

Using mouse knockout strategy, the authors uncovered a role for Borg5 in microvascular angiogenesis. In primary mouse cardiac endothelial cells, Borg5 interacts with septin cytoskeleton and colocalizes with perinuclear actomyosin fibers. The data presented suggest that Borg5 and septin regulate the actomyosin activity critical for persistent directional migration.
Abstract
The microvasculature is important for vertebrate organ development and homeostasis. However, the molecular mechanism of microvascular angiogenesis remains incompletely understood. Through studying Borg5 (Binder of the Rho GTPase 5), which belongs to a family of poorly understood effector proteins of the Cdc42 GTPase, we uncover a role for Borg5 in microvascular angiogenesis. Deletion of Borg5 in mice results in defects in retinal and cardiac microvasculature as well as heart development. Borg5 promotes angiogenesis by regulating persistent directional migration of the endothelial cells (ECs). In primary mouse cardiac ECs (MCECs), Borg5 associates with septins in the perinuclear region and colocalizes with actomyosin fibers. Both Borg5 deletion and septin 7 knockdown lead to a disruption of the perinuclear actomyosin and persistent directional migration. Our findings suggest that Borg5 and septin cytoskeleton spatially control actomyosin activity to ensure persistent directional migration of MCECs and efficient microvascular angiogenesis. Our studies reported here should offer a new avenue to further investigate the functions of Borg5, septin, and actomyosin in the microvasculature in the context of development and disease.
INTRODUCTION
The vertebrate cardiovascular system is the first organ system to form and function during embryogenesis. The extensive network of the circulatory system allows nutrients and oxygen to be efficiently delivered to, and waste to be removed from, cells within the organism. Endothelial cells (ECs) lining the blood vessels are the major players in angiogenesis. The development of the vasculature begins with vasculogenesis, in which ECs differentiated from vascular progenitor cells assemble into a primitive plexus; this is followed by a remodeling process to refine and expand the structures (Chung and Ferrara, 2010; Carmeliet and Jain, 2011; Herbert and Stainier, 2011). Subsequent recruitment of mural cells helps to stabilize the vessels and to further regulate their functions (Chung and Ferrara, 2010; Carmeliet and Jain, 2011; Herbert and Stainier, 2011). After the formation of the primordial vascular network, neovessels can also form from existing vessels through sprouting angiogenesis or intussusception (Chung and Ferrara, 2010; Carmeliet and Jain, 2011; Herbert and Stainier, 2011). Among these processes, sprouting angiogenesis has been studied most extensively. In response to external stimuli, ECs form angiogenic sprouts consisting of tip and stalk cells, which then undergo coordinated directional migration (Carmeliet and Jain, 2011; Herbert and Stainier, 2011). Migration of ECs and the subsequent lumen formation require dynamic organization of the cytoskeleton network (Lamalice et al., 2007; Iruela-Arispe and Davis, 2009). Among the four classes of cytoskeletons, actin and microtubules have well-established roles in various aspects of angiogenesis, whereas the involvement of intermediate filaments or the septin network is less clear (Bayless and Johnson, 2011).
As P‑loop GTPases, septins form heterocomplexes, which further polymerize into paired apolar filaments (Weirich et al., 2008; Mostowy and Cossart, 2012). Septin cytoskeleton has been found to associate with actin and microtubules in different contexts (Weirich et al., 2008; Mostowy and Cossart, 2012). Despite increasing evidence that septins play important roles during development and in adult homeostasis (Cao et al., 2009; Saarikangas and Barral, 2011), the function of septins in ECs is largely unknown. We have previously shown that Borg5 (Binder of the Rho GTPase 5), a Cdc42 effector protein, is up-regulated as mouse embryonic stem cells undergo differentiation (Vong et al., 2010). Borg5, also known as MSE55 (marrow stromal/EC protein with a molecular mass of 55,000 Da) and Cdc42ep1 (Cdc42 effector protein 1), belongs to a family of five related proteins (Borg1–5; Bahou et al., 1992; Joberty et al., 1999; Hirsch et al., 2001). Borg proteins bind directly to septins, and the binding is negatively regulated by Cdc42 (Joberty et al., 2001). Although Borg proteins have been shown to influence actin and septin cytoskeletons in tissue-cultured cells (Burbelo et al., 1999; Joberty et al., 1999, 2001; Hirsch et al., 2001; Sheffield et al., 2003), the physiological function of the Borg family of proteins during development remains elusive. By studying our newly created Borg5-null mice, we show that Borg5 regulates persistent directional migration of cardiac ECs to promote efficient microvascular angiogenesis.
RESULTS
Generating Borg5-null mice
We have previously shown that Borg5 is up-regulated during embryonic stem cell differentiation (Vong et al., 2010). In postnatal mouse tissues, Borg5 is preferentially expressed in the microvasculature of the brain and heart (Supplemental Figure S1, A and B). Within the heart, specific expression of Borg5 was found in the capillary ECs during embryonic and postnatal development (Figures 1A and S1B). This suggests that Borg5 may play a role in the cardiac microvasculature. To understand the physiological function of Borg5, we generated knockout mice (Figure 1B). On backcrossing five generations into the C57BL/6J (B6) background, the heterozygous mice were intercrossed to produce Borg5-null mice (Figure 1C). Immunofluorescence staining of mouse embryonic hearts (Figure 1D) and Western blot analyses of isolated primary ECs (Figure 1E) confirmed this mutant allele to be a null allele. Although Borg5-null embryos showed no gross abnormalities compared with their wild-type littermates (Figure 1F), about half of the null mice died between embryonic day 16.5 (E16.5) and postnatal day 0 (P0) (Figure 1G). Those that survived beyond P0 continued to develop but had reduced body weight from birth to adulthood (Figure S2, A–D). Despite the smaller size, both male and female Borg5-null adult mice are fertile.
