

A unique ex vivo mock cataract surgery model is used to study the role of vimentin in repair cell function during wound healing within a clinically relevant setting. Vimentin is found to be critical for the function of repair cells in directing the collective migration of the epithelium during wound healing.
Abstract
Mock cataract surgery provides a unique ex vivo model for studying wound repair in a clinically relevant setting. Here wound healing involves a classical collective migration of the lens epithelium, directed at the leading edge by an innate mesenchymal subpopulation of vimentin-rich repair cells. We report that vimentin is essential to the function of repair cells as the directors of the wound-healing process. Vimentin and not actin filaments are the predominant cytoskeletal elements in the lamellipodial extensions of the repair cells at the wound edge. These vimentin filaments link to paxillin-containing focal adhesions at the lamellipodial tips. Microtubules are involved in the extension of vimentin filaments in repair cells, the elaboration of vimentin-rich protrusions, and wound closure. The requirement for vimentin in repair cell function is revealed by both small interfering RNA vimentin knockdown and exposure to the vimentin-targeted drug withaferin A. Perturbation of vimentin impairs repair cell function and wound closure. Coimmunoprecipitation analysis reveals for the first time that myosin IIB is associated with vimentin, linking vimentin function in cell migration to myosin II motor proteins. These studies reveal a critical role for vimentin in repair cell function in regulating the collective movement of the epithelium in response to wounding.
INTRODUCTION
In response to injury, a repair process essential to the homeostasis and survival of an organism is quickly initiated to regenerate the damaged tissue. After wounding of an epithelial tissue, reepithelialization involves “collective migration” of the epithelial cells into the wounded area, a process that is regulated by “leader” cells at the wound edge (Friedl and Gilmour, 2009; Khalil and Friedl, 2010; Weijer, 2009; Walker et al., 2010). These leader cells have a mesenchymal-like phenotype, including front-to-back polarity (Revenu and Gilmour, 2009) and the extension of polarized membrane protrusions (filopodia, lamellipodia) in the direction of wound closure (Omelchenko et al., 2003; Pastor-Pareja et al., 2004; Poujade et al., 2007). Despite their highly migratory phenotype, leader cells remain integrally linked to the collectively migrating epithelium that follows them. Although leader cells are important to movement of the epithelial sheet to close the wound, the mechanisms by which they regulate this process are unclear.
Leader cells can emerge in tissue or culture conditions from a partial mesenchymal transition of the endogenous epithelium (EMT) or alternatively from cells with a mesodermal lineage, such as fibroblasts or fibrocytes. Such cells are believed to direct the collective cell migration/invasion of epithelial cancer cells (Gaggioli et al., 2007; Kitamura et al., 2007; Montell, 2008; Weijer, 2009; Khalil and Friedl, 2010), enabling the epithelial cells following behind them to retain their intrinsic characteristics as they are directed to move to a new site. In a mock cataract surgery wound-healing model, which provides the opportunity to examine the injury repair process in response to a clinically relevant wounding, the leader cells that direct epithelial wound healing are derived from a mesenchymal repair cell progenitor subpopulation innate to the epithelium (Walker et al., 2007, 2010). In response to injury, the repair cell progenitors are activated, expand in number, assume a migratory morphology, and move over the epithelial cells’ apical surfaces to reach the wound edge (Walker et al., 2010). Here repair cells have a requisite function in regulating wound closure (Walker et al., 2010).
A defining feature of repair cells in most wound-repair models, including mock cataract surgery, is their high expression of vimentin intermediate filaments (Walker et al., 2010). Vimentin is considered a hallmark of the mesenchymal phenotype, expressed by cells involved in migration (Mendez et al., 2010), wound healing (SundarRaj et al., 1992; Buisson et al., 1996), and metastasis (Hendrix et al., 1996). Migration is impaired in vimentin-deficient fibroblasts (Eckes et al., 1998), and wound healing is delayed in vimentin-null mice (Eckes et al., 2000). Despite the connection between vimentin-rich cells and both cell migration and wound repair (Eckes et al., 1998, 2000; Gilles et al., 1999; Rogel et al., 2011), much remains to be learned about the mechanisms by which vimentin regulates leader cell function in the wound repair process. Here we investigate the role of vimentin in repair cells during wound healing.
In studies with a mock cataract surgery model we discovered that vimentin was intrinsic to the migratory function of repair cells and to their role in ensuring normal wound closure. At the leading edge of the wound, the repair cells extend vimentin-rich, actin-poor lamellipodial processes. The vimentin filaments are linked to paxillin-rich focal adhesion contacts at the cells’ lamellipodial tips, a region also rich in the motor protein myosin IIB. Biochemical analysis provides the first evidence that the motor protein myosin IIB is complexed to vimentin filaments. Disrupting vimentin function by small interfering RNA (siRNA) knockdown or with withaferin A (WFA), a drug that interferes with vimentin function, blocks the ability of repair cells at the wound edge to extend lamellipodial processes and slows wound closure. These studies position vimentin as a crucial cytoskeletal mediator of repair cell function in the wound-healing process of lens epithelial cells in response to mock cataract surgery.
RESULTS
Leader/follower paradigm of epithelial wound healing in an ex vivo mock cataract surgery injury model
Ex vivo mock cataract surgery epithelial explants have distinct advantages over other culture models for investigating the regulatory mechanisms of wound repair (Walker et al., 2010). One of the most significant is the ability to examine and manipulate the natural wound-healing process within a cell's native microenvironment. Also central to the value of this particular wound model are the intrinsic properties of the lens itself—it is a self-contained organ surrounded by a thick basement membrane (the lens capsule), avascular, not innervated, with no associated stromal compartment. Because the lens is removed from the embryo before performing the mock cataract surgery, the cells involved in regulating the repair process are limited to those innate to the lens. The wounding associated with cataract surgery results from extraction of the differentiated lens fiber cells through a small opening in the anterior lens capsule (Figure 1A; Walker et al., 2010). This procedure leaves behind both the lens epithelium, which borders the fiber cells in the intact lens, and a subpopulation of mesenchymal repair cell progenitors whose progeny are essential for effective wound healing of the lens epithelium (Walker et al., 2010). For unobstructed imaging of the response of both the lens epithelium and its associated repair cells to injury, the wounded lens was flattened on a tissue culture substrate cell side up, forming a star-shaped explant (Figure 1B; T0). The epithelial cells surrounded a highly reproducible circular wound area of ∼3 × 106 μm2 (Figure 1, B and C; T0). The wound edge (leading edge) was the site where the extracted lens fiber cells had met the lens epithelium. In response to this mock cataract surgery, the epithelial cells migrated across the denuded area of the basement membrane to repair the wound area (Figure 1B, D1–D3).
FIGURE 1:

The lens ex vivo wound repair model. (A) Mock cataract surgery was performed on E15 chick embryo lenses ex vivo, and cuts were made in the anterior lens capsule to flatten the wounded lens epithelium (green) on a culture dish. (B) The wounded epithelium remained attached to its endogenous basement membrane—the lens capsule—in the OAZ, just adjacent to the wound area, the region of the basement membrane (BM) from which the lens fiber cells were removed and onto which the lens epithelial cells migrate to fill the wound area, which becomes the CMZ. The dashed white line represents the border of the OAZ and CMZ. Here, a single wounded lens explant is imaged from the time of injury (T0) through day 3 in culture. Bar, 500 μm. (C) Quantification of wound closure, completed within 3 d in culture over an average of six experiments. (D) High-magnification image of cells at the leading edge in the CMZ, similar to the region boxed in B. Arrows identify the repair cells at the leading edge and the follower cells of the lens epithelium, modeled on the right. Bar, 100 μm. (E) Model depicting the OAZ and the CMZ, which is composed of cells located at the leading edge and lens epithelial follower cells.
For ease of reference, the region of the capsule to which the lens epithelium is originally attached was named the original attachment zone (OAZ; Figure 1, A, B, and E). It is located on the projections of the star-shaped explant. The circular cell-free area of the basement membrane in the middle of the explant from where the lens fiber cells were removed, and onto which the injured lens epithelial cells must migrate to heal the wound, will be called the cell migration zone (CMZ; Figure 1, A, B, D, and E). As occurs after cataract surgery in vivo and is characteristic of the wound-healing processes of epithelia, the injured lens epithelial cells emerged from the OAZ and migrated collectively across the CMZ (Figure 1, B and D, modeled in E), paralleling the mechanism known as collective migration. The repair cell progenitors, which in the uninjured lens are located in small niches among the cells of the lens epithelium, give rise to a population of mesenchymal repair cells that migrate to the wound edge immediately upon injury and function as the leader cells in the repair process (Walker et al., 2010). At the leading edge of the CMZ the repair cells (Figure 1, D, and modeled in red, E) extended cellular protrusions along the lens basement membrane capsule in the direction of movement.
