland, OR), was used to validate MS/MS-based peptide and protein identifications. Xcalibur software (Thermo Scientific) was used to quantify the XICs of peptide ions. Detailed parameters for data processing were described previously (Tong et al., 2009). The amount of phosphopeptide was indicated by estimated stoichiometry, which is the ratio of the integrated XIC of the peptide containing individual sites of phosphorylation to the sum of all integrated XICs associated with peptides of the same sequence, a method that produces an approximate value since it does not factor in the individual MS response rates of the different peptide isoforms (Wu et al., 2006).
Wound-healing assay
Cells were seeded at high density in 35-mm wells before the introduction of plasmid DNA. At 24 h posttransfection, fresh complete medium was applied overnight. Cells were then incubated in serum-free medium supplemented with DMSO (vehicle) or AG 1478 for 6 h before a scratch was inflicted on the monolayer using a p10 pipette tip. Wells were rinsed with PBS to remove debris, and fresh medium containing vehicle or inhibitor was added before acquiring images at T = 0, 18, and 48 h on a Leica DMIRE2 using Volocity Software, version 5.3.2. Four technical replicates were captured per well and matched across time points using a premarked plate grid. Replicates were omitted from further analysis if the cellular monolayer surrounding the scratch was not confluent at T = 0. Three biological replicates were conducted per sample. Wound closure was assessed using TScratch software (Gebäck et al., 2009) and expressed as the change in area (Δ area = initial open image area – final open image area). We used Graph Pad Prism, version 5.0d, to perform one-way ANOVA on the averages of technical replicates, followed by Bonferroni's multiple comparison test.
Apoptosis detection
The relative abundance of cleaved PARP in both suspension and adherent cells after overnight serum starvation was assessed. Cells suspended in the growth medium were collected by centrifugation at 1100 rpm for 5 min. The pellet was washed in 1× PBS, recentrifuged, and lysed in parallel with adherent cells for immunoblotting.
Quantitative PCR
Histological sections and snap-frozen fragments of normal and cancerous brain were provided by the Brain Tumour Foundation of Canada Brain Tumour Tissue Bank. All samples used for RNA extraction were prescreened by a neuropathologist and deemed to be of good quality, indicating a high ratio of tumor cells to necrotic, cauterized, fibrotic, or hemorrhagic tissue. Glioma samples were recovered from the tumor centers; normal brain was acquired from lobectomy surgeries. Corresponding clinical information was provided for each case.
Between 50 and 100 mg per sample of frozen tissue was homogenized in 1 ml of TRIzol reagent (Invitrogen) using a Dounce homogenizer. RNA isolation was performed according to TRIzol product documentation, beginning with an optional centrifugation at 12,000 × g for 10 min to remove insoluble material. Pelleted RNA was resuspended in 20 μl of diethylpyrocarbonate-treated water, and concentration was determined using the NanoDrop 1000 (Thermo Scientific). To remove genomic DNA contamination, 3 μg of each RNA sample was reacted with DNase I Amplification Grade (Invitrogen), for a final approximate RNA concentration of 90 ng/μl. Treated RNA was then evaluated using the Agilent Bioanalyzer 2100 and companion software (Agilent Technologies, Mississauga, Canada) to determine RNA integrity number (RIN). Only samples with RIN >5.0 were used in subsequent quantitative real-time PCR.
To detect ShcD (accession number NM_203349) mRNA transcript, we used primers developed by Perlegen Sciences (2006), which span the junction between exons 11 and 12. Hypoxanthine phosphoribosyltransferase 1 (HPRT1; NM_000194) was selected as a reference gene based on its robust and consistent performance as an endogenous control for brain tissue and malignancy (Vandesompele et al., 2002; de Kok et al., 2004; Santos and Duarte, 2008; Cook et al., 2009). Primer sequences were obtained from Vandesompele et al. (2002) and span exons 6 and 7. Additional experimental validation was performed to ensure that HPRT levels were stable across the conditions of interest. To test primer efficiencies, input cDNA was serially diluted, and threshold values were compared via linear regression of ΔCT versus log(cDNA concentration). This yielded a slope of approximately zero, indicating equal efficiencies and enabling us to use the comparative CT method of relative quantification.
All RNA expression tests were performed at the Genomics Facility, University of Guelph. A High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Burlington, Canada) was used for first-strand cDNA synthesis, with components added to the following final concentrations: 1× reverse transcription buffer, 80 nmol of dNTPs, 1× random primers, 50 U of MultiScribe reverse transcriptase, made up to 20 μl with RNA template and water. The reaction was run at 25°C for 10 min, 37°C for 120 min, 85°C for 5 min, and 4°C indefinitely.