FIGURE 1:

Generation of Borg5-null mice. (A) The expression of Borg5 in mouse embryonic heart. Confocal images of wild-type E16.5 heart sections stained with Borg5 rabbit polyclonal antibodies together with antibodies to PECAM-1 or Nkx2.5. Borg5 was found exclusively in ECs labeled with PECAM-1 but not in cardiomyocytes labeled with Nkx2.5. Scale bars: 100 μm. (B) Gene-targeting strategy. The entire coding sequence of Borg5 (light gray arrows) together with part of the intron sequence was replaced by a neomycin-resistance gene (Neo). The noncoding regions of the exons are illustrated as dark gray bars. (C) Mouse tail PCR of the wild-type and knockout alleles produced DNA fragments of 454 base pairs and 578 base pairs, respectively. (D) Immunofluorescence staining of frozen heart sections from wild-type (+/+) and Borg5-null (−/−) embryos at E15.5 using the rabbit Borg5 antibody. No specific Borg5 staining was found in the Borg5-null (−/−) mouse heart. Scale bar: 50 μm. (E) Western blotting analysis of primary MCEC lysates using a Borg5 antibody generated in chicken. The lack of Borg5 protein in Borg5-null (−/−) MCECs confirms the correct targeting. Actin was used as a loading control. Similar results were obtained with the rabbit antibody for Borg5. (F) Wild-type and Borg5-null littermates at E16.5. No gross developmental defects were observed in the mutant. Scale bar: 1 cm. (G) Offspring of each genotype recovered from Borg5+/− mating pairs. A significant reduction of Borg5−/− mice was observed at P0 and at weaning. P0, total pups recovered at birth. P0 live, live pups recovered at birth. Statistical analysis by chi-square test, *, p < 0.05; **, p < 0.01.
The cardiovascular defects in mice lacking Borg5
Because Borg5 is highly expressed in the capillary ECs in the heart, we analyzed the cardiac microvasculature by platelet endothelial cell adhesion molecule (PECAM-1) staining. Compared with control littermates, the capillary density is reduced in the Borg5-null E14.5 to E16.5 hearts (Figure 2, A–E). We also examined the myocardium development by hematoxylin and eosin staining (H&E). Before the formation of a functional microcirculation, myocardium grows as a trabecular layer to maximize the diffusion of nutrient and oxygen. At this stage (E12.5), Borg5-null hearts are indistinguishable from those of the wild-type littermates (Figure 2, F and G). With the formation of the compact layer beginning at around E14.5, cardiac microvasculature becomes essential for the continuous growth of the myocardium. Indeed, we found that the ventricular walls of the Borg5-null hearts were significantly thinner compared with controls at E15.5 (Figure 2, H–J). No signs of anemia or liver discoloration were found in Borg5-null embryos, ruling out the possibility of hematopoietic defects (see Figure 1F). We also did not find obvious defects in the placenta, suggesting that placenta insufficiency was unlikely to be the cause of the observed lethality.
FIGURE 2:

Cardiovascular defects in Borg5-null embryos. (A–D) Borg5 deletion resulted in the reduction of PECAM-1–positive microvessels in the E14.5 and E16.5 hearts. (C′) and (D′) Enlarged regions from (C) and (D), respectively. Scale bars: 100 μm. (E) Quantification of capillary density. Five mutant animals and four wild-type littermate controls at E14.5/E15.5 or E16.5 were analyzed. (F–I) Histology of wild-type and Borg5-null hearts from E12.5 and E15.5 littermates, showing an overall thinner compact layer (CL) in the E15.5 mutant heart compared with the wild-type. (H′) and (I′) Enlarged regions from (H) and (I), respectively, showing the compact layer (CL) of the left ventricles. Scale bars: 200 μm. (J) Quantification of the thickness of ventricular walls (compact layer) normalized by the heart size measured by the longest axis in E15.5 embryos. Four Borg5-null embryos and four wild-type littermate controls were analyzed. Error bars: SEM. Statistical analysis was done by two-tailed Student's t test.
To further characterize the vasculature defects in the Borg5-null embryonic hearts, we perfused E16.5 embryos with fluorescently labeled isolectin β4 and performed two-photon confocal microscopy of the whole-mount hearts after fixation and clearing (Bryson et al., 2011; Hama et al., 2011). Three-dimensional reconstruction revealed a dense network of cardiac vasculature (Figure 3, A and B). Similar to the observation made with two-dimensional heart sections above, the number of three-dimensional capillary structures in a given heart volume was significantly reduced in Borg5-null hearts compared with control littermates (Figure 3C; see Materials and Methods for the measurement of capillary structures). Intriguingly, the Borg5-null vasculature was characterized by a lower surface area to volume ratio than that of control littermates (Figure 3, D–F). Because the high surface area to volume ratio is the hallmark of a capillary network, these data suggest that loss of Borg5 compromises proper capillary network formation.
FIGURE 3:

Three-dimensional reconstruction of the vascular network in the whole-mount wild-type (A) and Borg5-null (B) E16.5 hearts. Three regions within the microvasculature from each heart (>3 hearts per genotype) were randomly picked ((A′) and (B′), dashed regions were enlarged in (A″) and (B″) for better visualization) and the number of vascular structures (C), surface area of the vasculature (D), volume of the vasculature (E), each normalized by the heart volume, and vascular surface area to vascular volume ratio (F) are quantified. Details about the quantification can be found in Materials and Methods. Scale bars: 100 μm. Error bars: SE of the mean (SEM). Statistical analysis was done by two-tailed Student's t test.
We next analyzed whether Borg5-null mice have angiogenesis defects in other vascular beds. We found that there was a mild but significant reduction of the radial lengths of the capillary network in Borg5-null retinas at both postnatal day 2 (P2) and 6 (P6) compared with controls (Figure 4, A and B). Filopodia and branching-point densities, however, were not significantly affected in mutant retinas (Figure 4, C–F). These results suggest that Borg5 is involved in EC migration during angiogenesis, but it probably affects processes other than filopodia formation or branching morphogenesis. The milder vascular defects seen in the retina, as compared with the heart, could be due to different degrees of redundancy among Borg proteins in the two vascular beds.