The collective movement of the epithelium onto the denuded lens capsule in response to wounding began rapidly, detectable within a few hours of injury. Wound closure was extensive by 1 d postinjury, the open wound area measuring on average 996,000 μm2, representing 67% closure of the wound (Figure 1, B and C). The repair process was completed within 3 d. The classical properties of this ex vivo wound repair model and its similarity to the wound repair process in vivo provided a unique opportunity to investigate the molecular mechanisms that govern repair cell function in response to injury of an epithelium.
Vimentin intermediate filaments are the dominant cytoskeletal elements of repair cells at the wound edge
To elucidate the functional properties of repair cells responsible both for their highly motile phenotype and their role in directing the response of an epithelium to wounding, we investigated the subcellular organization of their cytoskeleton after lens injury. These studies included examination of the organization and distribution of microfilaments, microtubules, and vimentin intermediate filaments, cytoskeletal elements whose coordinate action have been implicated in cell migration and wound repair. After mock cataract surgery the injured lens explants were fixed at 1 d postinjury, labeled for vimentin, and double stained for F actin (Figure 2, A–C) or α-tubulin, a principal component of microtubules (Figure 2, D–F). The cells at the leading edge were imaged by confocal microscopy. Although actin filaments are the cytoskeletal element typically implicated in cell motility, surprisingly few actin filaments could be detected in the lamellipodial processes extended by the repair cells (Figure 2, B and C, arrow). In contrast, there was a relatively high concentration of actin filaments comprising the cytoskeleton of the collectively moving epithelial cells located just behind the repair cells at the wound edge (Figures 2, B and C). Unique to the highly motile repair cell population was their rich concentration of vimentin intermediate filaments, which extended throughout these cells and their lamellipodial extensions through to the lamellipodia tips (Figure 2, A, C, D, and F). Just behind the leading edge, more-stable vimentin intermediate filaments were predominant (Figure 2A, arrow), whereas near the tips of the extended lamellipodia, nonfilamentous vimentin particles and squiggles (short intermediate filaments) were also present (Figure 2A, arrowhead). Vimentin particles and squiggles are more-soluble forms of vimentin that can serve as precursors to mature polymerized vimentin filaments (Prahlad et al., 1998; Herrmann and Aebi, 2000) and also can have separate functions where they may participate in regulating the migratory function of repair cells during wound healing. The vimentin- rich protrusions extended at the leading edge by the repair cells were verified as lamellipodia by the high-level expression of the lamellipodial marker cortactin (Supplemental Figure S2). Microtubules were also present in the lamellipodia of these cells, often coincident with the vimentin filaments, but were most highly concentrated in the rearward aspects of the repair cells (Figure 2, D–F). These results showed that vimentin filaments were the principal cytoskeletal element of the repair cells at the leading edge and suggested that there was a principal role for vimentin filaments in directing movement of the mesenchymal repair cells during wound healing of the lens epithelium.
FIGURE 2:

Extension of vimentin-rich lamellipodia at the wound edge depends on microtubule function. At 1 d postinjury, wounded lens explants were immunofluorescence labeled for vimentin (A, D, red) and α-tubulin (E, green) or colabeled for F-actin using fluorescent phalloidin (B, green), overlaid in C and F. Confocal imaging focused at the wound edge revealed that the protruding processes of the repair cells had few actin filaments but were rich in vimentin intermediate filaments. Filamentous vimentin (arrow) was concentrated toward the rear aspects of the repair cells, whereas nonfilamentous vimentin particles and squiggles (arrowhead) were observed near the lamellipodia tips (A–C). α-Tubulin (E, green) was detected within lamellipodia coincident with vimentin but was more concentrated at the back end of the cells. Exposure of wounded lens explants to the microtubule depolymerization drug nocodazole (3 μM) caused disassembly of microtubules of the repair cells at the wound edge (H, red, compare to control, G, red). Vimentin filaments, which normally extend throughout the repair cells (control, I, green), were collapsed and aggregated when the microtubule network was depolymerized (nocodazole, J, green). Coincident with the loss of microtubules and vimentin-rich lamellipodia extensions, exposure to nocodazole had a significant inhibitory effect on wound closure, shown in phase contrast (K) and quantified as an average of three experiments (L). Bar, 20 μm (A–J), 500 μm (K). Images presented as confocal projected images. Arrows identify repair cell at the wound edge (A–F). Secondary antibody controls were performed, which demonstrated specificity of antibody staining (Supplemental Figure S1).
Microtubule function is required for extension of vimentin-rich lamellipodia by repair cells
Because the distribution of intermediate filaments in the cell is dependent on the microtubule network, the depolymerization of microtubules has been used as an approach to collapse intermediate filaments (Geuens et al., 1983; Forry-Schaudies et al., 1986). Exposure of injured lens epithelial explants to the microtubule depolymerization agent nocodazole from the time of wounding throughout the first day in culture effectively depolymerized microtubules and collapsed the vimentin cytoskeleton and the vimentin-rich lamellipodial protrusions normally extended by repair cells at wound edge (Figure 2, H and J). The dual effect of nocodazole on microtubules and vimentin intermediate filaments dramatically impaired wound closure (Figure 2, K and L), implicating both of these cytoskeletal elements in the wound repair process in the mock cataract surgery wound repair model.
Vimentin filaments of repair cells are linked to paxillin-rich focal adhesions
The discovery of vimentin-rich intermediate filaments as the primary cytoskeletal element in the lamellipodial processes of repair cells suggested that in these cells, intermediate filaments might be a primary link between adhesion plaques of these motile cells and the cytoskeleton. For these studies, paxillin was used to label these classical adhesion structures, as it is an integral component of integrin focal adhesion plaques (Figure 3, A and D, arrows). Wounded lens explant cultures were fixed at day 2 postinjury, immunolabeled for paxillin, costained for F-actin (Figure 3B) or vimentin (Figure 3E), and imaged by confocal microscopy. Prominent paxillin-rich focal adhesions were localized at the tips of the lamellipodial protrusions of the repair cells at the leading edge of the wound (Figure 3, A, C, D, and F, arrows). As shown earlier, little filamentous actin was extended into the lamellipodia or linked to paxillin focal adhesion plaques (Figure 3, B and C). Instead, significant colocalization was found between vimentin filaments and these focal adhesions, with vimentin intermediate filaments extending to and appearing to insert into the site of these focal adhesion structures (Figure 3, E and F, arrow and inset), revealing that vimentin intermediate filaments are the predominant cytoskeletal link to the paxillin-rich integrin adhesion plaques of repair cells.
FIGURE 3:

The relationship between α6β4, paxillin, and vimentin in the protrusive processes extended by repair cells at the wound edge. Vimentin (E, green) colocalized and extended to and inserted into prominent paxillin-rich adhesion plaques (A, D, red) at the tips of these protrusive processes, which contained little actin (B, green) filaments. (C) Overlay of paxillin and actin. (F) Overlay of paxillin and vimentin. Arrows identify paxillin-rich focal adhesions preferentially linked to vimentin. Inset, vimentin and paxillin colocalization at higher magnification (D–F). (G) Coimmunoprecipitation analysis of wounded explants at D1–D3 in culture examined in wounded explants after separation into OAZ and CMZ fractions. Samples were immunoprecipitated for paxillin and the immunoprecipitates immunoblotted for vimentin and paxillin. Blots included whole-cell lysates (WCLs) blotted for total vimentin and paxillin expression in each fraction. Results were quantified over three independent experiments and are represented as the ratio of vimentin to paxillin in each fraction (H). Results demonstrate that vimentin interacts with paxillin predominately in the CMZ, with the greatest interaction occurring at D2, a time of significant motility for wound closure (H). Cells were labeled for α6 integrin (I, red) or β4 integrin (L, red) and colabeled with vimentin (J, green) or F-actin (M, green), respectively, with overlays in K and N, at 1 d after injury. Both α6 integrin and β4 integrin were concentrated in the protrusive processes of the repair cells, into which vimentin intermediate filaments but few F-actin filaments were extended (arrows). All images are confocal projected images except insets of paxillin, vimentin, and overlay (D–F, insets) and α6 integrin, vimentin, and overlay (I–K). (A–F, and I–N) were taken at the leading edge of the CMZ. Bar, 20 μm, for inset, 5 μm. Secondary antibody controls were performed, which demonstrated specificity of antibody staining (Supplemental Figure S1).
