

Calpain-mediated proteolysis of the thin filament pointed-end–capping protein tropomodulin results in actin subunit association onto pointed ends and increased thin filament lengths in two different murine models of Duchenne muscular dystrophy. This mechanism affects different skeletal muscles in a use- and disease severity–dependent manner.
Abstract
Duchenne muscular dystrophy (DMD) induces sarcolemmal mechanical instability and rupture, hyperactivity of intracellular calpains, and proteolytic breakdown of muscle structural proteins. Here we identify the two sarcomeric tropomodulin (Tmod) isoforms, Tmod1 and Tmod4, as novel proteolytic targets of m-calpain, with Tmod1 exhibiting ∼10-fold greater sensitivity to calpain-mediated cleavage than Tmod4 in situ. In mdx mice, increased m-calpain levels in dystrophic soleus muscle are associated with loss of Tmod1 from the thin filament pointed ends, resulting in ∼11% increase in thin filament lengths. In mdx/mTR mice, a more severe model of DMD, Tmod1 disappears from the thin filament pointed ends in both tibialis anterior (TA) and soleus muscles, whereas Tmod4 additionally disappears from soleus muscle, resulting in thin filament length increases of ∼10 and ∼12% in TA and soleus muscles, respectively. In both mdx and mdx/mTR mice, both TA and soleus muscles exhibit normal localization of α-actinin, the nebulin M1M2M3 domain, Tmod3, and cytoplasmic γ-actin, indicating that m-calpain does not cause wholesale proteolysis of other sarcomeric and actin cytoskeletal proteins in dystrophic skeletal muscle. These results implicate Tmod proteolysis and resultant thin filament length misspecification as novel mechanisms that may contribute to DMD pathology, affecting muscles in a use- and disease severity–dependent manner.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a hereditary muscle-wasting disorder afflicting ∼1 in 3500 males. DMD is caused by absence of the costameric protein dystrophin from the sarcolemma of skeletal muscle fibers, leading to sarcolemmal mechanical instability, membrane rupture during muscle contraction, pathological influx of extracellular Ca2+ into the muscle fiber interior, resultant hyperactivity of Ca2+-dependent proteases, and widespread misregulated proteolysis of muscle cytoskeletal proteins (Blake et al., 2002; Rahimov and Kunkel, 2013). Key players in the progression of Ca2+-dependent proteolysis in dystrophic muscle are the calpains, a ubiquitously expressed and well-conserved family of Ca2+-dependent cysteine proteases (Tidball and Spencer, 2000). Total calpain levels are elevated in the muscles of dystrophin-deficient mdx mice (a well-characterized animal model of DMD) as compared with wild-type (WT) mice, and this increase is primarily driven by elevated concentrations of m-calpain (Spencer et al., 1995). Moreover, myotubes differentiated from mdx myoblasts, isolated mdx muscle fibers, and intact mdx soleus muscles all exhibit heightened calpain-mediated proteolysis when compared with their WT counterparts (Turner et al., 1993; Alderton and Steinhardt, 2000; McCarter and Steinhardt, 2000; Gailly et al., 2007). Proteolysis in mdx myotubes can be attenuated by experimentally lowering external [Ca2+], treatment with Ca2+ leak channel antagonists, or treatment with calpain inhibitors, but inhibitors of lysosomal and proteosomal pathways have no effect (Alderton and Steinhardt, 2000). Despite the clear role of calpains in establishing the dystrophic phenotype, the relative contributions of the various proteolytic pathways to muscle structure and in vivo progression of DMD, as well as the relative utilities of these pathways as therapeutic targets, are highly controversial (Combaret et al., 1996; Briguet et al., 2008; Selsby et al., 2010, 2012; Childers et al., 2011; Wadosky et al., 2011).
Given the complex regulatory mechanisms that govern calpain activity in vivo, direct evidence implicating specific intrasarcomeric calpain substrates in the pathogenesis and progression of DMD-induced muscle wasting is elusive. One potential intrasarcomeric target of calpain-mediated proteolysis in dystrophic muscle is the tropomodulin (Tmod) family of actin filament pointed-end–capping proteins. Tmods are dynamic caps that regulate actin subunit association and dissociation from pointed ends in a tropomyosin-dependent manner (Weber et al., 1994, 1999), thereby controlling actin filament lengths and stability in a diverse assortment of cell types (Yamashiro et al., 2012). In the case of skeletal muscle sarcomeres, two sarcomeric Tmod isoforms (Tmod1 and Tmod4) localize to either side of the M-line, along the periphery of the H-zone, where they cap the pointed (free) ends of the thin filaments (Fowler et al., 1993; Almenar-Queralt et al., 1999; Gokhin et al., 2010, 2012; Gokhin and Fowler, 2011b). Sarcomeric Tmods maintain thin filament stability and correctly specified thin filament lengths in Drosophila and vertebrate cardiac muscles via their interactions with terminal tropomyosins and their ability to regulate actin subunit exchange at pointed ends (Gregorio et al., 1995; Sussman et al., 1998; Littlefield et al., 2001; Littlefield and Fowler, 2008; Mardahl-Dumesnil and Fowler, 2001; Mudry et al., 2003). In skeletal muscle, sarcomeric Tmods are proposed to fine-tune the lengths of the nebulin-free distal segments of the thin filaments, independent of the lengths of the nebulin-coated proximal segments of the thin filaments (Gokhin and Fowler, 2013). Given Tmods’ sterically accessible location within the sarcomere, as well as the fact that calpains can proteolyze a host of other actin-binding proteins in both muscle and nonmuscle cells (Carafoli and Molinari, 1998; Goll et al., 2003), we hypothesized that sarcomeric Tmods may be calpain substrates in DMD, with proteolysis of Tmods leading to misspecified thin filament lengths in dystrophic skeletal muscle. Such a mechanism would represent a novel and physiologically relevant defect in DMD because myofilament lengths determine the sarcomere length ranges at which muscles can establish actomyosin cross-bridges and produce contractile force (Gokhin and Fowler, 2013).
To directly address the question of whether calpain-mediated proteolysis of Tmods contributes to altered thin filament lengths and disease pathogenesis in DMD, we studied the responses of Tmods to calpain-mediated proteolysis in vitro and in dystrophic muscles in vivo. In this study, we show that both Tmod1 and Tmod4 are equally sensitive to calpain-mediated proteolysis in vitro, but Tmod1 is about an order of magnitude more sensitive to calpain-mediated proteolysis than Tmod4 when associated with myofibrils in situ. In mdx mice, an increase in m-calpain levels in dystrophic soleus muscle is associated with dramatic loss of Tmod1 (but not Tmod4) from the thin filament pointed ends, resulting in ∼11% increase in thin filament lengths as determined by distributed deconvolution (DDecon) analysis of fluorescence images. We also examined Tmod isoform loss in mdx/mTR mice, a more severe mouse model of DMD lacking both dystrophin and the telomerase RNA component, characterized by short telomeres and dramatically impaired regenerative capacity of intramuscular stem cells (Sacco et al., 2010). In mdx/mTR mice, Tmod1 disappears from the thin filament pointed ends in both tibialis anterior (TA) and soleus muscles, whereas Tmod4 additionally disappears from soleus muscle, resulting in thin filament length increases of ∼10 and ∼12% in the TA and soleus, respectively. Of note, comparison of mdx and mdx/mTR soleus muscles showed that graded losses of sarcomeric Tmods do not result in correspondingly graded increases in thin filament lengths, suggesting the existence of a ceiling on maximum thin filament length in vivo. Collectively these results delineate a novel mechanism contributing to skeletal muscle dysfunction in DMD in which calpain-mediated proteolysis of Tmods leads to actin subunit addition at thin filament pointed ends and increased thin filament lengths, and the types of muscles affected by this mechanism depend on the severity of DMD pathology.