FIGURE 4:
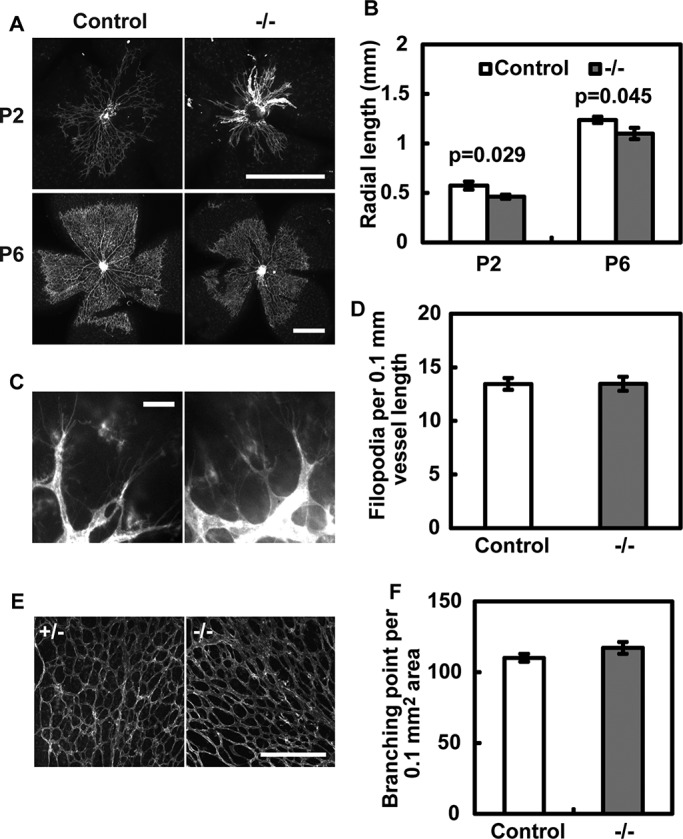
Defective retinal angiogenesis in Borg5-null mice. (A and B) Loss of Borg5 causes a significant reduction in the radial length of retinal blood vessels at P2 and P6. Scale bar: 0.5 mm. (C and D) Filopodia formation in P2 retinas was not significantly affected by the loss of Borg5. Scale bar: 10 μm. (E and F) Branch density in P6 retinas was not significantly affected by the loss of Borg5. Scale bar: 200 μm. Error bars: SEM. Statistical analysis was done by two-tailed Student's t test.
Borg5 is required for persistent directional migration of ECs during in vitro angiogenesis
To further study the role of Borg5 in EC migration, we utilized a heart explant model to assay for angiogenesis (Yue and Tomanek, 2001). In this assay, cardiac ECs migrate out of the heart explants into three-dimensional collagen gel and form extensive angiogenic sprouts consisting of multiple cells after 6 d of culture (Figure 5A). Consistent with a role for Borg5 in EC migration, sprouts formed by Borg5-null hearts were significantly shorter compared with control explants (Figure 5, A and B). To monitor cell migration behavior in real time, we utilized live imaging by confocal microscopy during day 4 to day 5 of the culture (Figure 5C). Individual nuclei within the sprouts were visualized by SYTO-16 dye, which has been shown to preferentially label ECs (Arima et al., 2011). Computer-assisted automatic tracking was used to quantify single-cell behavior as the cells traveled from the heart explant into the extracellular matrix. Interestingly, Borg5-null ECs displayed a significant reduction in persistent directional migration as measured by the straightness of the cell track, which is quantified as the distance of cell displacement divided by the migration path (Figure 5, D and E). The migration speed (path/time) of Borg5-null ECs was also significantly reduced compared with control (Figure 5F). The EC identity was confirmed by isolectin β4 staining at the end of each experiment (see Figure 5A). These findings show that Borg5 regulates persistent directional migration of ECs during in vitro angiogenesis.
FIGURE 5:

Borg5 regulates persistent directional migration of the cardiac ECs. (A) Heart explant angiogenesis assay. Wild-type and Borg5-null heart explants were stained with isolectin β4 to label ECs after 6 d of culture. Three-dimensional images were obtained by confocal microscopy and reconstructed in Imaris. Regions in the white dashed squares were enlarged to the right of each image for better visualization. Scale bars: 0.5 mm. (B) Quantification of sprout length. (C) Examples of automated tracking of EC migration from heart explants during day 4 to day 5 of culture. EC nuclei were labeled with SYTO-16 green fluorescent dye. Tracked nuclei are highlighted in white, and individual tracks are displayed as time color-coded lines. Time is expressed as h:min:s. Scale bar: 100 μm. (D) An illustration of individual cell displacement and path. (E) Quantification of persistent directional migration (straightness, displacement/path). (F) Quantification of migration speed (path/time). Error bars: SEM. Statistical analysis was done by two-tailed Student's t test.
Borg5 controls the localization of the septin filaments along the perinuclear actin fibers in primary mouse cardiac ECs
To find clues about how Borg5 promotes persistent directional migration during angiogenesis in the heart, we isolated primary mouse cardiac ECs (MCECs) from adult hearts (Lim and Luscinskas, 2006). The isolated Borg5-null MCECs showed no defect in proliferation in vitro (Figure S3, A and B). In addition, immunofluorescence imaging of MCECs attached to the coverslips revealed that both the wild-type and Borg5-null MCECs expressed the EC-specific marker VE-cadherin and were able to form adherens junctions (Figure S3, C and D). In the scratch wound–healing assay, centrosome and Golgi complex reorientation is an important early step in establishing polarity. Immunofluorescence staining of the centrosome and Golgi revealed that the isolated Borg5-null and wild-type MCECs exhibited similar centrosome/Golgi positioning after scratch wounding (Figure S3, E–H). Thus the reduced persistent directional migration in Borg5-null MCECs observed in the previously discussed heart explant assay is unlikely to be due to defects in the ability of MCECs to reorientate their centrosomes for directional migration.