To further examine the association between paxillin and vimentin in the wound-repair cultures, we performed coimmunoprecipitation analysis (immunoprecipitate [IP], paxillin; blot, vimentin; Figure 3G). For this study, the CMZ and the OAZ were separated by microdissection to examine these regions separately by biochemical analysis, providing a powerful approach for studying migration-specific molecular interactions. OAZ and CMZ regions of the ex vivo wounded explants were analyzed at days 1, 2, and, 3 in culture. Vimentin was discovered linked to paxillin predominately in the CMZ, the zone of migration (Figure 3, G and H), most highly at day 2 postinjury, the same time point at which paxillin adhesion plaques were so prominently observed by immunolocalization analysis (Figure 3, A and D). The association between vimentin and paxillin was significantly diminished by day 3 postinjury, consistent with a time when wound closure typically was completed and leader cell function no longer required (Figure 3H). These results confirm the role of vimentin intermediate filaments as the cytoskeletal elements linked to paxillin focal adhesion plaques during the period of greatest cell migration to repair the wound.
Integrin α6β4 enriched in lamellipodial tips of repair cells at the wound edge
Integrin α6β4 is distinct among most integrin cell-matrix adhesion receptors for its ability to link to intermediate filaments (Gomez et al., 1992; Rezniczek et al., 1998). Initially discovered as the integrin receptor of hemidesmosomes (Carter et al., 1990; Stepp et al., 1990), α6β4 integrin has since also been assigned a role in cell motility, first identified in cells associated with disease states such as cancer (Rabinovitz et al., 1995). We performed immunolocalization analysis for both α6 and β4 integrin subunits in the lens wound repair cultures at 1 d postinjury to investigate whether α6β4 could function as the cell-surface receptor linked to vimentin intermediate filaments in the repair cells at the leading edge of the wounded lens epithelium. Wounded mock cataract surgery cultures were immunostained for α6 or β4 integrin and costained for vimentin or colabeled with F-actin. The results show that the vimentin-rich repair cells were high expressers of both α6 and β4 integrins (Figure 3, I–N, arrows), which highly localized to the lamellipodial protrusions of repair cells at the leading edge of the wound and were often punctate in appearance. Vimentin filaments were enriched in lamellipodial extensions also rich in α6 integrin (Figure 3K), a region with few F-actin filaments (Figure 3M). The punctate staining observed for both α6 and β4 integrins in these cells (Figure 3, I and L) could reflect both more discrete types of adhesion structures and integrin trafficking. Integrin recycling that is associated with directed cell migration has been shown to involve vimentin function (Ivaska et al., 2005; Fortin et al., 2010). These data suggested that vimentin filament function in the repair cells was linked to its interaction with α6β4 integrin in the cells’ lamellipodial extensions.
Vimentin solubility increased in migratory cells during lens wound healing
Intermediate filaments were first described for their function as stable, rope-like structures that provide resistance to mechanical stress (Lazarides, 1980; Kim and Coulombe, 2007). It is now understood that intermediate filaments are in fact dynamic structures (Prahlad et al., 1998; Helfand et al., 2004; Eriksson et al., 2009) and that vimentin can exist in different states of assembly and subcellular organization (Kim and Coulombe, 2007; Goldman et al., 2008, 2012). Precursors to mature polymerized vimentin filaments include detergent-soluble nonfilamentous vimentin particles and short intermediate filament structures (squiggles) that can form functional pools with distinct cellular roles (Ivaska et al., 2007; Kim and Coulombe, 2007; Goldman et al., 2012). For instance, in response to nerve cell injury, soluble vimentin mediates the long-distance retrograde transport of phosphorylated Erk from the axon to the cell body (Perlson et al., 2005). Here we examined whether there were dynamic changes in the state of vimentin organization associated with cell migration in response to injury by examining whether there were increases in vimentin solubility. To accomplish this analysis, we examined the state of vimentin solubility/insolubility at the time of injury (T0) and compared it with that in both the OAZ and the region of active migration (CMZ) at day 1 (D1) postinjury (modeled in Figure 1D). Vimentin solubility was determined by its detergent extractability. Tissue samples were extracted sequentially with 1) Triton X-100, a mild, nonionic detergent, to isolate the most soluble forms of vimentin, 2) Triton X-100/octylglucoside (TX/OG) to isolate membrane proteins, including integrin receptors, in lipid rafts (Brown and Rose, 1992) and their associated cytoskeleton, and 3) a 4% SDS-containing buffer that solubilizes the highly detergent-insoluble (HI) cytoskeletal fraction, including the classical, highly stable intermediate filament population (Leonard et al., 2011). At the time of injury (T0) the majority of the vimentin filaments (60%) were present in a highly detergent-insoluble (HI) form; the remainder was divided between the highly soluble and lipid raft–linked fractions (Figure 4A). Vimentin solubility changed dramatically in the zone of cell migration in response to injury, with only ∼33% of vimentin remaining highly insoluble and the majority of vimentin (67%) moving to the soluble, detergent-extractable fractions (Figure 4C, 45% TX soluble, 22% TX/OG soluble). Of interest, the most significant increase in solubility was in the lipid raft fraction. Lipid raft–associated vimentin is known to function as a scaffold on which to recruit and localize signaling complexes (Runembert et al., 2002; Sprenger et al., 2006; Meckes et al., 2013). Therefore, the increase in the lipid raft–associated vimentin in the CMZ suggested a specific function for vimentin at these specialized microdomains in signaling required for cell migration. The results of these studies involving changes in solubility of vimentin in response to wounding demonstrated that dynamic changes in vimentin intermediate filament organization are involved in regulating the function of this cytoskeletal element in cell migration in response to wounding.
FIGURE 4:
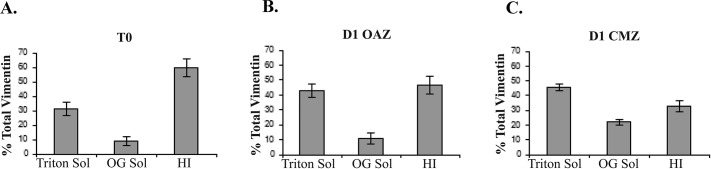
Vimentin solubility increased within the CMZ in response to injury. Ex vivo wounded explants were collected immediately after injury (time 0) and at 1 d postinjury (D1) after separating by microdissection into OAZ and CMZ domains (A–C). Samples were then sequentially extracted in Triton X-100, which solubilizes cytoplasmic and membrane-associated proteins, and TX/OG, which isolates membrane-associated proteins and lipid rafts, and TX/OG-resistant pools of vimentin were solubilized in SDS (4%). The SDS fraction is referred to as the highly insoluble population (HI). In contrast to the SDS, both the Triton and TX/OG fractions represent soluble pools of vimentin. The amount of soluble vimentin increased from T0 to D1 postinjury within the cell population, migrating across the CMZ (C). At T0, 60% of vimentin filaments were present in the HI fraction, whereas only ∼33% of vimentin remained HI in the CMZ (A, C). Within the CMZ the majority of vimentin remained soluble (45% TX soluble and 22% TX/OG soluble) (C). The percentage total amount of vimentin in each fraction was quantified over five independent experiments.
Knockdown of vimentin prevents formation of lamellipodial protrusions at the wound edge and suppresses wound closure
High-level expression of the vimentin intermediate filament protein is a prominent feature of the repair cell population that modulates wound closure in the mock cataract surgery wound repair model (Walker et al., 2010). This prominence of vimentin is a feature of the repair cells both before injury, when they are present in niches as precursor cells, and after their activation in response to wounding (Walker et al., 2010). These repair cells migrate quickly to the wound edge (Walker et al., 2010), where the studies presented here show that they extend vimentin-rich, F-actin–poor lamellipodia along the basement membrane capsule exposed by the injury (Figure 2, A–C). These results suggested that as a defining feature of repair cells at the wound edge, vimentin could be required for extension of these lamellipodial processes and, more important, the function of these vimentin-rich repair cells in wound closure. To investigate the possibility of a functional role for the vimentin intermediate filament protein in the repair process, we used a vimentin siRNA knockdown approach. For this study we knocked down vimentin expression at the time of wounding in the mock cataract surgery ex vivo wound repair culture model. This siRNA approach effectively suppressed vimentin expression (Figure 5A), and as a result the cells at the leading edge of the wound were prevented from extending lamellipodial protrusions (Figure 5B), and wound closure was suppressed (Figure 5, C and D). These results provide strong evidence that vimentin is essential for lamellipodial protrusion formation by the vimentin-rich cells at the wound edge and for closure of a wound created by mock cataract surgery in our wound repair model.