RESULTS
Sarcomeric Tmods are proteolytic substrates of m-calpain
Actin cytoskeletal proteins are frequently calpain cleavage substrates under a wide variety of pathological conditions (Carafoli and Molinari, 1998; Goll et al., 2003), but it is unknown whether sarcomeric proteins, such as Tmod1 and Tmod4, are also calpain cleavage substrates. To directly test whether m-calpain can proteolyze Tmod1 and/or Tmod4 in situ, we prepared isolated skeletal myofibrils from WT TA and extensor digitorum longus (EDL) muscles and treated these myofibrils with 40 μg/ml m-calpain activated by millimolar Ca2+. Immunostaining of m-calpain–treated myofibrils revealed disappearance of both Tmod1 and Tmod4 from the pointed ends of phalloidin-stained thin filaments in virtually all myofibrils examined, whereas the striated staining patterns of F-actin and α-actinin were unaffected (Figure 1, A and B). Western blotting confirmed depletion of Tmod1 and Tmod4 from the m-calpain–treated myofibrils, with no change in total actin levels, and Coomassie blue staining revealed no gross changes in myofibrillar protein composition resulting from m-calpain treatment (Figure 1C). Note that Tmod depletion determined by immunofluorescence appeared more striking than Tmod depletion determined by Western blotting, which may be due to either the narrower detection range of immunofluorescence or the inability of immunofluorescence to detect any Tmod protein that may have been extracted or solubilized from the washed myofibrils. We also examined myofibrils treated with either Ca2+ or m-calpain alone, as well as m-calpain in the presence of Ca2+ and a specific inhibitor (calpeptin). As expected, these control treatments had no effect on the localizations or protein levels of Tmod1 and Tmod4 (Figure 1, A–C).
FIGURE 1:

Sarcomeric Tmods show differential sensitivities to m-calpain–induced proteolysis in situ. (A, B) Isolated skeletal myofibrils from TA and EDL muscles from 2-mo-old WT mice were treated with 10 mM Ca2+, 40 μg/ml m-calpain, 40 μg/ml m-calpain activated by 10 mM Ca2+, or 40 μg/ml m-calpain activated by 10 mM Ca2+ in the presence of 10 μg/ml calpeptin. Myofibrils were then immunostained for either (A) Tmod1 or (B) Tmod4, immunostained for α-actinin, and phalloidin-stained for F-actin. Bars, 1 μm. (C) Coomassie blue–stained gel and Western blots of myofibrils treated as indicated, probed using antibodies against m-calpain, Tmod1, Tmod4, and actin. (D) Isolated skeletal myofibrils were treated with increasing concentrations of m-calpain activated by 10 mM Ca2+. Myofibrils were then subjected to Western blotting and probed using antibodies against Tmod1, Tmod4, α-actinin, and actin. (E) Quantification of Western blots for Tmod1 and Tmod4. Error bars reflect mean ± SEM of n = 3 independent experiments.
Next we tested whether Tmod1 and Tmod4 have differential sensitivities to m-calpain–induced proteolysis in situ by treating myofibrils with increasing concentrations of m-calpain and performing Western blots for Tmod1 and Tmod4. Quantitation of these Western blots showed that myofibrillar Tmod1 had ∼10-fold greater sensitivity to m-calpain–induced proteolysis as compared with myofibrillar Tmod4 (Figure 1, D and E). This result contrasts with the similar sensitivities of purified recombinant Tmod1 and Tmod4 to m-calpain–induced proteolysis in vitro (Figure 2).
FIGURE 2:
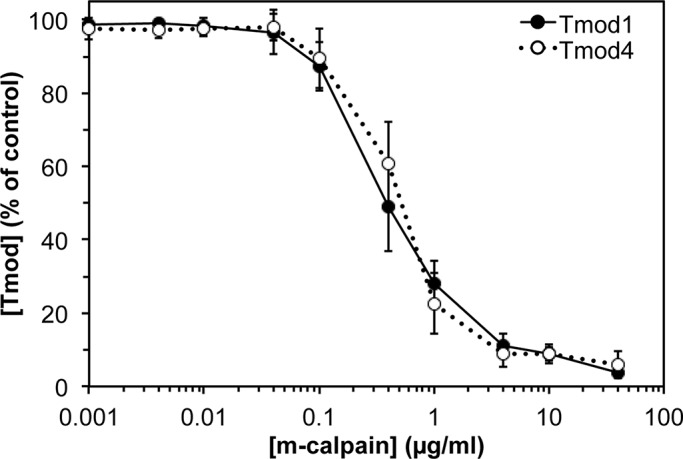
Purified sarcomeric Tmods show identical sensitivities to m-calpain–induced proteolysis in vitro. Constant amounts of recombinant human Tmod1 or mouse Tmod4 were treated with increasing concentrations of m-calpain activated by 10 mM Ca2+ for 1 h at room temperature. Mixtures were then electrophoresed on SDS–PAGE gels, stained with Coomassie blue, and densitometrically quantified. Error bars reflect mean ± SEM of n = 3 independent experiments.
The differential susceptibilities of Tmod1 and Tmod4 to m-calpain–induced proteolysis in situ may be due to different conformations of Tmod1 and Tmod4 at the thin filament pointed ends, leading to differential accessibility of proteolytic epitopes; this may be mediated by sarcomeric Tmods’ differential affinities for striated muscle tropomyosins (Gokhin et al., 2010). Alternatively, the apparent lesser susceptibility of Tmod4 to m-calpain–induced proteolysis may be due to a greater abundance of Tmod4 than Tmod1 in TA and EDL myofibrils. To distinguish between these possibilities, we performed quantitative Western blotting of isolated skeletal muscle myofibrils prepared from TA and EDL muscles and determined the molar ratio of sarcomeric Tmods (Supplemental Figure S1). In three separate experiments, the ratio of Tmod4 to Tmod1 varied from 8.6 to 9.6 (Table 1), indicating that Tmod4 is almost an order of magnitude more abundant than Tmod1 on the thin filament pointed ends. In these experiments, the molar ratio of actin to total sarcomeric Tmods varied from 187 to 208, corresponding to ∼2 Tmods/pointed end (Table 1). This result is somewhat greater than the 1.2–1.6 Tmods/pointed end previously determined via quantitative immunoprecipitation of Tmods from isolated myofibrils using a rabbit polyclonal antibody prepared against Tmod1 (Fowler et al., 1993). This discrepancy is most likely due to the fact that the immunoprecipitations were performed before the identification of Tmod4 (Almenar-Queralt et al., 1999), and the Tmod1 antibody failed to completely immunoprecipitate Tmod4. We conclude that the apparent lesser susceptibility of Tmod4 to m-calpain–induced proteolysis in situ is due to the greater abundance of Tmod4 than Tmod1 in TA and EDL myofibrils. Moreover, these results collectively identify Tmod1 and Tmod4 as novel proteolytic substrates of m-calpain, implicating these Tmods as proteolytic targets of endogenous m-calpain in DMD-induced muscle degeneration.