Next we examined Borg5 localization in primary MCECs. We found that Borg5 appears as a prominent filamentous patch above the nucleus and coaligns with the actin cytoskeleton in the majority (∼60%) of MCECs (see organized perinuclear septin patch in Figure 6A for an example, and Figure S4A for the Z series). Septin 7 (Sept7), a previously identified Borg5-interacting partner (Joberty et al., 2001), is also present in these perinuclear structures (Figures 6A and S4A). Borg5 and Sept7 colocalize throughout the primary MCECs with a Pearson correlation coefficient of 0.64 and 0.63 in single confocal sections (Figure 6B) and in three-dimensional reconstructed images (Figure S4B), respectively. Coimmunoprecipitation analyses of endogenous proteins from primary MCECs further confirmed the interaction between Borg5 and Sept7 (Figure 6C). Consistent with a previous report that Borg protein had no effect on septin heterodimer formation in vitro (Sheffield et al., 2003), the interaction between septin 2 (Sept2) and Sept7 was preserved in the Borg5-null MCECs (Figure 6C). However, loss of Borg5 resulted in a near depletion of the perinuclear septin patch (Figure 6, A and D), suggesting that, although Borg5 is not required for septin heterocomplex formation, it is required for the proper localization and organization of perinuclear septin patch in MCECs.
FIGURE 6:

Borg5 and Sept7 colocalize to and regulate perinuclear actin fibers in primary MCECs. (A) Confocal images of wild-type (+/+) and Borg5-null (−/−) MCECs at the wound edge. Examples of three different patterns of perinuclear septin filament and actin patch are shown, including organized, disorganized, and no clear structure (none), as indicated. Nuclei in the merged images were labeled with 4′,6-diamidino-2-phenylindole (blue). Scale bar: 25 μm. (B) Colocalization between Borg5 and Sept7 in confocal sections as quantified by Pearson's coefficient. Borg5-null cells were included to control for nonspecific background staining by Borg5 antibody. (C) Coimmunoprecipitation of endogenous Borg5 by Sept7 antibodies. Note that the interaction between Sept2 and Sept7 was not affected by Borg5 deletion. (D and E) Quantification of wild-type and Borg5-null MCECs with different septin (D) or actin (E) patterns. See (A) for examples of different patterns. (F) Quantification of colocalization between Sept7 and F-actin in the perinuclear region of wild-type and Borg5-null MCECs. (G) Quantification of cells with different perinuclear actin patterns after Sept7 knockdown. All images and quantifications are for cells at the wound edge. Error bars: SEM. Statistical analysis was done by two-tailed Student's t test.
Because Borg5 and Sept7 colocalize with a segment of parallel actin bundles in the perinuclear region, we also examined the perinuclear actin fibers in the wild-type and Borg5-null MCECs. While ∼60% of wild-type cells exhibited organized perinuclear actin fibers, only ∼5% of Borg5-null cells retained such structures (Figure 6, A and E). The colocalization of septin filaments and F-actin in the perinuclear region was also reduced upon Borg5 deletion, as judged by the Pearson correlation coefficiency (Figure 6F). Similarly, reduction of Sept7 in MCECs by RNA interference (RNAi) reduced the parallel actin fibers from ∼60 to ∼20% (Figures 6G and S4, C and D). These data are consistent with previous findings that septin and actin regulate each other (Kinoshita et al., 2002; Schmidt and Nichols, 2004).
To determine the generality of the actin-associated perinuclear Borg5/septin patches, we examined primary human umbilical cord ECs and several mouse cell lines, including NIH 3T3 (fibroblasts), C3H10T1/2 (mesenchymal stem cells), NMuMG (mammary gland epithelial cells), and an immortalized MCEC line (Barbieri and Weksler, 2007). We found that none of these cells contained the characteristic Borg5/septin patch aligning with parallel actin fibers right above or around the nucleus, as seen in the primary MCECs (Figure S5). Therefore the formation and/or integrity of the perinuclear filamentous septin patches are restricted to certain cell types, such as the primary MCECs.
Borg5 and Sept7 regulate persistent directional migration through myosin II
Previous studies have found that parallel actomyosin bundles in mouse embryonic fibroblasts preferentially localize to the interior region of the cell (Lo et al., 2004). These bundles align with the direction of cell migration and are required for the persistence of migration (Lo et al., 2004). When fibroblasts were cultured on micropatterned tracks, contractile actomyosin in the middle section of a cell appeared to couple the propulsive anterior and the resistive tail to allow these regions to undergo coordinated movement (Guo and Wang, 2012). Because Sept2, which forms a complex with Sept7, has been shown to bind to and activate myosin (Joo et al., 2007), the actin fibers associated with Borg5 and Sept7 might be involved in proper actomyosin contraction to prevent random migration. Immunofluorescence staining of phosphorylated myosin light chain (ppMLC), which labels activated myosin II, revealed that active myosin II localized along the Borg5-associated actin fibers in wild-type MCECs (Figure 7A). Importantly, Borg5-null MCECs, which exhibited a significant loss of the perinuclear septin patch and parallel actin fibers (see Figure 6, D and E), also showed a corresponding loss of organized ppMLC staining in the perinuclear region (Figure 7, B and C). A similar disruption was observed in MCECs treated with Sept7 small interfering RNA (siRNA; Figure 7D).
FIGURE 7:

Borg5 and Sept7 regulate actomyosin organization in primary MCECs. (A) Examples of ppMLC localization in MCECs at the wound edge. In addition to peripheral localization, ppMLC is also found on the Borg5-coated parallel actin fibers. Scale bar: 25 μm. (B) Quantification of perinuclear ppMLC organization in wild-type and Borg5-null MCECs. See (A) for examples of different patterns. (C) Quantification of perinuclear ppMLC intensity in wild-type and Borg5-null MCECs. (D) Quantification of perinuclear ppMLC organization in MCECs treated with Sept7 siRNA. (E–G) Quantification of persistent migration (straightness) of wild-type and Borg5-null MCECs (E) and MCECs treated with Sept7 siRNA (F) or myosin II inhibitor Y27632 (G) using scratch wound–healing migration assay. Error bars: SEM. Statistical analysis was done by two-tailed Student's t test.