FIGURE 5:

Knockdown of vimentin prevented lamellipodial extension at the wound edge and slowed wound closure. A siRNA approach was used to knock down vimentin expression (SiVim) in the ex vivo wounded explants to determine the effects on extension of lamellipodial processes at the wound edge and wound closure. Control cultures were transfected with siControl nontargeting siRNAs (SiC). (A) Immunoblot analyses show a reduction in vimentin expression. Results were quantified over four independent wounded ex vivo explants for each treatment and are represented as a ratio of vimentin to GAPDH. (B, high magnification) Protrusion formation at the leading edge is significantly inhibited in SiVim compared with SiC-treated ex vivo wounded explants, shown in phase contrast imaging. (C) Wound closure for SiC-treated and SiVim-transfected ex vivo wounded explants, in phase contrast imaging. Transfection of SiVim significantly inhibited wound closure compared with SiC-transfected ex vivo wounded explants. (D) Wound closure quantified for six independent wounded ex vivo explants for each treatment.. Bar, 500 μm (phase images), except for high magnification in B, 250 μm.
Inhibition of vimentin with WFA impairs repair function and slows wound repair
Type III intermediate filament proteins such as vimentin have been identified as potent in vivo druggable targets for WFA, a steroidal lactone, as in the case of tumor suppression (Lahat et al., 2010), corneal fibrosis, and glaucoma (Bargagna-Mohan et al., 2012, 2013). WFA can covalently bind to vimentin, causing aggregation of intermediate filaments, altering filament distribution and disrupting filament function (Bargagna-Mohan et al., 2007). Of interest, soluble forms of vimentin are highly sensitive to WFA (Bargagna-Mohan et al., 2013). We exposed newly wounded mock cataract surgery cultures to WFA as an alternative mechanism to investigate the role of vimentin in this wound repair model. Immunofluorescence analysis at 1 d postinjury verified that WFA had a significant effect on the organization of the vimentin intermediate filaments in repair cells and on repair cell morphology. In the presence of WFA the vimentin filaments aggregated and clustered around the nucleus. In response to collapse of the vimentin filament network and subsequent loss of tensegrity, repair cells became highly rounded and failed to maintain their mesenchymal shape (Figure 6A). The dose-dependent effect of WFA on repair cell localization was determined after 24-h exposure to the drug beginning at the time of injury. The cells were labeled for both vimentin and F-actin and imaged by confocal microscopy. Z-stacks were collected and the results displayed both as a single optical x-y plane (Figure 6B, bottom) and in an orthogonal view (Figure 6B, top). Treatment with 1.5 μM WFA had only minimal effect on the repair cells, whereas a dose of 2.5 μM WFA and higher caused significant cell rounding, and the repair cells accumulated and piled up near the wound edge. At the two higher concentrations of WFA (2.5 and 3.5 μM), as with the vimentin siRNA knockdown studies, the repair cells failed to move onto and extend lamellipodia along the wounded area of the lens basement membrane capsule (Figure 6B). This phenomenon was seen best in the orthogonal view (Figure 6B).
FIGURE 6:

Disruption of vimentin function with WFA impaired extension of vimentin-rich lamellipodia by repair cells at the wound edge and slowed wound healing. (A) Immunostaining for vimentin (red) in wounded explants exposed to 3.5 μM WFA demonstrates that this drug alters the intermediate filament networks of the repair cells and their cellular phenotype. The cells appear rounded, and their vimentin filaments are aggregated around the nucleus. (B) To determine the dose-dependent effect of WFA on repair cells, wounded lens explants were imaged at the wound edge by confocal microscopy after immunostaining for vimentin (red) and costaining for F-actin (green). Orthogonal cuts through Z-stacks were collected to examine the organization of the repair cells at the wound edge. The lowest concentration tested, 1.5 μM WFA, had the least effect on repair cell morphology and their ability to extend lamellipodia along the basement membrane. WFA 2.5 μM induced rounding and piling up of the vimentin-rich repair cells at the wound edge, and the greatest effect on repair cell shape and phenotype at the wound edge is observed at 3.5 μM WFA. Repair cells in control wounded lens explants remain organized as a monolayer and extend their lamellipodia along the basement membrane in the direction of migration (dimethyl sulfoxide). (C, D) The effect of WFA on the organization of microfilament and microtubule cytoskeletal networks was examined by labeling the cells for F-actin using fluorescent phalloidin (green) or α-tubulin (red). Both F-actin and microtubules maintain a high level of organization in the presence of WFA in both repair cells and lens epithelial cells. The lack of effect of WFA on these other cytoskeletal elements is highlighted by the fact that actin remains organized in a cortical distribution in the lens epithelial cells (C, arrow). Changes in the distribution of these cytoskeletal elements within repair cells correspond to changes in cell shape caused by WFA treatment (C and D, arrowhead). (E) Wound closure for control wounded explants compared with wounded explants treated with 3.5 μM WFA, shown in phase contrast imaging. (F) WFA treatment affects wound closure in wounded explants in a dose-dependent manner. Although no effect on wound closure is observed at 1.5 μM WFA, which had little effect on repair cell morphology and ability to extend lamellipodia along the basement membrane (see B), wound closure was slowed with 2.5 μM WFA treatment, and the greatest inhibition was observed at 3.5 μM WFA, quantified for three independent experiments (F). Bar, 20 μm (A–D), 500 μm (phase images). Secondary antibody controls were performed, which demonstrated specificity of antibody staining (Supplemental Figure S1).
Because vimentin intermediate filaments can interact with other cytoskeletal filaments, including microtubules and microfilaments (Chang and Goldman, 2004), it is not surprising that exposure of some cell types to WFA has been reported to change the organization of F-actin and/or microtubules, together with its effects on their vimentin filaments (Bargagna-Mohan et al., 2007; Grin et al., 2012). Therefore, for our studies, it was important to determine whether the effects of WFA on the vimentin filaments of cells in the wounded explant cultures also affected the organization of other cytoskeletal filaments. We found that there was little effect on the organization of either F-actin or microtubules (stained for α-tubulin) when the wounded cultures were exposed to WFA (Figure 6, C and D). Particularly striking was the ability of the cortical actin filaments of the lens epithelial cells to maintain both their integrity and their distribution in the presence of WFA (Figure 6C, arrow). Similarly, the integrity of actin filaments (Figure 6C, arrowhead) and microtubules (Figure 6D) was maintained in the repair cells at the wound edge despite the dramatic change in their cell shape that accompanied collapse of their vimentin intermediate filaments. These results support the conclusion that in the ex vivo wound repair cultures that we study here, WFA functioned as a vimentin-specific cytoskeletal inhibitor.
The effect of WFA on vimentin had a significant effect on wound closure, suppressing the collective migration of the lens epithelium across the wounded area of the lens capsule (Figure 6E). The effects of WFA on wound closure were very similar to those obtained after vimentin siRNA knockdown. The effect of WFA on wound repair was dose dependent (Figure 6F). The lowest concentration tested, 1.5 μM WFA, which had a minimal effect on repair cell morphology (Figure 6B), did not impair wound closure (Figure 6F). Higher concentrations of WFA that both collapsed the intermediate filament network and rounded the repair cells (Figure 6B) slowed wound closure, with the greatest effect at the highest concentration tested (Figure 6, E and F). Given that WFA has been reported to induce apoptosis in certain cell types (Bargagna-Mohan et al., 2007; Lahat et al., 2010; Grin et al., 2012), we examined whether the effects of this drug on wound closure were related to an induction of cell death in the repair cells. Few apoptotic cells were detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay in the region of wound repair in WFA-treated, wounded explant cultures, similar to untreated controls (Supplemental Figure S3). Therefore the suppression of wound closure in the presence of WFA was most likely due to the altered behavior of repair cells when they fail to extend along the wound edge. Because the soluble pool of vimentin is highly sensitive to WFA, these results could reflect a particular role for this dynamic vimentin population that our studies showed is enhanced in the cell population migrating to fill the wound area. Because before the identification of WFA as a vimentin inhibitor acrylamide was used as a classical approach to disrupt vimentin function (Durham et al., 1983; Eckert, 1985; Sager, 1989), wounded lens explants also were exposed to acrylamide for 24 h from the time of injury. Acrylamide was found to inhibit wound healing in a dose-dependent manner (Supplemental Figure S4). Together the vimentin siRNA knockdown and WFA results show, for the first time, the importance of vimentin to repair of cell function in the regulation of wound closure of an injured epithelium.