TABLE 1:
Stoichiometry of sarcomeric Tmods associated with thin filaments in mouse TA and EDL myofibrils.
| Experiment | Tmod1/gel sample (ng/μl)a | Tmod4/gel sample (ng/μl)a | Actin/gel sample (ng/μl)b | Tmod4/Tmod1 (mol/mol) | Actin/sarcomeric Tmods (mol/mol) | Sarcomeric Tmods/pointed endc |
|---|---|---|---|---|---|---|
| 1 | 0.49 ± 0.03 | 4.25 ± 0.35 | 985 ± 45 | 8.6 | 208 | 1.8 |
| 2 | 0.36 ± 0.05 | 3.26 ± 0.22 | 722 ± 15 | 8.8 | 187 | 2.1 |
| 3 | 0.39 ± 0.03 | 3.84 ± 0.43 | 780 ± 68 | 9.6 | 193 | 2.1 |
aAmounts of sarcomeric Tmods were determined by quantitative Western blotting and densitometry of skeletal muscle myofibrils (1–8 μl of gel sample) electrophoresed alongside purified recombinant protein standards (0.25–4 ng) on the same gel. Tmod1 or Tmod4 protein standards were mixed with Tmod1−/− or Tmod4−/− TA muscle lysates, respectively, to equalize the effects of endogenous non-Tmod proteins on the Western transfer efficiencies of endogenous vs. recombinant purified Tmods. SDs reflect three or four different myofibril volumes from different lanes on the same blot (Supplemental Figure S1).
bAmounts of actin were determined by densitometry of Coomassie blue–stained gels containing skeletal muscle myofibrils (1–8 μl of gel sample) electrophoresed alongside rabbit skeletal muscle actin standards (0.25–4 μg) on the same gel. SDs correspond to three or four different myofibril volumes from different lanes on the same gel (Supplemental Figure S1).
cNumbers of Tmods/pointed end were calculated based on thin filament length of 1.11 μm and 13 actin subunits per 37 nm of thin filament (Fowler et al., 1993; Gokhin et al., 2010).
Soleus muscles from mdx mice exhibit proteolysis of Tmod1 and increased thin filament lengths
DMD pathogenesis is characterized by sarcolemmal fragility, membrane rupture during muscle contraction, influx of extracellular Ca2+ into the muscle fiber interior, and hyperactivity of Ca2+-dependent proteases, including calpains (Blake et al., 2002). It was previously shown that leg muscle from the mdx mouse model of DMD exhibits elevated concentrations of m-calpain (Spencer et al., 1995), although a systematic comparison of different muscle types was not performed. Therefore we used Western blots to measure m-calpain levels in TA, EDL, and soleus muscles from WT and mdx mice; these muscles were chosen because they reflect a diversity of muscle fiber recruitment levels and architectures (Burkholder et al., 1994). Results show that mdx soleus muscle, but not mdx TA or EDL muscle, exhibits greater than twofold increase in m-calpain levels as compared with WT muscle (Figure 3, A and B). Thus, elevated m-calpain levels in DMD muscles appear to be primarily characteristic of heavily recruited, slow-twitch muscles such as the soleus.
FIGURE 3:

Elevated levels of m-calpain are associated with decreased levels of sarcomeric Tmods in dystrophic muscles. (A) Western blots of homogenates of TA, EDL, and soleus muscles from 2-mo-old WT and mdx mice were probed using antibodies against m-calpain, Tmod1, and Tmod4. GAPDH was used as a loading control. (B) Quantification of Western blots. Error bars reflect mean ± SEM of n = 4 lanes/genotype within a single blot. (C) Western blots of homogenates of TA and soleus muscles from 2-mo-old WT and mdx/mTR mice were probed using antibodies against m-calpain, Tmod1, and Tmod4. GAPDH was used as a loading control. (D) Quantification of Western blots. Error bars reflect mean ± SEM of n = 3 lanes/genotype within a single blot. (E) RT-PCR of Tmod1 and Tmod4 mRNA transcripts from homogenates of TA, EDL, and soleus muscles from 2-mo-old WT and mdx mice. GAPDH was used as a loading control. (F) Quantification of RT-PCR. Error bars reflect mean ± SEM of n = 3 lanes/genotype within a single gel. *p < 0.05.
To determine whether increased m-calpain levels in mdx soleus muscle are associated with proteolysis of sarcomeric Tmods in vivo, we immunostained longitudinal cryosections of TA and soleus muscles from postnecrotic (2-mo-old) WT and mdx mice for Tmod1 and Tmod4. In WT TA and soleus muscles, both Tmod1 and Tmod4 exhibited their well-established striated staining patterns of periodic doublets, corresponding to the pointed ends of phalloidin-stained thin filaments (Figures 4 and 5). Tmod1 and Tmod4 did not colocalize with m-calpain, which localizes to Z-line-flanking and M-line stripes (Supplemental Figure S2), possibly associated with the sarcoplasmic reticulum (SR; Gokhin and Fowler, 2011a). Of note, Tmod1 did not exhibit a striated staining pattern in mdx soleus muscle, instead showing a diffuse and punctate pattern consistent with proteolytic degradation (Figure 4B). By contrast, Tmod4 staining appeared to be unaffected in mdx soleus muscle, where it showed a pattern of striations at the thin filament pointed ends, indistinguishable from WT soleus muscle (Figure 5B). To confirm these results, we performed Western blots for Tmod1 and Tmod4. In agreement with the immunostaining data, Tmod1 showed ∼40% decrease in protein levels in mdx soleus muscle, but Tmod1 levels were unchanged in mdx TA and EDL muscles (Figure 3, A and B). Similarly, Tmod4 levels were unchanged in mdx TA, EDL, and soleus muscles (Figure 3, A and B). Reverse transcription (RT)-PCR analysis showed identical Tmod1 and Tmod4 mRNA levels in WT and mdx muscles, indicating that reduced Tmod1 protein levels in mdx soleus muscle were not due to reduced transcription of the Tmod1 gene (Figure 3, E and F). We conclude that elevated m-calpain levels in mdx soleus muscle are associated with proteolytic degradation of Tmod1. This finding is consistent with the greater proteolytic sensitivity of myofibrillar Tmod1 as compared with Tmod4 in situ (Figure 1).
FIGURE 4:

Soleus muscle from mdx mice and both TA and soleus muscles from mdx/mTR mice exhibit loss of Tmod1 from thin filament pointed ends. (A, B) Longitudinal cryosections of (A) TA and (B) soleus muscles from 2-mo-old WT, mdx, and mdx/mTR mice were immunostained for Tmod1 and α-actinin and phalloidin-stained for F-actin. P, thin filament pointed ends; Z, Z-line. Bars, 1 μm.
FIGURE 5:

Soleus muscle from mdx/mTR mice exhibits loss of Tmod4 from thin filament pointed ends. (A, B) Longitudinal cryosections of (A) TA and (B) soleus muscles from 2-mo-old WT, mdx, and mdx/mTR mice were immunostained for Tmod4 and α-actinin and phalloidin-stained for F-actin. P, thin filament pointed ends; Z, Z-line. Bars, 1 μm.
Pointed-end capping by Tmod1 restricts actin subunit association and dissociation from the pointed ends of actin filaments (Weber et al., 1994, 1999), and the extent of capping by Tmod1 is inversely related to thin filament length in sarcomeres of Drosophila and vertebrate cardiac muscles (Gregorio et al., 1995; Sussman et al., 1998; Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001; Gokhin and Fowler, 2011b). Therefore we speculated that depletion of Tmod1 from mdx soleus muscle would allow actin subunit addition from the cytoplasmic pool onto the thin filament pointed ends, leading to increased thin filament lengths. To test this, we performed DDecon analysis of line scans from fluorescence images to measure thin filament lengths from the breadth of the phalloidin signal and the distances of sarcomeric Tmods from the Z-line (Littlefield and Fowler, 2002; Gokhin and Fowler, 2013). Consistent with normal Tmod immunostaining and protein levels in mdx TA muscle, thin filament lengths in mdx TA muscle were indistinguishable from those in WT TA muscle (Table 2). On the other hand, thin filament lengths in mdx soleus muscle were ∼12% greater than in WT soleus muscle (1.39 ± 0.10 vs. 1.24 ± 0.06 μm, respectively; Table 2), consistent with reduced Tmod1 levels in mdx soleus muscle. We conclude that calpain-mediated proteolysis of Tmod1 in mdx soleus muscle leads to thin filament elongation from pointed ends and increased thin filament lengths.