We then tested whether Borg5, Sept7, and ppMLC were required for persistent directional migration of MCECs during in vitro scratch wound–healing migration. We found that Borg5-null cells exhibited a similar reduction in persistent directional migration as observed in the three-dimensional assay above (Figure 7E; compare with Figure 5F). However, the Borg5-null cells migrated faster than wild-type cells in two-dimensional (Figure S4E), contrary to our finding in three-dimensional (see Figure 5F). Cell migration behavior can differ in two-dimensional and three-dimensional models (Even-Ram and Yamada, 2005; Petrie et al., 2009; Shih and Yamada, 2010). The above analyses show that the persistent directional migration behavior of MCECs in three-dimensional, but not migration speed, can be modeled in two-dimensional cultures. We therefore focused on using the two-dimensional model to study the role of Sept7 and ppMLC in persistent directional migration of MCECs. We found that persistent directional migration of MCECs was significantly reduced upon Sept7 knockdown or myosin II inhibition by Y27632 (Figure 7, F–G). These results are consistent with the idea that Borg5 and septin cytoskeleton regulate the positioning and organization of the actomyosin fibers in MCECs, which is important for persistent directional migration of MCECs in vitro. Because immortalized mouse embryonic fibroblasts derived from septin 9 knockout embryos also exhibit reduced directional migration compared with controls (Fuchtbauer et al., 2011), septins may regulate persistent directional migration in different cell types.
DISCUSSION
Angiogenesis requires highly coordinated morphogenesis of ECs, which would involve proper coupling of different cytoskeletal networks within the EC. By studying Borg5, we have uncovered a previously unappreciated actomyosin structure that contains Borg5 and septins in MCECs. Because the binding of Borg5 to septins is disrupted by Cdc42-GTP (Joberty et al., 2001), it is possible that a dynamic interaction between Borg5 and septins regulated by Cdc42 might facilitate the positioning and organization of the contractile actomyosin fibers above the nucleus of MCECs. Our observation that ∼60% of wild-type MCECs exhibit the Borg5/septin-associated actin fibers is consistent with the idea that this structure is dynamically assembled and disassembled.
A previous study found that overexpressing a fragment of Borg3 resulted in aggregation of septin cytoskeleton but no effect on actin cytoskeleton (Joberty et al., 2001). However, there were many cases in which overexpression of a dominant-negative mutant caused a phenotype that differed from the loss-of-function studies. We show that either genetic deletion of Borg5 or reduction of septin in the primary MCECs resulted in the disruption of the perinuclear actomyosin fiber, reduction of myosin II activity (as judged by ppMLC staining), and diminished persistent directional migration of MCECs in vitro. On the basis of these correlative data, we suggest that Borg5 functions with septins to control proper organization and activation of actomyosin in MCECs, which in turn ensures persistent directional migration. Other adherent cell types, such as fibroblasts, also contain actin networks in the interior region of the cell, which is important for the persistent directional migration (Lo et al., 2004). While it is likely that actomyosin fibers control migration through interactions with the focal adhesion sites, as septin/actin are localized underneath the nuclei in these cells, it has also been proposed that contractile actomyosin in the middle section of a cell plays a role in coupling the propulsive anterior and the resistive tail enabling these regions to undergo coordinated movement (Guo and Wang, 2012). Therefore it remains possible that the perinuclear septin/actomyosin machinery in primary MCECs could be involved in similar functions in regulating persistent directional migration. However, as the primary MCECs may be unique in relying on Borg5 to organize the perinuclear actomyosin, we stress that further studies geared at addressing how Borg5 regulates the dynamic organization and activity of the fibers would be required to establish a direct functional relationship between Borg5 and septin/actomyosin in cell migration. Unfortunately, the primary MCECs only grow three passages before undergoing senescence and are refractory to plasmid transfection or lentiviral infection. These technical difficulties have thus far made it extremely challenging to dissect whether and how Borg5 regulates MCEC migration through septins and actomyosin. However, our efforts in combining mouse knockout studies with in vitro analyses of primary MCECs represent an important step forward in understanding the mechanism of microvascular angiogenesis during heart development.
Although Borg5 was identified two decades ago as the first member of the Borg family of proteins, the in vivo function of any of the Borg proteins has remained elusive. We report here a function of Borg5 in microvascular ECs. In addition, our preliminary observations suggest that Borg1, 2, and 4 are also expressed in the endothelium. Interestingly, Borg proteins have been found only in vertebrates. The development and function of large and sophisticated organs unique to the vertebrates require an extensive circulatory system. It is therefore tempting to speculate that Borg proteins could be evolved to facilitate the function of microvascular ECs unique to vertebrate animals. As a family of highly conserved GTPases in eukaryotes, septins form a unique cytoskeleton. Although mutations in septins have been linked to many human diseases (Saarikangas and Barral, 2011), the underlying mechanisms are still not clear. Our findings suggest a physiological function of septins in the cardiovascular system.
Abnormalities in the microvasculature not only cause developmental defects but are also associated with human diseases such as cancer, coronary microvascular dysfunction, and diabetes (Camici and Crea, 2007; Langenkamp and Molema, 2009; Wiernsperger and Rapin, 2012). Future studies utilizing mouse models that are relevant to human diseases should provide important insights into how Borg5 and other members of the family could be involved in disease-related capillary angiogenesis.