SYBR Green reactions were prepared for quantitative analysis on a StepOnePlus real-time PCR thermocycler (Applied Biosystems). Reagents and their final concentrations were 1× SYBR Green PCR Master Mix (Applied Biosystems), 150 nM each forward and reverse primers, 5 μl of a 2× dilution cDNA template, and sterile water, for a final reaction volume of 20 μl. A no-template control was run for each primer pair in each experiment. The amplification program consisted of an initial 10-min hold at 95°C, followed by 40 cycles of denaturation (95°C for 15 s) and annealing/extension (60°C for 1 min). All runs were followed by melt curve analysis to detect dissociation of the amplicon. A dynamic range test was performed on a dilution series of RNA, revealing that 300 ng of input template was suitable. This concentration was used in all subsequent reactions. Two technical replicates of each biological sample were run.
The 2−ΔΔCT method of Livak and Schmittgen (2001) was used to express fold induction: ΔCT = target CT − normalizer CT or ShcD CT − HPRT CT. Average values obtained for malignant (n = 14) and benign (n = 2) groups were compared via t test (GraphPad Prism, version 6.0c, La Jolla, CA). ΔΔCT = (ΔCT experimental) − (ΔCT calibrator) or (ShcD ΔCT astrocytoma) − (ShcD ΔCT normal). Fold induction values for each sample relative to wild type are presented graphically.
Fluorescence immunohistochemistry
Paraffin-embedded tissue sections matched to snap-frozen tumor samples were provided by the Brain Tumour Foundation of Canada Brain Tumour Tissue Bank. Staining was performed according to antibody manufacturer's recommendations (Cell Signaling Technologies). Briefly, the sections were rehydrated in xylene, 100% EtOH, and 95% EtOH, and antigen was retrieved using a 1 mM EDTA buffer (pH 8.0) at 90°C for 15 min (Endoh et al., 2009). Samples were blocked for 60 min at room temperature and incubated overnight with anti-pEGFR and anti-tubulin sera. Goat anti-rabbit Alexa Fluor 488– and goat anti-mouse Alexa Fluor 594–conjugated secondary antibodies were applied for 60 min, and the slides were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen) and imaged on the confocal microscope.
Supplementary Material
Acknowledgments
We are indebted to Michaela Strüder Kypke and Graham Smith for microscopy training and mentorship, as well as to Graham Smith, Steve Hawley, Derek Clouthier, and Anita Acai for subcloning GFP-tagged constructs (ShcB, ShcC, ShcD-Y6F, ShcD-PTB*, and ShcD-SH2*). This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (Discovery Grant RG327372 to N.J.), the Brain Tumour Foundation of Canada (to N.J.), and the Canadian Institutes of Health Research (to M.M.). N.J. is the recipient of a Natural Sciences and Engineering Research Council of Canada University Faculty Award and a Tier Two Canada Research Chair. M.K.B.W. is a recipient of a Canadian Institutes of Health Research Vanier Canada Graduate Scholarship and a University of Guelph Brock Doctoral Scholarship, and previously received an Ontario Genomics Institute Summer Fellowship.
Abbreviaions used:
- CH
collagen homology
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Erk
extracellular-signal-regulated kinase
- Grb2
growth factor receptor–bound protein 2
- MAPK
mitogen-activated protein kinase
- PLC
phospholipase C
- PTB
phosphotyrosine binding
- RTK
receptor tyrosine kinase
- SH2
Src homology 2
- Shc
Src homology and collagen
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-08-0434) on January 15, 2014.
M.K.B.W. provided Figures 1A, 2, 3A, and 4–7, Supplemental Figures S1 and S2, and Supplemental Table S1. J.T. contributed Figure 3B and C, Supplemental Figure S2, and Table 1. S.L.T. produced Figure 1D.
The authors declare no competing interests.