TABLE 2:
Thin filament lengths in 2-mo-old mice determined by DDecon analysis of fluorescence images.
| TA | Soleus | ||||||
|---|---|---|---|---|---|---|---|
| WT | mdx | mdx/mTR | WT | mdx | mdx/mTR | ||
| Phalloidin | Mean ± SD (μm) | 1.00 ± 0.04 | 1.01 ± 0.07 | 1.10 ± 0.05* | 1.15 ± 0.03 | 1.28 ± 0.09* | 1.29 ± 0.06* |
| Min-max (μm) | 0.94–1.09 | 0.86–1.11 | 0.98–1.18 | 1.06–1.23 | 1.11–1.47 | 1.14–1.46 | |
| n | 87 | 107 | 67 | 61 | 52 | 50 | |
| Tmod | Mean ± SD (μm) | 1.06 ± 0.04 | 1.08 ± 0.02 | 1.16 ± 0.06* | 1.24 ± 0.06 | 1.39 ± 0.10* | n.d. |
| Min-max (μm) | 0.98–1.13 | 1.03–1.12 | 1.05–1.28 | 1.07–1.34 | 1.15–1.58 | n.d. | |
| n | 68 | 86 | 60 | 54 | 64 | n.d. | |
| Nebulin M1M2M3 | Mean ± SD (μm) | 0.93 ± 0.03 | 0.93 ± 0.05 | 0.94 ± 0.07 | 0.92 ± 0.04 | 0.93 ± 0.04 | 0.94 ± 0.05 |
| Min-max (μm) | 0.88–0.98 | 0.86–1.03 | 0.85–1.06 | 0.82–0.98 | 0.85–1.01 | 0.86–1.05 | |
| n | 35 | 47 | 27 | 36 | 45 | 32 | |
Parameters correspond to the breadth of phalloidin staining and the distances of Tmod and nebulin M1M2M3 from the Z-line. In WT TA, mdx TA, and WT soleus muscles, Tmod lengths were determined from both Tmod1 and Tmod4 immunostaining. In mdx soleus and mdx/mTR TA muscles, Tmod lengths were determined from only Tmod4 immunostaining, due to the absence of striated Tmod1 immunostaining. In mdx/mTR soleus muscle, Tmod lengths were not determined (n.d.) due to the lack of striated Tmod1 and Tmod4 immunostaining. n, number of myofibrils.
*Significant difference when compared with WT (p < 0.01).
TA and soleus muscles from mdx/mTR mice exhibit proteolysis of Tmod1 and Tmod4 and increased thin filament lengths
The usefulness of the mdx mouse as an animal model of DMD is restricted by its mild phenotype and normal lifespan, which are believed to be due to a host of mouse-specific compensatory responses, such as sustained postnatal up-regulation of utrophin (Law et al., 1994; Deconinck et al., 1997; Grady et al., 1997) and robust proliferative capacity of intramuscular stem cells to replace necrotic fibers (Sacco et al., 2010). Hence we examined Tmod isoform loss and measured thin filament lengths in mdx/mTR mice, a more severe animal model that recapitulates human DMD more accurately. In addition to lacking dystrophin, mdx/mTR mice lack the telomerase RNA component, leading to short telomeres and stem cell exhaustion during muscle regeneration (Sacco et al., 2010). Immunostaining of longitudinal cryosections of mdx/mTR TA muscles revealed disappearance of Tmod1 from the thin filament pointed ends, reflecting a more severe phenotype than in mdx TA muscle, where Tmod1 localization was normal (Figure 4A). Loss of Tmod1 from postnecrotic mdx/mTR TA muscle was due to DMD-induced proteolysis, and not absence of Tmod1 during myofibril assembly or muscle development, because TA muscles from prenecrotic (2-wk-old) mdx/mTR mice exhibited normal Tmod1 localization and correspondingly normal thin filament lengths (Supplemental Figure S3 and Supplemental Table S1). Tmod1 was also lost from the thin filament pointed ends in mdx/mTR soleus muscle, as in mdx soleus muscle (Figure 4B). Tmod4 showed normal localization in mdx/mTR TA muscle, similar to mdx TA muscle, although slight attenuation of the Z-line-flanking, SR-associated Tmod4 striations was observed (Figure 5A). By contrast, Tmod4 completely disappeared from the thin filament pointed ends in mdx/mTR soleus muscle, reflecting a more severe phenotype than in mdx soleus muscle, where Tmod4 localization was normal (Figure 5B). Western blots confirmed that decreased protein levels of sarcomeric Tmods were associated with elevated m-calpain levels in TA and soleus muscles from mdx/mTR mice (Figure 3, C and D) to a greater extent than in mdx soleus muscles (Figure 3, A and B). Collectively these observations indicate that mdx/mTR muscles exhibit more severe calpain-mediated proteolysis of sarcomeric Tmods than mdx muscles, consistent with more pronounced DMD disease severity in mdx/mTR mice.
Enhanced proteolysis of sarcomeric Tmod isoforms in mdx/mTR muscles as compared with mdx muscles led us to speculate that mdx/mTR muscles would also exhibit more pronounced increases in thin filament lengths. To test this, we used DDecon analysis to measure thin filament lengths in mdx/mTR muscles. Consistent with proteolysis of Tmod1 in mdx/mTR TA muscle, thin filament lengths were ∼10% longer in mdx/mTR TA muscle than in WT muscle, unlike mdx TA muscle, where Tmod1 was not degraded (Table 2). Of interest, only Tmod1 was proteolyzed in mdx soleus muscle, whereas both Tmod1 and Tmod4 were proteolyzed in mdx/mTR soleus muscle, yet both mdx and mdx/mTR soleus muscles showed nearly identical increases in thin filament length of ∼12% (Table 2). This indicates that disappearance of two sarcomeric Tmod isoforms does not necessarily result in longer thin filament lengths than disappearance of a single sarcomeric Tmod isoform, and thin filament length is not a simple inverse function of pointed-end capping by sarcomeric Tmods in skeletal muscle. Collectively our DDecon measurements show that calpain-mediated proteolysis of sarcomeric Tmods leads to increased thin filament lengths in mdx/mTR TA and soleus muscles.
The N-terminal M1M2M3 domain of nebulin is normally localized in dystrophic skeletal muscle
In skeletal muscle thin filaments, nebulin extends from its C-terminus in the Z-line to its N-terminal M1M2M3 domain at the end of the proximal segment, whereas the distal segment extends beyond the nebulin M1M2M3 domain and is capped by sarcomeric Tmods (Gokhin and Fowler, 2013). It is uncertain whether the increases in thin filament lengths in dystrophic muscle are due to longer distal segments, longer proximal segments, or a combination of both. To distinguish among these possibilities, we immunostained longitudinal cryosections of TA and soleus muscles from WT, mdx, and mdx/mTR mice for the nebulin M1M2M3 domain and measured the distances of the nebulin M1M2M3 domain from the Z-line. In TA and soleus muscles of all three genotypes, the nebulin M1M2M3 domain localized in striations slightly proximal to the pointed ends of the thin filaments (Figure 6), as expected (Castillo et al., 2009; Gokhin et al., 2010, 2012; Gokhin and Fowler, 2013). The nebulin M1M2M3 striations were ∼0.92–0.94 μm from the Z-line in both TA and soleus muscles of all three genotypes, and no significant differences among the genotypes were detected (Table 2). Therefore the lengths of the nebulin-coated proximal segments are unchanged in mdx and mdx/mTR muscles, and the overall increases in thin filament lengths observed in these muscles are driven entirely by increases in the lengths of the nebulin-free, Tmod-capped distal segments.
FIGURE 6:

The N-terminal M1M2M3 domain of nebulin exhibits normal localization in mdx and mdx/mTR muscles. (A, B) Longitudinal cryosections of (A) TA and (B) soleus muscles from 2-mo-old WT, mdx, and mdx/mTR mice were immunostained for nebulin M1M2M3 and α-actinin and phalloidin-stained for F-actin. Note that F-actin extends further beyond nebulin M1M2M3 in soleus muscle than in TA muscle, consistent with longer distal segments in slow muscles and shorter distal segments in fast muscles (Gokhin and Fowler, 2013). P, thin filament pointed ends; Z, Z-line. Bars, 1 μm.