MATERIALS AND METHODS
Borg5 knockout mouse
DNA fragments for homologous recombination arms of Borg5 targeting vectors were amplified from the BAC clone RP23-37M4 derived from the genomic DNA of the C57BL/6J mouse. The 4-kb 5′ arm was amplified by PCR with primers 5′-TAGGATGCGGCCGCTGAAGTTTTTCCTGGACTTCCCATCC-3′ and 5′-TAGGACAAGCTTAGGGAGTCTGTGGGTTCTCTCTCTGC-3′ and then inserted into the NotI/HindIII sites of pYZ1932, a gene-targeting vector containing PGK-NeoR and HSV-tk (full information is available upon request). The 2-kb 3′ arm was amplified with primers 5′-TACCACTTAATTAAAGACCTGCCTCCCATACCTGATTGTT-3′ and 5′-TAGGAAGGCGCGCCGAATGGAGTCATCTGGGATGGTGAAG-3′ and then inserted into the PacI/AscI sites. The targeting vectors were linearized with NotI and electroporated into V6.5 mouse embryonic stem cells (ESC). After selection with 400 μg/ml G418 (Invitrogen, Carlsbad, CA) and 2 μM ganciclovir (G2536; Sigma-Aldrich, St. Louis, MO), ESC clones were screened for homologous recombination of the flanking regions of the 3′ recombination arm from the genomic DNA by PCR with primers 5′-GGCTCTATGGCTTCTGAGGCGGAAAG-3′ and 5′-GGCTCTGGGAAGCTGGAGTCAGAATC-3′. A correctly targeted ESC clone was injected into mouse blastocysts to produce chimeras. Male chimeras with germ-line transmission were bred with wild-type C57BL/6J (Jackson Laboratory, Bar Harbor, ME) females to generate F1 heterozygous mutants, which were then backcrossed for four additional generations. The resulting Borg5+/− mice were intercrossed to produce Borg5−/− mice. Genotyping for Borg5 knockout mice was done by tail DNA PCR using a mixture of three primers (ZL-412: 5′-ACTCCAGGGTAGGGATCCTTTTA-3′; ZL-413: 5′-GTTGCTGAGGAAGGACGTATCTC-3′; ZL-415: 5′-AAAGCGAAGGAGCAAAGCTG-3′). All mouse procedures were in accordance with the guidelines of the Carnegie Institution Animal Care and Use Committee.
Antibodies and labeling reagents
Generation of pericentrin antibodies and Borg5 rabbit and chicken antibodies were described previously (Liu and Zheng, 2009; Vong et al., 2010). All the other antibodies and molecules used in this study were commercially available, including α-tubulin (T9026, 1:1000; Sigma-Aldrich), actin (A3853, 1:200; Sigma-Aldrich), GM130 (610822, 1:200; BD Biosciences, Franklin Lakes, NJ), isolectin β4 (I21412, 1:200; Invitrogen), Nkx2.5 (sc-8697, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA), PECAM-1 (550274, 1:10; BD Biosciences), phalloidin (A22287 or A12379, 1:200; Invitrogen), phospho-MLC (3674, 1:200; Cell Signaling Technology, Danvers, MA), Sept7 (18991, 1:1000; IBL, Minneapolis, MN), and VE-cadherin (sc-6458, 1:50; Santa Cruz Biotechnology).
Histology and immunohistochemistry
For embryonic mice, pregnant females from timed mating were killed by cervical dislocation and the embryos were dissected and fixed in 10% Formalin for 24–48 h at 4°C. For postnatal mice, hearts were dissected and fixed in the same manner as above for 48 h. Following dehydration and paraffin embedding, 5 μm sections were cut and stained with H&E (Surgipath, Buffalo Grove, IL).
For immunohistochemistry of the tissue, mouse hearts were dissected and fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) overnight at 4°C; this was followed by equilibration in 30% sucrose overnight at 4°C and embedding in optimal cutting temperature compound. Transverse sections (embryonic hearts) or cross-sections (postnatal hearts) of 10-μm thickness were obtained with a Leica CM3050S cryostat. The sections were blocked in Tris-buffered saline (TBS) with 2% bovine serum albumin (BSA), 0.25% casein, and 0.2% Triton X-100 for 30 min; this was followed by incubation with primary antibodies diluted in TBS with 1% BSA and 0.04% Tween-20 overnight at 4°C and secondary antibodies for 1 h at room temperature.
Capillary density was quantified blindly by manual counting of PECAM-1–labeled ECs and normalized by the number of total nuclei. Thickness of the ventricular wall was measured as the compact layer in the left ventricle normalized by the long axis of the heart. Animals from more than three litters were analyzed.
Retinas from P2 and P6 mouse pups were processed according to a published protocol (Pitulescu et al., 2010). At least six retinas from more than three litters were analyzed.
Three-dimensional reconstruction of cardiac vasculature
E16.5 embryos were perfused with isolectin β4 as previously described (Bryson et al., 2011). The dissected hearts were then fixed in 4% PFA overnight at 4°C; this was followed by clearing in ScaleA2 for 2 wk at 4°C (Hama et al., 2011). A middle region of each ventricle was imaged using a two-photon confocal laser-scanning microscope (Leica TCS SP5 MP) equipped with a 25× water lens at 1024 × 1024 (xy) and 0.5-μm intervals (z axis). Three-dimensional images were reconstructed in Imaris software (Bitplane Scientific Software, Zurich, Switzerland). For quantification of the vascular structures, three 150 × 150 × 200 (xyz) regions within the capillary network (avoiding the large arteries and veins) were randomly picked and surfaces were built. The number of structures, surface area, and volume were automatically calculated by the software. More than three hearts per genotype were analyzed.
Heart explant culture and imaging
E14.5 hearts were dissected out, and the ventricles were cut into halves. The top half was then embedded between two layers of neutralized 1.5 mg/ml collagen gel (354236; BD Biosciences) containing culture medium and 10% fetal bovine serum (FBS) in eight-well chambered coverglass (155411; Thermo Fisher Scientific, Rochester, NY). After overnight culture, an equal volume (0.2 ml) of DMEM containing 20% FBS and 20 ng/ml VEGF/FGF2 (Peprotech, Rocky Hill, NJ) was added, and the culture was continued for an additional 2 d. Then 0.1 ml medium with 10 ng/ml VEGF/FGF2 and 1:1000 SYTO-16 dye (S7578; Invitrogen) was added. After one more day of culture, the explants were imaged by time-lapse confocal microscopy at 10-min intervals for 1–2 d in a live-cell chamber with CO2 and humidity controls. Three-dimensional images were reconstructed in Imaris, and individual nuclei were automatically tracked based on the SYTO-16 signal. The straightness (displacement divided by path; see Figure 5D for an illustration) was averaged from more than 30 individual cells in each experiment, and statistical analysis was based on results from three independent experiments. The heart explants were fixed after 6 d of culture and stained with isolectin β4 to confirm the EC identity of the migratory cells. Images were obtained using confocal microscopy. The length of three randomly picked sprouts from each explant were measured from maximum projections, and results from three independent experiments were averaged.