Myosin II is linked to vimentin in repair cells
Nonmuscle myosin II motor proteins are viewed as master regulators of migration, implicated in protrusion extension, adhesion dynamics, and polarization of migrating cells (Vicente-Manzanares et al., 2009), typically in conjunction with actin filaments. Because our data indicated that vimentin intermediate filaments played a central role in regulating repair cell function in response to injury, we investigated whether there might be a coordinate role for myosin II with vimentin. For these studies we examined both myosin IIA and IIB isoforms, as each has distinct motor activities, subcellular locations, and functions in migrating cells (Even-Ram and Yamada, 2007; Vicente-Manzanares et al., 2009; Wang et al., 2011). The distribution of myosin IIA and IIB in repair cells was examined by coimmunostaining wounded cultures for these two myosin II isoforms. These studies showed a prominence of myosin IIB over myosin IIA in the repair cells at the wound edge (Figure 7, A–C). These two isoforms had distinct patterns of distribution. In contrast to myosin IIB, myosin IIA was highly localized along stress fibers, reflecting its known function with F-actin (Figure 7B, arrows).
FIGURE 7:

Novel association between myosin II and vimentin during wound healing. To determine the distribution of myosin IIA and IIB in repair cells, wounded explants D1 postinjury were coimmunostained for myosin IIA (B, green) and myosin IIB (A, red; overlaid in C). Distinct patterns of distribution for these two isoforms were observed. Myosin IIA localized to stress fibers (arrows), whereas myosin IIB was predominately expressed throughout the cytoplasm of repair cells at the wound edge. Coimmunoprecipitation analysis at D1 postinjury revealed a previously unknown association between myosin II and vimentin (D). Lysates were immunoprecipitated (IP) for vimentin (vim) and immunoblotted for vimentin, myosin IIA (MyoIIA), or myosin IIB (MyoIIB), and the results are quantified (E) from three independent experiments. Whereas both myosin IIA and IIB were linked to vimentin, the greatest association occurred between myosin IIB and vimentin (E). Expression of myosin IIA, myosin IIB, and vimentin in WCLs is shown by direct immunoblotting (D). To examine the distribution of myosin IIB and vimentin in repair cells, D1 postinjury wounded lens explants were colabeled for myosin IIB (F, red), and vimentin (G green; overlaid in H). Myosin IIB was highly expressed in repair cells rich in vimentin located at the wound edge (H, white arrows). The boxed area is shown at higher magnification as an inset. Bars, 20 μm. A–H are shown as single optical sections. Secondary antibody controls were performed, which demonstrated specificity of antibody staining (Supplemental Figure S1).
To investigate the possibility of vimentin-linked myosin II function, we performed coimmunoprecipitation analysis (IP, vimentin; blot, myosin II [A or B]) at 1 d after injury, a time of active cell migration. The results revealed a previously unknown linkage of myosin II with vimentin (Figure 7D) and showed that this phenomenon predominantly reflected the association of vimentin with the myosin IIB isoform (Figure 7, D and E). Further evidence of a vimentin-linked myosin IIB role was the finding that myosin IIB and vimentin were highly colocalized in the repair cell population at the wound edge (Figure 7H, arrows and inset). The findings reveal a previously unappreciated association of myosin II motor proteins with vimentin in repair cells responding to injury, suggesting a role for myosin II motor activity in vimentin function during wound healing.
DISCUSSION
Our previous studies revealed the existence of a subpopulation of repair cell progenitors, innate to the lens epithelium, that were activated in response to the injury inflicted by performing a mock cataract surgery (Walker et al., 2010). The repair cells, rich in vimentin, quickly traveled to the wound edge to modulate the repair process (Walker et al., 2010). The present study adds these repair cells to the list of cells that can function as leader cells at the wound edge in response to injury. We also show that vimentin is essential to repair cell function, including the ability to adopt and maintain a mesenchymal morphology at the wound edge, extend lamellipodia along the basement membrane, and modulate the repair process. A high concentration of vimentin filaments stretched out to the tips of the lamellipodia of the repair cells, similar to the findings of Helfand et al. (2011), and nonfilamentous vimentin particles were enriched in these lamellipodia regions. Nonfilamentous vimentin is a soluble form of this intermediate filament protein, which has particular importance to our study, as soluble intermediate filament proteins are associated with dynamic cell processes such as wound closure and invasion (Windoffer et al., 2011). An unusual property of the lamellipodia of the repair cell population is the finding that vimentin filaments also extended to the tips of lamellipodia, where they were linked to paxillin in focal adhesion plaques. The fact that surprisingly few actin filaments were present in these lamellipodial extensions likely provides the need for vimentin filament association with adhesion complexes, in contrast to more-classical models of actin-based lamellipodial movement. Extension of vimentin throughout the repair cells required microtubule function. The role of actin filaments and microtubules in focal adhesion dynamics is well studied, but much less is understood about the role of intermediate filaments at these adhesion complexes. Vimentin intermediate filament–linked focal adhesions have been detected in cell types such as fibroblasts (Bershadsky et al., 1987) and endothelial cells (Gonzales et al., 2001; Kreis et al., 2005; Bhattacharya et al., 2009).
Vimentin can regulate both the structure and function of focal adhesions (Eckes et al., 1998; Tsuruta and Jones, 2003). For instance, vimentin regulates the size of focal adhesions and stabilization of cell-matrix adhesion strength in endothelial cells under shear stress (Tsuruta and Jones, 2003). The formation of intermediate filament–linked focal adhesions may be specific to cell type or adapted under conditions in which cells are subjected to stress and/or mechanical forces. Studies show that intermediate filaments can be recruited to focal adhesions in a process dependent on microtubules (Bhattacharya et al., 2009) and that vimentin filaments can be delivered along microtubules to focal adhesions, where they are assembled (Helfand et al., 2002; Bhattacharya et al., 2009). These studies are consistent with our finding that vimentin functions at the leading edge in lamellipodia, including in the establishment of repair cell morphology, properties that depend on microtubules. Our results suggest that vimentin-linked focal adhesions in the lamellipodia protrusions may provide unique properties to repair cells that are related to mechanical and motile functions required in their response to injury.
Vimentin-knockout mice exhibit defects in wound repair; however, complete loss of vimentin protein in these cells also results in dramatic effects on the organization of the actin cytoskeleton (Eckes et al., 1998), making it difficult to separate the role of these individual cytoskeletal systems in the wound-healing process. An siRNA knockdown approach allowed us to target the vimentin intermediate filament protein population just at the time of wounding, allowing us to identify the immediate effects of loss of vimentin function. In this study it is most likely that we are affecting the soluble, less stable pool of vimentin. The knockdown of vimentin impaired wound closure and the cells at the leading edge of the wound failed to extend lamellipodia. This finding revealed an essential role for vimentin in repair cell function in wound closure. Altering vimentin function through the use of a vimentin inhibitor confirmed the siRNA-knockdown finding. For this study we used WFA, a plant-derived inhibitor that can bind to vimentin and block intermediate filament function (Bargagna-Mohan et al., 2007). Soluble vimentin is particularly sensitive to WFA (Bargagna-Mohan et al., 2013). In some cultured cell types, the effect of WFA on vimentin filament organization has indirect effects on the organization of actin microfilaments and microtubules (Bargagna-Mohan et al., 2007; Grin et al., 2012). However, we observed no similar effects of WFA on the repair cells in the wounded explant cultures, making the mock cataract surgery–wounded explants a good system in which to use WFA to study the role of vimentin in wound repair. WFA altered vimentin organization in the repair cells in these explants without affecting the integrity of either actin filaments or microtubules. Dramatic changes in repair cell shape and behavior were observed when the wounded cultures were exposed to WFA, with repair cells failing to extend lamellipodial processes. This effect interfered with repair cell function in wound healing and slowed wound closure. The WFA results paralleled those of vimentin siRNA knockdown in this mock cataract surgery model, showing that vimentin intermediate filaments are essential to repair cell function in regulating wound closure.