SR-associated actin cytoskeletal proteins are normally localized in dystrophic skeletal muscle
Tmod3, an SR-associated Tmod isoform, has been shown to structurally compensate for the absence of Tmod1 in Tmod1−/− skeletal muscle by dissociating from its SR compartment and relocating to the thin filament pointed ends, thereby maintaining normal thin filament lengths (Gokhin et al., 2010; Gokhin and Fowler, 2011a). To test whether SR-associated Tmod3 may also compensate for depletion of Tmod1 and/or Tmod4 in dystrophic muscles via a similar relocation-based mechanism, we immunostained longitudinal cryosections of TA and soleus muscles from WT, mdx, and mdx/mTR mice for Tmod3. Surprisingly, Tmod3 localized to Z-line-flanking and M-line stripes (Figure 7A) in TA muscles of all three genotypes, indicative of Tmod3’s normal localization in the SR (Gokhin and Fowler, 2011a). In soleus muscles of all three genotypes, Tmod3 also localized to Z-line-flanking stripes, with fainter M-line localization (Figure 7B). Examination of highly stretched myofibrils in all three genotypes did not reveal any Tmod3 associated with the thin filament pointed ends that might have been obscured by the bright Tmod3 fluorescence at the M-line (unpublished data). Thus, in contrast to Tmod1−/− skeletal muscle, Tmod3 fails to cap Tmod1-free thin filament pointed ends and compensate for depletion of sarcomeric Tmods in dystrophic muscle.
FIGURE 7:

Tmod3 exhibits normal, SR-associated localization in mdx and mdx/mTR muscles. (A, B) Longitudinal cryosections of (A) TA and (B) soleus muscles from 2-mo-old WT, mdx, and mdx/mTR mice were immunostained for Tmod3 and α-actinin and phalloidin-stained for F-actin. M, M-line; Z, Z-line. Bars, 1 μm.
Tmod3 caps the pointed ends of cytoplasmic γ-actin (γcyto-actin) filaments and interacts with nonmuscle tropomyosins and small ankyrin 1.5 to mechanically stabilize the SR membrane (Gokhin and Fowler, 2011a). Dystrophic muscles exhibit dramatic up-regulation of γcyto-actin, presumably as a compensatory fortification response to sarcolemmal fragility (Hanft et al., 2006, 2007). On the basis of these observations, we speculated that depletion of sarcomeric Tmods from dystrophic muscle would result in incorporation of excess γcyto-actin into the longer thin filaments, as observed in myofibrils from γcyto-actin–overexpressing muscle (Jaeger et al., 2009). To test this, we immunostained longitudinal cryosections of TA and soleus muscles from WT, mdx, and mdx/mTR mice for γcyto-actin. In TA muscles of all three genotypes, γcyto-actin localized to Z-line-flanking and M-line stripes (Figure 8A), consistent with its previously observed localization in the SR (Gokhin and Fowler, 2011a). In soleus muscles of all three genotypes, γcyto-actin localized to a wide I-band stripe and lacked M-line localization (Figure 8B), revealing the presence of different γcyto-actin architectures in TA versus soleus muscles, which remain unaltered in mdx and mdx/mTR mice. Of note, γcyto-actin staining was markedly brighter in TA and soleus muscles from mdx and mdx/mTR mice (Figure 8), consistent with increased γcyto-actin levels in dystrophic muscles (Hanft et al., 2006, 2007). Both increased γcyto-actin and correspondingly increased Tmod3 levels were confirmed by Western blotting (Supplemental Figure S4). However, γcyto-actin was not enriched at the Z-line or H-zone periphery in mdx or mdx/mTR muscles (Figure 8), indicating no incorporation of γcyto-actin into the barbed or pointed ends of the thin filaments, respectively. We conclude that depletion of sarcomeric Tmods from dystrophic skeletal muscle does not enable incorporation of γcyto-actin into the thin filaments.
FIGURE 8:

γcyto-Actin exhibits normal, SR-associated localization in mdx and mdx/mTR muscles. (A, B) Longitudinal cryosections of (A) TA and (B) soleus muscles from 2-mo-old WT, mdx, and mdx/mTR mice were immunostained for γ-actin and α-actinin and phalloidin-stained for F-actin. M, M-line; Z, Z-line. Bars, 1 μm.
DISCUSSION
We identified calpain-mediated proteolysis of sarcomeric Tmods and resultant elongation of thin filaments from their pointed ends in skeletal muscles from murine models of DMD. Two factors influence this mechanism: 1) disease severity (low vs. high in mdx vs. mdx/mTR muscles, respectively), and 2) muscle recruitment and use (infrequent vs. frequent in TA vs. soleus muscles, respectively). By systematically analyzing Tmod isoform localizations and levels, we showed that mdx soleus and mdx/mTR TA muscles lose only Tmod1 via calpain-mediated proteolysis, whereas mdx/mTR soleus muscle loses both Tmod1 and Tmod4 (summarized in Figure 9). Thus dystrophic muscle provides a useful experimental system in which to study thin filament length regulation by sarcomeric Tmods and suggests that misspecification of thin filament length may contribute to muscle pathology in DMD. Note that the observed relationship between elevated m-calpain levels and Tmod proteolysis is correlative and not necessarily causative. Other proteolytic pathways may also contribute to Tmod proteolysis in DMD.
FIGURE 9:

Model of pointed-end capping by sarcomeric Tmods and thin filament elongation in dystrophic skeletal muscle. TA and soleus muscles differ by their fiber type distributions, recruitment levels, and thin filament (distal segment) lengths. In mdx mice (mild dystrophy), soleus muscle exhibits depletion of Tmod1 and thin filament elongation. In mdx/mTR mice (severe dystrophy), TA muscle exhibits depletion of Tmod1, soleus muscle exhibits depletion of both Tmod1 and Tmod4, and both TA and soleus exhibit thin filament elongation. For simplicity, one actin strand, one nebulin molecule, one tropomyosin polymer, and one Tmod molecule are depicted for each thin filament. Also for simplicity, equal proportions of Tmod1 and Tmod4 are depicted for each WT sarcomere.
Implications for thin filament length regulation in skeletal muscle
In Drosophila and vertebrate cardiac muscles, thin filaments lack nebulin, and thin filament lengths are inversely related to the extent of pointed-end capping by sarcomeric Tmods, with greater capping yielding shorter lengths and reduced capping yielding longer lengths (Gregorio et al., 1995; Sussman et al., 1998; Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001; Gokhin and Fowler, 2011b). By contrast, in skeletal muscle, thin filaments contain nebulin and are divided into two structurally distinct segments: 1) a constant-length, nebulin-coated proximal segment anchored at the Z-line, and 2) a variable-length, nebulin-free distal segment capped by sarcomeric Tmods at the H-zone periphery (Gokhin and Fowler, 2013). This study identifies mdx and mdx/mTR mice as the first in vivo models of Tmod isoform depletion leading to thin filament elongation in skeletal muscle, demonstrating that the lengths of the nebulin-free distal segments of skeletal muscle thin filaments are regulated by sarcomeric Tmods via a similar inverse manner as the nebulin-free thin filaments in Drosophila and vertebrate cardiac muscles. A corollary of this conclusion is that the nebulin-free thin filaments in Drosophila and vertebrate cardiac muscles are effectively “ultralong distal segments” lacking nebulin-coated proximal segments (Gokhin and Fowler, 2013).