Primary MCEC culture and siRNA transfection
Primary MCECs were isolated from 4- to 7-wk-old sex-matched littermates according to a previously published method (Lim and Luscinskas, 2006). Briefly, hearts from individual mice were digested with 5 ml 0.2% collagenase I (LS004196; Worthington) for 45 min; this was followed by positive sorting with PECAM-1 antibody (553370; BD Biosciences) coupled to magnetic beads (11035; Invitrogen). Cells were washed with the isolation medium containing DMEM (11995; Invitrogen), 20% FBS (Invitrogen), and penicillin/streptavidin (15140; Invitrogen), and then cultured in growth medium containing the isolation medium plus nonessential amino acids (11140; Invitrogen), gentamicin/amphotericin B (R01510; Invitrogen), 0.1 mg/ml heparin (H3393; Sigma-Aldrich), 20 mM HEPES (H0887; Sigma-Aldrich), and 50 μg/ml ECGS (BT-203; Biomed Tech) until near confluence in gelatin-coated plates before a second round of sorting with ICAM-2 antibody (553325; BD Biosciences). Passage 1 (outgrowth of cells after second sorting) or passage 2 cells were used in experiments. Incorporation of 5-ethynyl-2′-deoxyuridine (EdU) was detected using an EdU detection kit (Invitrogen) after incubating the cells with 10 μM EdU overnight.
For siRNA transfection, passage 1 cells were seeded in a six-well plate and transfected next day at 40% confluence with Lipofectamine RNAiMax in serum-free medium for 3 h; this was followed by feeding with fresh growth medium. Cells were cultured for 2 more days before being replated on glass coverslips for immunofluorescence staining as described below. Control (5′-CGUACGCGGAAUACUUCGA-3′), Sept7 siRNA1 (5′-GACAAUAGUUGAUACUCCA-3′; Li et al., 2012), and siRNA2 (5′-AGAAAGCUAGCAGCAGUGAC-3′; Kinoshita et al., 2002) oligos were purchased from Dharmacon and used at a final concentration of 100 nM.
Immunofluorescence staining of MCECs
For immunofluorescence staining, passage 2 MCECs or MCECs treated with siRNA for 2 d were detached from culture plates by 0.05% trypsin-EDTA for 5 min, pelleted, and resuspended in the isolation medium. A 50-μl drop containing 1 × 105 cells was placed on the inside surface of the lid of a Petri dish (bacteria grade). A 12-mm round glass coverslip that had been freshly treated with 0.1% gelatin (37ºC for 1 h) was then inverted on top of the drop of MCECs. The lid was then carefully inverted and placed back on the Petri dish. To prevent drying of the cells, we added 5–10 ml PBS into the dish. After overnight incubation, coverslips with cells attached were removed from the lid, scratched with a 200-μl wide-orifice pipette tip, and incubated in 24-well plates containing 0.5 ml of the isolation medium for 6 h before fixation for immunofluorescence staining.
Cells were fixed with 4% PFA for 15 min and permeabilized in TBS containing 0.1% Triton X-100 for 20 min. After blocking with 4% BSA, coverslips were incubated with primary antibodies overnight at 4°C, followed by secondary antibodies for 1–2 h at room temperature. Confocal images were obtained with a Leica TCS SP5. Colocalization and image intensity were measured in Leica Application Suite Advanced Fluorescence (LAS AF). For three-dimensional colocalization measurement, confocal sections were reconstructed in Imaris and then analyzed.
Wound-healing migration assay
MCECs were cultured to confluence in the growth medium before being scratched with a 200-μl wide-orifice pipette tip. The growth medium was then replaced with the isolation medium to discourage cell proliferation. When applicable, Y27632 (5 μM) was supplemented in the medium immediately after the scratch. Wound-healing migration was monitored live at 10-min intervals overnight. Individual cells at the migration front (excluding those undergoing mitosis) were manually traced using MetaMorph software. The straightness of migration is determined by the displacement distance of cells divided by path. More than 30 individual cells were tracked in each experiment, and statistical analysis was based on results from three independent experiments.
Immunoprecipitation
Primary MCECs were harvested in lysis buffer (25 mM Tris, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 1 mM EDTA, 0.5% Triton X-100) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN). Clarified cell lysates were incubated with 1 μg of rabbit anti-Sept7 antibody (IBL) for 1 h on ice, and then precipitated by protein G agarose (Invitrogen) at 4°C for 2 h. After washes, the immunoprecipitates were analyzed by Western blotting with chicken anti-Borg5 antibody (1:2000) or goat anti-Sept2 antibody (1:200; Santa Cruz Biotechnology).
Statistical analysis
Each experiment was repeated at least three times. Results are reported as mean ± SEM. Data were analyzed by Student's t test unless otherwise indicated, and differences were considered significant when p < 0.05.
Supplementary Material
Acknowledgments
We thank Ona Martin, Rong Chen, and Xiaohong Ma for technical support; Youngjo Kim for designing the Borg5 knockout screening primers; Elias T. Spiliotis for advice on septin antibodies; and Chen-Ming Fan and members of the Zheng lab for helpful comments. The research was supported by National Heart, Lung, and Blood Institute intramural research (C.L.), R01 GM056312, and the Howard Hughes Medical Institute (Q.P.V., Z.L., and Y.Z.).
Abbreviations used:
- Borg5
Binder of the Rho GTPase 5
- BSA
bovine serum albumin
- EC
endothelial cell
- EdU
5-ethynyl-2′-deoxyuridine
- ESC
embryonic stem cells
- H&E
hematoxylin and eosin
- MCEC
mouse cardiac EC
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- ppMLC
phosphorylated myosin light chain
- RNAi
RNA interference
- TBS
Tris-buffered saline
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-09-0543) on January 22, 2014.
*Present address: St. Jude Children's Research Hospital, Memphis, TN 38105.