REFERENCES
- Batzer AG, Rotin D, Ureña JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14:5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A, et al. Cytohesins are cytoplasmic ErbB receptor activators. Cell. 2010;143:201–211. doi: 10.1016/j.cell.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Inda MM, Vandenberg S, Cheng S-Y, Nagane M, Hadwiger P, Tan P, Sah DWY, Cavenee WK, Furnari FB. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene. 2011;31:4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263–274. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- Cai Z, Zhang H, Liu J, Berezov A, Murali R, Wang Q, Greene MI. Targeting erbB receptors. Semin Cell Dev Biol. 2010;21:961–966. doi: 10.1016/j.semcdb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook NL, Vink R, Donkin JJ, van den Heuvel C. Validation of reference genes for normalization of real-time quantitative RT-PCR data in traumatic brain injury. J Neurosci Res. 2009;87:34–41. doi: 10.1002/jnr.21846. [DOI] [PubMed] [Google Scholar]
- de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2004;85:154-159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- Endoh H, et al. Immunohistochemical analysis of phosphorylated epidermal growth factor receptor might provide a surrogate marker of EGFR mutation. Lung Cancer. 2009;63:241–246. doi: 10.1016/j.lungcan.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Fagiani E, et al. RaLP, a new member of the Src homology and collagen family, regulates cell migration and tumor growth of metastatic melanomas. Cancer Res. 2007;67:3064–3073. doi: 10.1158/0008-5472.CAN-06-2301. [DOI] [PubMed] [Google Scholar]
- Gan Y, Shi C, Inge L, Hibner M, Balducci J, Huang Y. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene. 2010;29:4947–4958. doi: 10.1038/onc.2010.240. [DOI] [PubMed] [Google Scholar]
- Gebäck T, Schulz MMP, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- Goldkorn T, Balaban N, Matsukuma K, Chea V, Gould R, Last J, Chan C, Chavez C. EGF-receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am J Respir Cell Mol Biol. 1998;19:786–798. doi: 10.1165/ajrcmb.19.5.3249. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Toyoda M, Shibuya M. Tyrosine phosphorylation sites at amino acids 239 and 240 of Shc are involved in epidermal growth factor-induced mitogenic signaling that is distinct from Ras/mitogen-activated protein kinase activation. Mol Cell Biol. 1997;17:1824–1831. doi: 10.1128/mcb.17.4.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SP, Wills MKB, Rabalski AJ, Bendall AJ, Jones N. Expression patterns of ShcD and Shc family adaptor proteins during mouse embryonic development. Dev Dyn. 2011;240:221–231. doi: 10.1002/dvdy.22506. [DOI] [PubMed] [Google Scholar]
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- Jones N, Hardy WR, Friese MB, Jorgensen C, Smith MJ, Woody NM, Burden SJ, Pawson T. Analysis of a Shc family adaptor protein, ShcD/Shc4, that associates with muscle-specific kinase. Mol Cell Biol. 2007;27:4759–4773. doi: 10.1128/MCB.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KMV, Pawson T. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev. 2000;14:1132–1145. [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald-Obermann JL, Pike LJ. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J Biol Chem. 2009;284:13570. doi: 10.1074/jbc.M109.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders E, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Mukasa A, Wykosky J, Ligon KL, Chin L, Cavenee WK, Furnari F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci USA. 2010;107:2616–2621. doi: 10.1073/pnas.0914356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- O'Bryan JP, Songyang Z, Cantley L, Der CJ, Pawson T. A mammalian adaptor protein with conserved Src homology 2 and phosphotyrosine-binding domains is related to Shc and is specifically expressed in the brain. Proc Natl Acad Sci USA. 1996;93:2729–2734. doi: 10.1073/pnas.93.7.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pelicci G, et al. A family of Shc related proteins with conserved PTB, CH1 and SH2 regions. Oncogene. 1996;13:633–641. [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Giuseppe Pelicci P. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Troglio F, Bodini A, Melillo RM, Pettirossi V, Coda L, De Giuseppe A, Santoro M, Pelicci PG. The neuron-specific Rai (ShcC) adaptor protein inhibits apoptosis by coupling Ret to the phosphatidylinositol 3-kinase/Akt signaling pathway. Mol Cell Biol. 2002;22:7351–7363. doi: 10.1128/MCB.22.20.7351-7363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlegen Sciences Modulation of skin color. 2006. U.S. Patent Application 20070148664. Available at: http://patents.justia.com/patent/20070148664 (accessed September 2008)
- Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene. 2001;20:6322–6330. doi: 10.1038/sj.onc.1204776. [DOI] [PubMed] [Google Scholar]