Intriguingly, comparison of WT, mdx, and mdx/mTR soleus muscles reveals that graded losses of sarcomeric Tmod isoforms do not result in correspondingly graded increases in thin filament lengths. Namely, mdx soleus (lacking Tmod1) and mdx/mTR soleus (lacking both Tmod1 and Tmod4) have statistically indistinguishable thin filament lengths of 1.28 ± 0.09 and 1.29 ± 0.06 μm, respectively. The additional loss of Tmod4 from mdx/mTR soleus without a further increase in thin filament length, as compared with mdx soleus, where only Tmod1 is lost, also suggests unique functions for Tmod1 and Tmod4 in sarcomeres. Whereas Tmod1 is clearly important for length regulation, Tmod4 appears to be less important and may instead play a role in regulating actomyosin contractility (see later discussion). Previous studies using quantitative immunoprecipitation with pan-Tmod antibodies led to the conclusion that there are 1.2–1.6 Tmods/thin filament in rat psoas muscles (Fowler et al., 1993). However, based on the quantitative Western blotting experiments presented here, Tmod1 is an order of magnitude less abundant than Tmod4 in sarcomeres, and ∼2 Tmods cap each thin filament in mouse skeletal muscles. Nevertheless, loss of Tmod1 in the mdx soleus can still result in an increase in average thin filament length, even when the protein levels and immunostaining pattern of the more abundant Tmod4 isoform remain unchanged. Calculations of sarcomeric Tmod concentrations based on 2 Tmods/pointed end (this study), a mouse skeletal muscle α-actin concentration of 893 μM (Hanft et al., 2006), and subcellular fractionation of mouse skeletal muscle showing ∼50% of Tmod1 and Tmod4 associated with myofibrils (Gokhin and Fowler, 2011a) yield a total sarcomeric Tmod concentration of ∼9.2 μM and a Tmod1 concentration of just 0.92 μM. Therefore, even when ∼50% of the Tmod1 is degraded in the mdx soleus, total Tmod is reduced by just 5%. Thus it is surprising that the more abundant Tmod4 fails to occupy the vacant ends formerly occupied by Tmod1 in the mdx soleus. This is not because the cytosolic concentration of sarcomeric Tmods (∼1.84 μM) is below the Kd of Tmod1 or Tmod4 binding to tropomyosin-coated actin filaments (∼10 pM; Almenar-Queralt et al., 1999; Weber et al., 1999). A consequence of this is that the cytosolic subpopulations of Tmod1 and Tmod4 are unlikely to freely exchange with the thin filament pointed-end populations. We speculate that, instead, thin filament–associated Tmod4 dynamically shuttles among adjacent pointed ends to regulate thin filament lengths within a sarcomere, similar to a proposed mechanism of Tmod1 dynamics in cardiac myocytes (Littlefield et al., 2001). Therefore, when Tmod1 is depleted, a reduced ratio of total Tmod to pointed ends can lead to actin subunit addition and longer thin filaments in skeletal muscle, similar to observations in cardiac and Drosophila muscle (Gregorio et al., 1995; Sussman et al., 1998; Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001; Bai et al., 2007).
Our findings further show that uniformity of thin filament lengths is well regulated in the mdx/mTR soleus, even when both Tmod1 and Tmod4 are depleted and thin filament lengths increase by ∼12%. This implies the existence of additional structural and/or regulatory constraints that restrict uncontrolled elongation of thin filaments from their pointed ends. Two such potential constraints are 1) limited availability of actin monomers in the cytosol (Shimizu and Obinata, 1986) and 2) pointed ends interacting with as-yet-unidentified M-line or H-zone constituents (Gokhin and Fowler, 2013). It is noteworthy that mdx/mTR soleus muscle also exhibits occasional patches of myofibril degeneration and disorganization, and we cannot exclude the possibility that any “overly long” thin filaments that appear in the mdx/mTR soleus may be transient or unstable, subsequently disassembling via ADF/cofilin-mediated actin depolymerization (Nagaoka et al., 1995; Ono and Ono, 2002; Miyauchi-Nomura et al., 2012) or some other disassembly mechanism. Such disassembly would be reminiscent of—but not identical to—observations in cultured cardiomyocytes, in which antibody inhibition of Tmod1/tropomyosin binding leads to thin filament disassembly (Mudry et al., 2003).
In Tmod1−/− mouse skeletal muscle, thin filament lengths are normal because Tmod3 structurally compensates for the absence of Tmod1 by dissociating from its SR-associated compartment and redistributing to the thin filament pointed ends (Gokhin et al., 2010; Gokhin and Fowler, 2011a). However, Tmod3 fails to compensate for depletion of Tmod1 (and Tmod4) in dystrophic muscle. The most likely explanation for this discrepancy is that Tmod3 can only assemble in the complete absence of Tmod1 during muscle development, prior to Tmod3 associating with its binding partners in the SR. It appears that, in Tmod1−/− muscle, absence of Tmod1 eliminates the competition between Tmod1 and Tmod3 for the striated muscle tropomyosins at the pointed ends (Gokhin et al., 2010), thereby enabling Tmod3 to aberrantly assemble onto the thin filament pointed ends. By contrast, in adult mdx and mdx/mTR muscle, Tmod3 is already associated with the pointed ends of γcyto-actin filaments in the SR-associated cytoskeletal network before the onset of muscle damage (Gokhin and Fowler, 2011a). Thus, even as contraction-induced sarcolemmal rupture leads to Ca2+ influx, calpain activation, and proteolytic depletion of sarcomeric Tmods in mdx and mdx/mTR muscles, Tmod3’s interactions with its SR-associated binding partners may render it unable to translocate to the thin filament pointed ends. Indeed, stable association of Tmod3 with the SR is supported by its resistance to proteolysis in mdx and mdx/mTR muscles.
Implications for DMD pathology
Thin filament elongation due to depletion of sarcomeric Tmods in mdx and mdx/mTR muscles indicates that secondary, myofibril-intrinsic changes are likely to contribute to muscle weakness in DMD, occurring in conjunction with the primary changes directly resulting from dystrophin deficiency (i.e., sarcolemmal fragility and impaired lateral force transmission). First, sliding filament theory predicts that thin filament elongation due to depletion of sarcomeric Tmods will result in a widening and rightward shift in the sarcomere length–tension relationship and increased optimum sarcomere length for force production in mdx and mdx/mTR muscles (Granzier et al., 1991; Gokhin and Fowler, 2013). However, physiological experiments have shown no such changes in mdx muscles (Coulton et al., 1988). The most likely explanation for this discrepancy is that the thin filament length changes observed in mdx and mdx/mTR muscles are too small to induce measurable changes in the sarcomere length–tension curve, especially given the experimental variability associated with conventional physiological assays. Indeed, physiological analyses of skeletal muscles from perch fish, nebulin−/− mice, and human nemaline myopathy patients have shown that thin filament length changes of as much as 0.30 μm may be required to significantly alter the sarcomere length–tension curve (Granzier et al., 1991; Gokhin et al., 2009; Ottenheijm et al., 2009, 2010). Such thin filament length changes are markedly greater than the thin filament length changes of ∼0.15 μm observed in this study.
Second, depletion of sarcomeric Tmods may inhibit the formation of productive actomyosin cross-bridges in mdx and mdx/mTR muscles. Skinned fiber mechanics and x-ray diffraction studies of Tmod1−/− muscles reveal reduced tropomyosin strand movement and actomyosin cross-bridge recruitment during thin filament activation (Ochala et al., 2014). Although the biophysical mechanism is not understood, Tmod1’s interactions with F-actin and/or terminal tropomyosins at the pointed end may mediate a long-range conformational change in the thin filament that facilitates cross-bridge recruitment. Tmod4 may also play its own as-yet-undetermined role in regulating thin filament activation and/or cross-bridge recruitment, distinct from Tmod1. Thus we might expect sarcomeric Tmod-depleted fibers from mdx and mdx/mTR muscles to also exhibit attenuated thin filament activation and impaired cross-bridge recruitment. Future studies will be required to explore this prediction experimentally using skinned fiber mechanics with mdx/mTR TA muscles, as soleus muscles are unsuitable for skinned fiber mechanics. However, based on this prediction, it is tempting to speculate that increasing actomyosin contractility via pharmacological interventions (e.g., troponin activators) may ameliorate muscle weakness in DMD. Such a strategy should be approached with caution, because increased actomyosin contractility has the potential to exacerbate sarcolemmal damage in dystrophic muscle, thereby canceling out any potentially beneficial effect of the increased actomyosin contractility. Preclinical studies using dystrophic mouse models are underway in order to resolve this dilemma (Miciak et al., 2013).