Intermediate filaments were classically viewed only as stable cytoskeletal structures whose principal function was to provide cells with mechanostability and resistance to mechanical stress (Lazarides, 1980; Kim and Coulombe, 2007), whereas it is now appreciated that vimentin intermediate filaments are dynamic structures (Prahlad et al., 1998; Helfand et al., 2004; Eriksson et al., 2009), with their state of assembly regulated by targeted phosphorylation events (Ivaska et al., 2007; Sihag et al., 2007). Differences in the assembly state of the vimentin filaments can affect vimentin function. It has been hypothesized that active free pools of vimentin (soluble vimentin) have functions distinct from those of large, rope-like vimentin filaments (Perlson et al., 2006; Kim and Coulombe, 2007; Lahat et al., 2010; Goldman et al., 2012). These detergent-soluble populations are likely to be heterogeneous and contain short vimentin filaments that serve as building blocks for the larger, more mechanostable forms—the freer forms of vimentin likely including monomers, dimers, or tetramers. The soluble form of vimentin has been implicated as a long-distance messenger (Perlson et al., 2005). Data also show increased levels of soluble vimentin in disease states, as in sarcoma tumor cell lines, a property correlated with increased sensitivity to WFA (Lahat et al., 2010), highlighting a potential function for soluble vimentin in disease processes. Expression of vimentin is often correlated with the metastatic state (Hendrix et al., 1996). Nonfilamentous vimentin particles and squiggles, considered soluble forms of vimentin, were concentrated near the tips of lamellipodial extensions of the repair cells, where they can function to regulate the cell's migratory behavior. We also observed an increase in the soluble pool of vimentin in regions of active migration in response to injury. Accordingly, the dramatic effects on wound closure with both siRNA knockdown and WFA are likely to reflect the specific targeting of the vimentin soluble pool. We hypothesize that it is the soluble forms of vimentin filaments that play a critical role in regulating repair cell response to injury during wound healing.
The finding that vimentin filaments had motile function suggested that these filaments would be associated with motor proteins, as is typical of other cytoskeletal elements involved in directing cell motility. We discovered that the motor protein myosin II, a principal regulator of actin filament–based cell movement, may also play a role in vimentin-based movement. Both myosin II isoforms, myosin IIA and IIB, localized to repair cells at the leading edge, but myosin IIB was predominant and was linked biochemically to vimentin in wounded lens cultures undergoing active repair. Myosin IIA and IIB have distinct roles in migration and distinct kinetic properties (Even-Ram and Yamada, 2007; Vicente-Manzanares et al., 2009; Wang et al., 2011); myosin IIB has a higher affinity for ADP and duty ratio, which refers to its ability, under force-generating situations, to bind to actin longer than myosin IIA (Kovacs et al., 2003; Wang et al., 2003; Vicente-Manzanares et al., 2009). These properties allow myosin IIB to exert tension for longer periods of time than myosin IIA (Kovacs et al., 2003; Wang et al., 2003; Vicente-Manzanares et al., 2009). Myosin II participates not only in the cellular response to mechanical stimuli, but also in the ability of a cell to sense pliability of its environment (Beningo et al., 2001, 2006; Chen, 2008; Vicente-Manzanares et al., 2009). The myosin IIB isoform is associated with acquisition of a mesenchymal phenotype and, in mammary cells treated with transforming growth factor-β to induce an EMT, its expression was required for their transmigration and invasion (Beach et al., 2011). These properties poise myosin II motor proteins with unique mechanical sensing and force-related properties and suggest that the myosin IIB isoform, in association with vimentin, has an important function in repair cells at the wound edge. To our knowledge, this is the first demonstration of a role for myosin II motor proteins with vimentin for cell migration in an injury model. This suggests the importance of furthering our understanding of the role of myosin II in regulation of vimentin function. In summary, the present study adds significant knowledge about how repair cells can perform unique functions in regulating the wound-healing process that revolves on vimentin function in cell migration.
MATERIALS AND METHODS
Ex vivo epithelial injury model preparation
To prepare ex vivo epithelial injury explants, lenses were removed from the eyes of embryonic day (E) 15 chicken embryo (Truslow Farms, Chestertown, MD, and B&E Eggs, York Springs, PA) by dissection (Walker et al., 2007, 2010). Then an incision was made in the anterior lens capsule—the thick basement membrane that surrounds the lens—from which the lens fiber cell mass was removed by hydroelution (Walker et al., 2007, 2010). This procedure was modified from a human lens capsular bag model developed to study posterior capsular opacification (Liu et al., 1996). This process, in which the lens epithelium remains tightly adherent to the capsule, mimics cataract surgery. The principal wound edge (leading edge) of the epithelium borders the area where the fiber cells had been attached (model, Figure 1). Cuts were made in the anterior region of this tissue, creating additional wound edges that allowed the explants to be flattened and pinned to the culture dish cell-side-up (Figure 1). The response of the lens epithelium to wounding within their native microenvironment was followed by microscopic imaging. Movement of the lens epithelial cells along the posterior aspects of the capsule was tracked using an SMZ800 (Nikon, Tokyo, Japan) or AZ100 (Nikon) microscope and imaging software (NIS Elements; Nikon). The ex vivo epithelial explants were cultured in Media 199 (Invitrogen, Carlsbad, CA) containing 1% penicillin/streptomycin (Mediatech-Cellgro, Manassas, VA) and 1% l-glutamine (Mediatech-Cellgro) with 10% fetal calf serum (Invitrogen). Microtubules were depolymerized by growing injury explants in the presence of 3 μM nocodazole (Sigma-Aldrich, St. Louis, MO). To perturb vimentin function, ex vivo explants were grown in the presence of 1.5–3.5 μM withaferin A (Tocris, Ellisville, MO) or acrylamide (1–2 mM; Bio-Rad, Hercules, CA). Inhibitors were dissolved in dimethyl sulfoxide and added to the culture medium. In all experiments, cultures were incubated in the presence or absence (vehicle controls) of inhibitor. Fresh inhibitor was added each day. Wound area was calculated over time using NIS Elements software. Measurements were exported and graphed in Excel (Microsoft, Redmond, WA).
Western blot analysis
Samples were extracted and lysed in TX/OG buffer (44.4 mM n-octyl β-d glucopyranoside, 1% Triton X-100, 100 mM NaCl, 1 mM MgCl2, 5 mM EDTA, 10 mM imidazole) containing 1 mM sodium vanadate, 0.2 mM H2O2, and a protease inhibitor cocktail (Sigma-Aldrich). For sequential extraction, samples were prepared as described (Leonard et al., 2011). Briefly, studies using sequential extraction samples were extracted first in Triton (1% Triton X-100, 100 mM NaCl, 1 mM MgCl2, 5 mM EDTA, 10 mM imidazole, pH 7.4), then in Triton/OG, and finally in 2× sample buffer (125 mM Tris-HCl, 4% SDS, 20% glycerol, 2% β-mercaptoethanol, 0.5% bromophenol blue). Protein concentrations were determined with an assay (BCA Assay; Pierce, Rockford, IL). Proteins were separated on Tris-glycine gels (Novex, San Diego, CA), electrophoretically transferred to membrane (Immobilon-P; Millipore, Billerica, MA), and immunoblotted as described previously (Walker and Menko, 1999). For detection, ECL reagent (Amersham Life Sciences, Arlington Heights, IL) was used. Immunoblots were scanned, and densitometry analysis was performed (1D software; Eastman Kodak, Rochester, NY). For each protein analyzed, the densitometry results first were normalized to control, and then the ratio of each protein to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was calculated for each sample. All gels were run under reducing conditions. Antibodies used for Western blotting included vimentin (monoclonal antibody [mAb]; Developmental Studies Hybridoma Bank, Iowa City, IA), vimentin (polyclonal) antibody (a generous gift from Paul FitzGerald, University of California, Davis, CA), myosin IIB (mAb; Developmental Studies Hybridoma Bank), myosin IIA (polyclonal; Sigma-Aldrich), GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA), and paxillin (mAb; BD Transduction Laboratories, Franklin Lakes, NJ).
Immunoprecipitation techniques
For immunoprecipitation studies, equal amounts of protein were used from ex vivo epithelial explants extracted in TX/OG from different days in culture or between treated and untreated samples. For immunoprecipitations, samples were incubated at 4°C sequentially with primary antibody and TrueBlot Immunoprecipitation beads (eBioscience, San Diego, CA) and the immunoprecipitates subjected to SDS/PAGE as described previously and earlier (Walker et al., 2002). Antibodies used included vimentin (Developmental Studies Hybridoma Bank) and paxillin (BD Transduction Laboratories). Immunoblots were scanned and densitometric analysis was performed using Kodak 1D software. To standardize these results for the paxillin IP, quantitative analysis was performed. For each protein analyzed, the densitometry results were normalized to D1 OAZ, and then the ratio of each coprecipitated protein to the original immunoprecipitated protein was calculated for each sample. For vimentin IP, the ratio of myosin IIA versus myosin IIB associated with vimentin was calculated for each sample.
siRNA knockdown of vimentin
Vimentin was silenced in ex vivo wounded lens explant cultures using a pool of four siRNA duplexes designed as a custom SMARTpool plus by Dharmacon Research (Lafayette, CO). As a negative control, ex vivo explants were transfected with siControl (siC) nontargeting siRNAs (Dharmacon). For maximum knockdown of vimentin, wounded lens explant cultures were transfected at time of wounding and again at 24 h after injury using Lipofectamine 2000 (Invitrogen) and 30–100 nM pool siRNA to vimentin or nontargeting siRNA control, according to manufacturer's protocol.