REFERENCES
- Arima S, Nishiyama K, Ko T, Arima Y, Hakozaki Y, Sugihara K, Koseki H, Uchijima Y, Kurihara Y, Kurihara H. Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development. 2011;138:4763–4776. doi: 10.1242/dev.068023. [DOI] [PubMed] [Google Scholar]
- Bahou WF, Campbell AD, Wicha MS. cDNA cloning and molecular characterization of MSE55, a novel human serum constituent protein that displays bone marrow stromal/endothelial cell-specific expression. J Biol Chem. 1992;267:13986–13992. [PubMed] [Google Scholar]
- Barbieri SS, Weksler BB. Tobacco smoke cooperates with interleukin-1β to alter β-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo. FASEB J. 2007;21:1831–1843. doi: 10.1096/fj.06-7557com. [DOI] [PubMed] [Google Scholar]
- Bayless KJ, Johnson GA. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J Vasc Res. 2011;48:369–385. doi: 10.1159/000324751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson JL, Coles MC, Manley NR. A method for labeling vasculature in embryonic mice. J Vis Exp. 2011;56:e3267. doi: 10.3791/3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Snow DM, Bahou W, Spiegel S. MSE55, a Cdc42 effector protein, induces long cellular extensions in fibroblasts. Proc Natl Acad Sci USA. 1999;96:9083–9088. doi: 10.1073/pnas.96.16.9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- Cao L, Yu W, Wu Y, Yu L. The evolution, complex structures and function of septin proteins. Cell Mol Life Sci. 2009;66:3309–3323. doi: 10.1007/s00018-009-0087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2010;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- Even-Ram S, Yamada KM. Cell migration in 3D matrix. Curr Opin Cell Biol. 2005;17:524–532. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Fuchtbauer A, Lassen LB, Jensen AB, Howard J, Quiroga Ade S, Warming S, Sorensen AB, Pedersen FS, Fuchtbauer EM. Septin9 is involved in septin filament formation and cellular stability. Biol Chem. 2011;392:769–777. doi: 10.1515/BC.2011.088. [DOI] [PubMed] [Google Scholar]
- Guo WH, Wang YL. A three-component mechanism for fibroblast migration with a contractile cell body that couples a myosin II-independent propulsive anterior to a myosin II-dependent resistive tail. Mol Biol Cell. 2012;23:1657–1663. doi: 10.1091/mbc.E11-06-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue-Sawano A, Miyawaki A. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch DS, Pirone DM, Burbelo PD. A new family of Cdc42 effector proteins, CEPs, function in fibroblast and epithelial cell shape changes. J Biol Chem. 2001;276:875–883. doi: 10.1074/jbc.M007039200. [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Davis GE. Cellular and molecular mechanisms of vascular lumen formation. Dev Cell. 2009;16:222–231. doi: 10.1016/j.devcel.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Perlungher RR, Macara IG. The Borgs, a new family of Cdc42 and TC10 GTPase-interacting proteins. Mol Cell Biol. 1999;19:6585–6597. doi: 10.1128/mcb.19.10.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Perlungher RR, Sheffield PJ, Kinoshita M, Noda M, Haystead T, Macara IG. Borg proteins control septin organization and are negatively regulated by Cdc42. Nat Cell Biol. 2001;3:861–866. doi: 10.1038/ncb1001-861. [DOI] [PubMed] [Google Scholar]
- Joo E, Surka MC, Trimble WS. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev Cell. 2007;13:677–690. doi: 10.1016/j.devcel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. Self- and actin-templated assembly of mammalian septins. Dev Cell. 2002;3:791–802. doi: 10.1016/s1534-5807(02)00366-0. [DOI] [PubMed] [Google Scholar]
- Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Langenkamp E, Molema G. Microvascular endothelial cell heterogeneity: general concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell Tissue Res. 2009;335:205–222. doi: 10.1007/s00441-008-0642-4. [DOI] [PubMed] [Google Scholar]
- Li S, Ou XH, Wei L, Wang ZB, Zhang QH, Ouyang YC, Hou Y, Schatten H, Sun QY. Septin 7 is required for orderly meiosis in mouse oocytes. Cell Cycle. 2012;11:3211–3218. doi: 10.4161/cc.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YC, Luscinskas FW. Isolation and culture of murine heart and lung endothelial cells for in vitro model systems. Methods Mol Biol. 2006;341:141–154. doi: 10.1385/1-59745-113-4:141. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zheng Y. A requirement for epsin in mitotic membrane and spindle organization. J Cell Biol. 2009;186:473–480. doi: 10.1083/jcb.200902071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol Biol Cell. 2004;15:982–989. doi: 10.1091/mbc.E03-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 2012;13:183–194. doi: 10.1038/nrm3284. [DOI] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- Saarikangas J, Barral Y. The emerging functions of septins in metazoans. EMBO Rep. 2011;12:1118–1126. doi: 10.1038/embor.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Nichols BJ. Functional interdependence between septin and actin cytoskeleton. BMC Cell Biol. 2004;5:43. doi: 10.1186/1471-2121-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield PJ, Oliver CJ, Kremer BE, Sheng S, Shao Z, Macara IG. Borg/septin interactions and the assembly of mammalian septin heterodimers, trimers, and filaments. J Biol Chem. 2003;278:3483–3488. doi: 10.1074/jbc.M209701200. [DOI] [PubMed] [Google Scholar]
- Shih W, Yamada S. Myosin IIA dependent retrograde flow drives 3D cell migration. Biophys J. 2010;98:L29–L31. doi: 10.1016/j.bpj.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong QP, Liu Z, Yoo JG, Chen R, Xie W, Sharov AA, Fan CM, Liu C, Ko MS, Zheng Y. A role for borg5 during trophectoderm differentiation. Stem Cells. 2010;28:1030–1038. doi: 10.1002/stem.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Barral Y. The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol. 2008;9:478–489. doi: 10.1038/nrm2407. [DOI] [PubMed] [Google Scholar]
- Wiernsperger N, Rapin JR. Microvascular diseases: is a new era coming. Cardiovasc Hematol Agents Med Chem. 2012;10:167–183. doi: 10.2174/187152512800388885. [DOI] [PubMed] [Google Scholar]
- Yue X, Tomanek RJ. Effects of VEGF(165) and VEGF(121) on vasculogenesis and angiogenesis in cultured embryonic quail hearts. Am J Physiol Heart Circ Physiol. 2001;280:H2240–H2247. doi: 10.1152/ajpheart.2001.280.5.H2240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.