Although it has been established that calpain-mediated proteolysis contributes to skeletal muscle degeneration in DMD (Spencer and Tidball, 1992; Spencer et al., 1995; Tidball and Spencer, 2000), identifying the molecular mechanisms of calpain function in vivo has been challenging due to the paucity of confirmed calpain substrates and the widely variable amino acid sequences surrounding calpain cleavage sites (Carafoli and Molinari, 1998; Goll et al., 2003). Calpains generally favor cytoskeletal proteins, including F-actin–associated proteins, such as spectrin (reviewed in Czogalla and Sikorski, 2005), filamin (Wencel-Drake et al., 1991), and cortactin (Perrin et al., 2006). Calpain also proteolyzes utrophin (Courdier-Fruh and Briguet, 2006), a dystrophin homologue and actin-binding protein that compensates for dystrophin deficiency in mdx mouse muscle but not human DMD (Law et al., 1994; Deconinck et al., 1997; Grady et al., 1997; Galkin et al., 2002; Rybakova et al., 2002, 2006). These observations, in conjunction with our findings that m-calpain proteolyzes sarcomeric Tmods in mdx and mdx/mTR muscles, collectively suggest that F-actin remodeling is an important contributor to muscle dysfunction in DMD. Our findings also implicate Tmods as potential calpain cleavage targets in other pathological events characterized by heightened calpain activity, such as traumatic brain injury, stroke, and myocardial infarction (Carafoli and Molinari, 1998; Goll et al., 2003). Future studies will seek to examine such Tmod proteolysis and F-actin remodeling in human patients.
MATERIALS AND METHODS
Experimental animals
WT and mdx mice were obtained from the Jackson Laboratory (Bar Harbor, ME). The mdx/mTR mice have been described previously (Sacco et al., 2010). The mdx/mTR mice were bred for two generations to minimize telomere length while maintaining mouse viability, as described previously (Sacco et al., 2010; Mourkioti et al., 2013). Because DMD is an X-linked disorder, experiments were restricted to male mice. Mice were either 2 wk or 2 mo old at the time of killing, corresponding to time points before or after the onset of muscle necrosis, respectively (Anderson et al., 1988; Coulton et al., 1988). Mice were killed by isoflurane inhalation followed by cervical dislocation. All procedures were performed in accordance with animal care guidelines enforced by the Institutional Animal Care and Use Committee at the Scripps Research Institute.
Antibodies
Primary antibodies and dilutions were as follows: affinity-purified rabbit polyclonal anti-human Tmod1 (R1749bl3c, 3.1 μg/ml for cryosections, 0.31 μg/ml for isolated myofibrils; Gokhin et al., 2010), rabbit polyclonal antiserum to residues 340–359 of a human Tmod1 peptide (PA2211, 1:5000 for Western blots; Gokhin et al., 2012), rabbit polyclonal antiserum to chicken Tmod4 preadsorbed by passage through a Tmod1 Sepharose column (R3577bl3c, 1:25 for cryosections, 1:250 for isolated myofibrils, 1:2500 for Western blots; Gokhin et al., 2010), rabbit polyclonal antiserum to human Tmod3 preadsorbed by passage through a Tmod1 Sepharose column (R5168bl3c, 1:100 for cryosections, 1:1000 for Western blots; Gokhin et al., 2010), affinity-purified rabbit polyclonal anti–nebulin M1M2M3 domain (R1357L, 9.3 μg/ml for cryosections; a gift from Carol C. Gregorio, University of Arizona, Tucson, AZ), affinity-purified rabbit polyclonal anti-γcyto-actin (7577, 0.9 μg/ml for cryosections, 0.09 μg/ml for Western blots; a gift from James M. Ervasti, University of Minnesota, Minneapolis, MN), mouse monoclonal anti–m-calpain (107-82, 1:50 for cryosections, 1:1000 for Western blots; Thermo Fisher Scientific, Waltham, MA), mouse monoclonal anti–α-actinin (EA53, 1:100 for cryosections, 1:1000 for isolated myofibrils, 1:10,000 for Western blots; Sigma-Aldrich, St. Louis, MO), mouse monoclonal anti-actin (C4, 1:10,000 for Western blots; EMD Millipore, Billerica, MA), and mouse monoclonal anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1D4, 1:5000 for Western blots; Novus Biologicals, Littleton, CO). Secondary antibodies and dilutions were as follows: Alexa Fluor 488–conjugated goat anti-rabbit immunoglobulin G (IgG; 1:200 for cryosections, 1:1000 for isolated myofibrils; Life Technologies, Carlsbad, CA), Alexa Fluor 647–conjugated goat anti-mouse IgG (1:200 for cryosections, 1:1000 for isolated myofibrils; Life Technologies), 800CW-conjugated goat anti-rabbit IgG (1:20,000 for Western blots; LI-COR Biosciences, Lincoln, NE), and 680LT-conjugated goat anti-mouse IgG (1:20,000 for Western blots; LI-COR Biosciences).
Preparation and immunostaining of isolated myofibrils
Myofibril preparation procedures were modified from those described previously (Knight and Trinick, 1982; Castillo et al., 2009). Briefly, two TA and two EDL muscles were excised from mouse hindlimbs, stretched, pinned to cork, and relaxed overnight in ice-cold relaxing buffer (100 mM NaCl, 2 mM KCl, 2 mM MgCl2, 6 mM K3PO4, 1 mM ethylene glycol tetraacetic acid [EGTA], 0.1% glucose, pH 7.0) supplemented with protease inhibitor cocktail (1:1000; Life Technologies). TA and EDL tissues were pooled due to their functional similarity and nearly identical fiber type distributions (Burkholder et al., 1994). Muscles were then transferred to ice-cold rigor buffer (100 mM KCl, 10 mM K3PO4, 2 mM MgCl2, 1 mM EGTA, pH 7.0) for 10 min, placed in a conical tube containing 5 ml of rigor buffer and protease inhibitor cocktail (1:1000; Life Technologies), and homogenized with a Tekmar Tissumizer at 70% of maximum speed for 90 s. The homogenate was then centrifuged at 1000 × g for 5 min, the supernatant was decanted, and myofibrils were resuspended in ice-cold rigor buffer. Centrifugation/resuspension was repeated three times to clean the myofibril suspension (∼10:1 buffer:myofibrils after washing), which was then divided into aliquots, to which 10 mM CaCl2 and increasing concentrations of m-calpain (BioVendor, Asheville, NC) ± 10 μg/ml calpeptin (Enzo Life Sciences, Farmingdale, NY) were added. Myofibril suspensions were incubated for 1 h at room temperature, dropped onto poly-l-lysine–coated microscope slides (Polysciences, Warrington, PA), and allowed to adhere for 30 min. Adherent myofibrils were then fixed for 30 min in 4% paraformaldehyde (PFA) in rigor buffer, washed for 5 min in phosphate-buffered saline (PBS) plus 0.1% Triton X-100 (PBST), and then blocked for 1 h in 4% bovine serum albumin (BSA) plus 1% goat serum in PBST at room temperature. Myofibrils were then labeled with primary antibodies diluted in blocking buffer for 1 h at room temperature, washed for 5 min in PBST, and labeled with a fluorophore-conjugated secondary antibody mixture in blocking buffer for 1 h at room temperature. The secondary antibody mixture was supplemented with rhodamine–phalloidin (1:500; Life Technologies) to stain F-actin. Myofibrils were then washed again in PBST, preserved in Gel Mount aqueous mounting medium (Sigma-Aldrich), and coverslipped.