Immunofluorescence
Epithelial explants were immunostained as described (Walker et al., 2007, 2010). Briefly, explants were fixed in 3.7% formaldehyde in phosphate-buffered saline (PBS) and permeabilized in 0.25% Triton X-100 (Sigma-Aldrich) in PBS before immunostaining. Cells were incubated with primary antiserum followed by rhodamine-conjugated (Jackson ImmunoResearch Laboratories, West Grove, PA, and Millipore), fluorescein-conjugated (Jackson ImmunoResearch Laboratories), or Alexa Fluor 488–conjugated (Invitrogen-Molecular Probes, Carlsbad, CA) secondary antibodies. The following primary antibodies were used for the immunofluorescence studies: vimentin (mAb; Developmental Studies Hybridoma Bank), vimentin (polyclonal) antibody (a generous gift from Paul FitzGerald), paxillin (mAb; BD Transduction Laboratories, Franklin Lakes, NJ), β4 integrin (polyclonal) antibody as described in Sta Iglesia et al. (2000), α6 integrin (mAb; Covance, Princeton, NJ) myosin IIA (polyclonal, Sigma-Aldrich), myosin IIB (mAb; Developmental Studies Hybridoma Bank), α-tubulin (mAb; Developmental Studies Hybridoma Bank), and α-tubulin (polyclonal) antibody (Cell Signaling, Danvers, MA). Some explants were counterstained with Alexa Fluor 488–conjugated phalloidin, which binds filamentous actin, and TO-PRO 3, a nuclear stain (Invitrogen-Molecular Probes). TUNEL assay was performed according to manufacturer's protocol (Roche, Indianapolis, IN). Immunostained samples were examined with a confocal microscope (LSM 510; Zeiss, Thornwood, NY). Either single images or Z-stacks were collected and analyzed; the data presented represent single optical planes, projected images, or orthogonal sections imaged from apical to basal direction.
Supplementary Material
Acknowledgments
We thank Paul Fitzgerald (UC Davis School of Medicine) for kindly providing the vimentin polyclonal antibody. The monoclonal antibodies AMF17b (vimentin) developed by Alice B. Fulton (University of Iowa Carver College of Medicine), CMII 23 (myosin IIB) developed by Abigail Conrad and Gary Conrad (Kansas State University), and AA4.3 (α-tubulin) developed by Charles Walsh (University of Pittsburgh) were all obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology, Iowa City, IA. This work was supported by National Institutes of Health grants to A.S.M. (EY021784, EY014258, and EY014798) and J.L.W. (EY019571).
Abbreviations used:
- BM
basement membrane
- CMZ
cell migration zone
- EMT
epithelial mesenchymal transition
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HI
highly detergent-insoluble
- IP
immunoprecipitate
- OAZ
original attachment zone
- PBS
phosphate-buffered saline
- siC
siControl
- siRNA
small interfering RNA
- siVim
siVimentin
- Sol
soluble
- T0
time 0
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- TX
Triton X-100
- TX/OG
Triton X-100/octylglucoside
- WCL
whole cell lysate
- WFA
Withaferin A
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0900) on January 29, 2014.
REFERENCES
- Bargagna-Mohan P, Deokule SP, Thompson K, Wizeman J, Srinivasan C, Vooturi S, Kompella UB, Mohan R. Withaferin A effectively targets soluble vimentin in the glaucoma filtration surgical model of fibrosis. PLoS One. 2013;8:e63881. doi: 10.1371/journal.pone.0063881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P, et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P, et al. Corneal antifibrotic switch identified in genetic and pharmacological deficiency of vimentin. J Biol Chem. 2012;287:989–1006. doi: 10.1074/jbc.M111.297150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, et al. Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:17991–17996. doi: 10.1073/pnas.1106499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol. 2001;153:881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo KA, Hamao K, Dembo M, Wang YL, Hosoya H. Traction forces of fibroblasts are regulated by the Rho-dependent kinase but not by the myosin light chain kinase. Archiv Biochem Biophys. 2006;456:224–231. doi: 10.1016/j.abb.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky AD, Tint IS, Svitkina TM. Association of intermediate filaments with vinculin-containing adhesion plaques of fibroblasts. Cell Motil Cytoskeleton. 1987;8:274–283. doi: 10.1002/cm.970080308. [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Gonzalez AM, Debiase PJ, Trejo HE, Goldman RD, Flitney FW, Jones JC. Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength. J Cell Sci. 2009;122:1390–1400. doi: 10.1242/jcs.043042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Buisson AC, Gilles C, Polette M, Zahm JM, Birembaut P, Tournier JM. Wound repair-induced expression of a stromelysins is associated with the acquisition of a mesenchymal phenotype in human respiratory epithelial cells. Lab Invest. 1996;74:658–669. [PubMed] [Google Scholar]
- Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J Cell Biol. 1990;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol. 2004;5:601–613. doi: 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- Chen CS. Mechanotransduction—a field pulling together. J Cell Sci. 2008;121:3285–3292. doi: 10.1242/jcs.023507. [DOI] [PubMed] [Google Scholar]
- Durham HD, Pena SD, Carpenter S. The neurotoxins 2,5-hexanedione and acrylamide promote aggregation of intermediate filaments in cultured fibroblasts. Muscle Nerve. 1983;6:631–637. doi: 10.1002/mus.880060903. [DOI] [PubMed] [Google Scholar]
- Eckert BS. Alteration of intermediate filament distribution in PtK1 cells by acrylamide. Eur J Cell Biol. 1985;37:169–174. [PubMed] [Google Scholar]
- Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, Martin P. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- Eckes B, et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J Cell Sci. 1998;111:1897–1907. doi: 10.1242/jcs.111.13.1897. [DOI] [PubMed] [Google Scholar]
- Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, Goldman RD. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119:1763–1771. doi: 10.1172/JCI38339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even-Ram S, Yamada KM. Of mice and men: relevance of cellular and molecular characterizations of myosin IIA to MYH9-related human disease. Cell Adh Migr. 2007;1:152–155. doi: 10.4161/cam.1.3.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forry-Schaudies S, Murray JM, Toyama Y, Holtzer H. Effects of colcemid and Taxol on microtubules and intermediate filaments in chick embryo fibroblasts. Cell Motil Cytoskeleton. 1986;6:324–338. doi: 10.1002/cm.970060309. [DOI] [PubMed] [Google Scholar]
- Fortin S, Le Mercier M, Camby I, Spiegl-Kreinecker S, Berger W, Lefranc F, Kiss R. Galectin-1 is implicated in the protein kinase C epsilon/vimentin-controlled trafficking of integrin-beta1 in glioblastoma cells. Brain Pathol. 2010;20:39–49. doi: 10.1111/j.1750-3639.2008.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- Geuens G, de Brabander M, Nuydens R, De Mey J. The interaction between microtubules and intermediate filaments in cultured cells treated with Taxol and nocodazole. Cell Biol Int Rep. 1983;7:35–47. doi: 10.1016/0309-1651(83)90103-0. [DOI] [PubMed] [Google Scholar]
- Gilles C, Polette M, Zahm JM, Tournier JM, Volders L, Foidart JM, Birembaut P. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci. 1999;112:4615–4625. doi: 10.1242/jcs.112.24.4615. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Cleland MM, Murthy SN, Mahammad S, Kuczmarski ER. Inroads into the structure and function of intermediate filament networks. J Struct Biol. 2012;177:14–23. doi: 10.1016/j.jsb.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Grin B, Mendez MG, Kuczmarski ER. Intermediate filaments: versatile building blocks of cell structure. Curr Opin Cell Biol. 2008;20:28–34. doi: 10.1016/j.ceb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez M, Navarro P, Quintanilla M, Cano A. Expression of alpha 6 beta 4 integrin increases during malignant conversion of mouse epidermal keratinocytes: association of beta 4 subunit to the cytokeratin fraction. Exp Cell Res. 1992;201:250–261. doi: 10.1016/0014-4827(92)90272-a. [DOI] [PubMed] [Google Scholar]
- Gonzales M, Weksler B, Tsuruta D, Goldman RD, Yoon KJ, Hopkinson SB, Flitney FW, Jones JC. Structure and function of a vimentin-associated matrix adhesion in endothelial cells. Mol Biol Cell. 2001;12:85–100. doi: 10.1091/mbc.12.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grin B, Mahammad S, Wedig T, Cleland MM, Tsai L, Herrmann H, Goldman RD. Withaferin a alters intermediate filament organization, cell shape and behavior. PLoS One. 2012;7:e39065. doi: 10.1371/journal.pone.0039065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Chang L, Goldman RD. Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci. 2004;117:133–141. doi: 10.1242/jcs.00936. [DOI] [PubMed] [Google Scholar]