- Rojas M, Yao S, Lin YZ. Controlling epidermal growth factor (EGF)-stimulated Ras activation in intact cells by a cell-permeable peptide mimicking phosphorylated EGF receptor. J Biol Chem. 1996;271:27456–27461. doi: 10.1074/jbc.271.44.27456. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Okabayashi Y, Kido Y, Kimura S, Matsumura Y, Inushima K, Kasuga M. Shc phosphotyrosine-binding domain dominantly interacts with epidermal growth factor receptors and mediates Ras activation in intact cells. Mol Endocrinol. 1998;12:536–543. doi: 10.1210/mend.12.4.0094. [DOI] [PubMed] [Google Scholar]
- Sakai R, Henderson JT, O'Bryan JP, Elia AJ, Saxton TM, Pawson T. The mammalian ShcB and ShcC phosphotyrosine docking proteins function in the maturation of sensory and sympathetic neurons. Neuron. 2000;28:819–833. doi: 10.1016/s0896-6273(00)00156-2. [DOI] [PubMed] [Google Scholar]
- Santos ARA, Duarte CB. Validation of internal control genes for expression studies: effects of the neurotrophin BDNF on hippocampal neurons. J Neurosci Res. 2008;86:3684–3692. doi: 10.1002/jnr.21796. [DOI] [PubMed] [Google Scholar]
- Sato K, Kimoto M, Kakumoto M, Horiuchi D, Iwasaki T, Tokmakov AA, Fukami Y. Adaptor protein Shc undergoes translocation and mediates up-regulation of the tyrosine kinase c-Src in EGF-stimulated A431 cells. Genes Cells. 2000;5:749–764. doi: 10.1046/j.1365-2443.2000.00358.x. [DOI] [PubMed] [Google Scholar]
- Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:E1–E13. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinojima N, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol Cell Biol. 2006;26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler C, Beguinot L, Carpenter G. Individual epidermal growth factor receptor autophosphorylation sites do not stringently define association motifs for several SH2-containing proteins. J Biol Chem. 1994;269:12320–12324. [PubMed] [Google Scholar]
- Suenaga A, Hatakeyama M, Kiyatkin AB, Radhakrishnan R, Taiji M, Kholodenko BN. Molecular dynamics simulations reveal that Tyr-317 phosphorylation reduces Shc binding affinity for phosphotyrosyl residues of epidermal growth factor receptor. Biophys J. 2009;96:2278–2288. doi: 10.1016/j.bpj.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc Natl Acad Sci USA. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Taylor P, Jovceva E, St-Germain JR, Jin LL, Nikolic A, Gu X, Li ZH, Trudel S, Moran MF. Tandem immunoprecipitation of phosphotyrosine-mass spectrometry (TIPY-MS) indicates C19ORF19 becomes tyrosine-phosphorylated and associated with activated epidermal growth factor receptor. J Proteome Res. 2008;7:1067–1077. doi: 10.1021/pr7006363. [DOI] [PubMed] [Google Scholar]
- Tong J, Taylor P, Peterman SM, Prakash A, Moran MF. Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and phosphorylations at Ser1039 and Thr1041. Mol Cell Proteomics. 2009;8:2131–2144. doi: 10.1074/mcp.M900148-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco MY, Furia L, Dietze A, Fernandez Diaz L, Ronzoni S, Sciullo A, Simeone A, Constam D, Faretta M, Lanfrancone L. Cellular heterogeneity during embryonic stem cell differentiation to epiblast stem cells is revealed by the ShcD/RaLP adaptor protein. Stem Cells. 2012;30:2423–2436. doi: 10.1002/stem.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Gish GD, Lai VK, Stephens R, White MF, Kaplan D, Pawson T. Identification of residues that control specific binding of the Shc phosphotyrosine-binding domain to phosphotyrosine sites. Proc Natl Acad Sci USA. 1996b;93:963–968. doi: 10.1073/pnas.93.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Gish GD, Pawson T. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol. 1996a;6:1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Lai VK, Olivier JP, Gish GD, Stephens R, Kaplan D, Shoelson S, Pawson T. A conserved amino-terminal Shc domain binds to phosphotyrosine motifs in activated receptors and phosphopeptides. Curr Biol. 1995;5:404–412. doi: 10.1016/s0960-9822(95)00081-9. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:1–12. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, van Binnendijk EP, Hornsby CD, Van der Voort HT, Krozowski ZS, de Kloet ER, van Driel R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci. 1996;109:787–792. doi: 10.1242/jcs.109.4.787. [DOI] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- Wills MKB, Jones N. Teaching an old dogma new tricks: twenty years of Shc adaptor signalling. Biochem J. 2012;447:1–16. doi: 10.1042/BJ20120769. [DOI] [PubMed] [Google Scholar]
- Wu SL, Kim J, Bandle RW, Liotta L, Petricoin E, Karger BL. Dynamic profiling of the post-translational modifications and interaction partners of epidermal growth factor receptor signaling after stimulation by epidermal growth factor using extended range proteomic analysis (ERPA) Mol Cell Proteomics. 2006;5:1610–1627. doi: 10.1074/mcp.M600105-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.