- Helfand BT, et al. Vimentin organization modulates the formation of lamellipodia. Mol Biol Cell. 2011;22:1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mikami A, Vallee RB, Goldman RD. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J Cell Biol. 2002;157:795–806. doi: 10.1083/jcb.200202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Chu YW, Trevor KT, Seftor RE. Role of intermediate filaments in migration, invasion and metastasis. Cancer Metastasis Rev. 1996;15:507–525. doi: 10.1007/BF00054016. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Aebi U. Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr Opin Cell Biol. 2000;12:79–90. doi: 10.1016/s0955-0674(99)00060-5. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313:2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005;24:3834–3845. doi: 10.1038/sj.emboj.7600847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AA, Friedl P. Determinants of leader cells in collective cell migration. Integr Biol (Camb) 2010;2:568–574. doi: 10.1039/c0ib00052c. [DOI] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–1597. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Kitamura T, et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39:467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- Kovacs M, Wang F, Hu A, Zhang Y, Sellers JR. Functional divergence of human cytoplasmic myosin II: kinetic characterization of the non-muscle IIA isoform. J Biol Chem. 2003;278:38132–38140. doi: 10.1074/jbc.M305453200. [DOI] [PubMed] [Google Scholar]
- Kreis S, Schonfeld HJ, Melchior C, Steiner B, Kieffer N. The intermediate filament protein vimentin binds specifically to a recombinant integrin alpha2/beta1 cytoplasmic tail complex and co-localizes with native alpha2/beta1 in endothelial cell focal adhesions. Exp Cell Res. 2005;305:110–121. doi: 10.1016/j.yexcr.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Lahat G, et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One. 2010;5:e10105. doi: 10.1371/journal.pone.0010105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lazarides E. Intermediate filaments as mechanical integrators of cellular space. Nature. 1980;283:249–256. doi: 10.1038/283249a0. [DOI] [PubMed] [Google Scholar]
- Leonard M, Zhang L, Zhai N, Cader A, Chan Y, Nowak RB, Fowler VM, Menko AS. Modulation of N-cadherin junctions and their role as epicenters of differentiation-specific actin regulation in the developing lens. Dev Biol. 2011;349:363–377. doi: 10.1016/j.ydbio.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CS, Wormstone IM, Duncan G, Marcantonio JM, Webb SF, Davies PD. A study of human lens cell growth in vitro. A model for posterior capsule opacification. Invest Ophthalmol Vis Sci. 1996;37:906–914. [PubMed] [Google Scholar]
- Meckes DG, Jr, Menaker NF, Raab-Traub N. EBV LMP1 modulates lipid raft microdomains and the vimentin cytoskeleton for signal transduction and transformation. J Virol. 2013;87:1301–1311. doi: 10.1128/JVI.02519-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell DJ. Morphogenetic cell movements: diversity from modular mechanical properties. Science. 2008;322:1502–1505. doi: 10.1126/science.1164073. [DOI] [PubMed] [Google Scholar]
- Omelchenko T, Vasiliev JM, Gelfand IM, Feder HH, Bonder EM. Rho-dependent formation of epithelial “leader” cells during wound healing. Proc Natl Acad Sci USA. 2003;100:10788–10793. doi: 10.1073/pnas.1834401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Pareja JC, Grawe F, Martin-Blanco E, Garcia-Bellido A. Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell. 2004;7:387–399. doi: 10.1016/j.devcel.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Perlson E, Michaelevski I, Kowalsman N, Ben-Yaakov K, Shaked M, Seger R, Eisenstein M, Fainzilber M. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364:938–944. doi: 10.1016/j.jmb.2006.09.056. [DOI] [PubMed] [Google Scholar]
- Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A, Silberzan P. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci USA. 2007;104:15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitz I, Nagle RB, Cress AE. Integrin alpha 6 expression in human prostate carcinoma cells is associated with a migratory and invasive phenotype in vitro and in vivo. Clin Exp Metastasis. 1995;13:481–491. doi: 10.1007/BF00118187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenu C, Gilmour D. EMT 2.0: shaping epithelia through collective migration. Curr Opin Genet Dev. 2009;19:338–342. doi: 10.1016/j.gde.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Rezniczek GA, de Pereda JM, Reipert S, Wiche G. Linking integrin alpha6beta4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the beta4 subunit and plectin at multiple molecular sites. J Cell Biol. 1998;141:209–225. doi: 10.1083/jcb.141.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel MR, Soni PN, Troken JR, Sitikov A, Trejo HE, Ridge KM. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011;25:3873–3883. doi: 10.1096/fj.10-170795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runembert I, Queffeulou G, Federici P, Vrtovsnik F, Colucci-Guyon E, Babinet C, Briand P, Trugnan G, Friedlander G, Terzi F. Vimentin affects localization and activity of sodium-glucose cotransporter SGLT1 in membrane rafts. J Cell Sci. 2002;115:713–724. doi: 10.1242/jcs.115.4.713. [DOI] [PubMed] [Google Scholar]
- Sager PR. Cytoskeletal effects of acrylamide and 2,5-hexanedione: selective aggregation of vimentin filaments. Toxicol Appl Pharmacol. 1989;97:141–155. doi: 10.1016/0041-008x(89)90063-x. [DOI] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313:2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger RR, Fontijn RD, van Marle J, Pannekoek H, Horrevoets AJ. Spatial segregation of transport and signalling functions between human endothelial caveolae and lipid raft proteomes. Biochem J. 2006;400:401–410. doi: 10.1042/BJ20060355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sta Iglesia DD, Gala PH, Qiu T, Stepp MA. Integrin expression during epithelial migration and restratification in the tenascin-C-deficient mouse cornea. J Histochem Cytochem. 2000;48:363–376. doi: 10.1177/002215540004800306. [DOI] [PubMed] [Google Scholar]
- Stepp MA, Spurr-Michaud S, Tisdale A, Elwell J, Gipson IK. Alpha 6 beta 4 integrin heterodimer is a component of hemidesmosomes. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SundarRaj N, Rizzo JD, Anderson SC, Gesiotto JP. Expression of vimentin by rabbit corneal epithelial cells during wound repair. Cell Tissue Res. 1992;267:347–356. doi: 10.1007/BF00302973. [DOI] [PubMed] [Google Scholar]
- Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116:4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JL, Menko AS. alpha6 Integrin is regulated with lens cell differentiation by linkage to the cytoskeleton and isoform switching. Dev Biol. 1999;210:497–511. doi: 10.1006/dbio.1999.9277. [DOI] [PubMed] [Google Scholar]
- Walker JL, Wolff IM, Zhang L, Menko AS. Activation of SRC kinases signals induction of posterior capsule opacification. Invest Ophthalmol Vis Sci. 2007;48:2214–2223. doi: 10.1167/iovs.06-1059. [DOI] [PubMed] [Google Scholar]
- Walker JL, Zhai N, Zhang L, Bleaken BM, Wolff I, Gerhart J, George-Weinstein M, Menko AS. Unique precursors for the mesenchymal cells involved in injury response and fibrosis. Proc Natl Acad Sci USA. 2010;107:13730–13735. doi: 10.1073/pnas.0910382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JL, Zhang L, Zhou J, Woolkalis MJ, Menko AS. Role for α6 integrin during lens development: evidence for signaling through IGF-1R and ERK. Dev Dyn. 2002;223:273–284. doi: 10.1002/dvdy.10050. [DOI] [PubMed] [Google Scholar]
- Wang F, Kovacs M, Hu A, Limouze J, Harvey EV, Sellers JR. Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J Biol Chem. 2003;278:27439–27448. doi: 10.1074/jbc.M302510200. [DOI] [PubMed] [Google Scholar]
- Wang A, Ma X, Conti MA, Adelstein RS. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem Soc Trans. 2011;39:1131–1135. doi: 10.1042/BST0391131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijer CJ. Collective cell migration in development. J Cell Sci. 2009;122:3215–3223. doi: 10.1242/jcs.036517. [DOI] [PubMed] [Google Scholar]
- Windoffer R, Beil M, Magin TM, Leube RE. Cytoskeleton in motion: the dynamics of keratin intermediate filaments in epithelia. J Cell Biol. 2011;194:669–678. doi: 10.1083/jcb.201008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.