Preparation and immunostaining of muscle tissue cryosections
Mouse hindlimbs were pinned to cork, immersed in ice-cold relaxing buffer, relaxed for 24 h at 4°C, and then fixed in 4% PFA in relaxing buffer for 24 h at 4°C. TA and soleus muscles were then excised, cryoprotected in 30% sucrose in PBS for 4 h, embedded in Tissue-Tek OCT Compound (Sakura Finetek USA, Torrance, CA), and frozen on a metal block chilled in liquid N2. Muscles were divided into 12-μm-thick cryosections, mounted on slides, and stored at −20°C until use. Sections were washed in PBST, permeabilized for 15 min in PBS plus 0.3% Triton X-100, and blocked for 2 h in 4% BSA plus 1% goat serum in PBST. Tissues were labeled with primary antibodies diluted in blocking buffer overnight at 4°C, washed three times in PBST, and then labeled with a fluorophore-conjugated secondary antibody mixture in blocking buffer for 2 h at room temperature. The secondary antibody mixture was supplemented with rhodamine–phalloidin (1:100; Life Technologies) to stain F-actin. Tissues were then washed three times in PBST, preserved in Gel Mount aqueous mounting medium (Sigma-Aldrich), and coverslipped.
Confocal imaging
Images of single optical sections were collected on a Bio-Rad Radiance 2100 laser-scanning confocal microscope (Bio-Rad, Hercules, CA) mounted on a Nikon TE2000-U microscope (Nikon, Melville, NY) using a 100× (1.4 numerical aperture) Plan-Apochromat oil-objective lens at room temperature (zoom = 3). Bio-Rad LaserSharp 2000 software was used during image collection. Images were processed with Photoshop (Adobe, San Jose, CA), and image figures were constructed in Adobe Illustrator. Supplemental Figure S5 shows diagrams of sarcomeres indicating the expected locations of the various fluorescently labeled proteins in Figures 4–8.
Thin filament length measurements
DDecon was used to measure distances of Tmod1 and Tmod4 from the Z-line as well as the breadth of the F-actin (phalloidin) signal across the Z-lines of adjacent half-sarcomeres (I-Z-I arrays; Littlefield and Fowler, 2002). Analysis was performed using a DDecon plug-in originally developed for ImageJ (National Institutes of Health, Bethesda, MD) by Ryan S. Littlefield (University of Washington, Seattle, WA) and modified by Zhen Ren (The Scripps Research Institute, La Jolla, CA). This plug-in is available for public download at http://www.scripps.edu/fowler/. The DDecon plug-in generates the best fit of a model intensity distribution function for a given thin filament component (Tmod or F-actin) to an experimental one-dimensional myofibril fluorescence intensity profile (line scan) of three thin filament arrays obtained for each fluorescent probe (anti-Tmod or phalloidin; Littlefield and Fowler, 2002). Model fitting of background-corrected line scans was optimized by an iterative fitting procedure that minimizes the error between the observed line-scan intensities and the modeled intensities, using a multivariate line-fitting algorithm, as described previously (Littlefield and Fowler, 2002). Distances were calculated by converting pixel sizes into micrometers based on each image's magnification factor (25.6 pixels/μm).
Western blotting
For Western blots of whole-muscle homogenates, TA, EDL, and soleus muscles were dissected in ice-cold PBS and homogenized with a Tekmar Tissumizer in 10 volumes of PBS supplemented with protease inhibitor cocktail (1:1000; Life Technologies). For Western blots of isolated myofibrils, myofibril suspensions were prepared as described under Preparation and immunostaining of isolated myofibrils. Protein standards for SDS–PAGE and Coomassie blue staining to determine actin concentrations in isolated myofibrils were prepared from rabbit skeletal muscle actin (Cytoskeleton, Denver, CO). Protein standards for quantitative Western blots were prepared by spiking increasing volumes of Tmod1−/− (Gokhin et al., 2010) or Tmod4−/− TA muscle homogenates with increasing amounts of recombinant purified human Tmod1 or mouse Tmod4 proteins (Yamashiro et al., 2010), respectively. This approach equalized the effects of endogenous non-Tmod proteins on the Western transfer efficiencies of endogenous versus recombinant purified Tmods. (The Tmod4−/− mouse was made from mouse embryonic stem cells with a lacZ/neo insertion in exon 2 of the Tmod4 gene, and its phenotype will be described in detail elsewhere.)
Muscles, myofibrils, and purified protein preparations were solubilized in an equal volume of 2× SDS sample buffer and boiled for 5 min. Proteins were separated via SDS–PAGE on 4–20% Tris-glycine gradient minigels for 1 h at 200 V and transferred to nitrocellulose (pore size, 0.2 μm), as described previously (Gokhin et al., 2010). Blots were stained with 0.2% Ponceau S in 3% trichloroacetic acid to verify protein transfer, blocked for 2 h in 4% BSA plus 1% goat serum in PBS at room temperature, and then incubated in primary antibodies diluted in Blitz buffer (4% BSA, 10 mM NaHPO4, 150 mM NaCl, 1 mM EDTA, 0.2% Triton X-100, pH 7.4) overnight at 4°C. After washing in PBST, blots were incubated in secondary antibodies diluted in Blitz for 1 h at room temperature. After washing again in PBST, bands were visualized using a LI-COR Odyssey infrared imaging system and densitometrically quantified using ImageJ (with normalization to GAPDH, where appropriate).
RT-PCR
Total RNA was extracted from frozen TA, EDL, and soleus muscles using TRIzol reagent (Qiagen, Valencia, CA) and reverse transcribed using oligo(dT) primers and M-MLV reverse transcriptase (Life Technologies), according to the manufacturer's instructions. Primers were as follows: Tmod1 sense, 5′-CAACGCCATGATGAGCAAC-3′; Tmod1 antisense, 5′-CATCGGTAGAACACGTCCAG-3′; Tmod4 sense, 5′-GATGCGGTAGAGATGGAGATG-3′; Tmod4 antisense, 5′-TCTCTTCTTTTGCTGACGACG-3′; GAPDH sense, 5′-GGGCATCTTGGGCTACACT-3′; and GAPDH antisense, 5′-GAGCAATGCCAGCCCCG-3′. PCR was then performed using a Bio-Rad T100 thermal cycler. An initial hold at 95°C for 1 min was followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 20 s. After a final hold at 72°C for 5 min, reaction products were electrophoresed on agarose gels, stained with EtBr, and visualized using a MultiImage Light Cabinet (Alpha Innotech, San Leandro, CA). Tmod1 and Tmod4 band intensities were densitometrically quantified using ImageJ with normalization to GAPDH.
In vitro proteolysis
Recombinant human Tmod1 and mouse Tmod4 were purified as described previously (Yamashiro et al., 2010). Increasing concentrations of m-calpain were mixed with 3 μg of Tmod1 or Tmod4 in a final volume of 10 μl, and mixtures were then incubated for 1 h at room temperature, solubilized in an equal volume of 2× SDS sample buffer, and boiled for 5 min. Proteins were separated via SDS–PAGE on 4–20% Tris-glycine gradient minigels for 1 h at 200 V. Gels were then stained with Coomassie blue for 10 min, destained overnight, scanned at 600 dpi, and densitometrically quantified using ImageJ.
Statistics
Data are presented as either mean ± SD or mean ± SEM, where appropriate. Differences between two groups were detected using Student's t test. Differences between three groups were detected using one-way analysis of variance with post hoc Fisher's PLSD tests. Statistical significance was defined as p < 0.05. Statistical analysis was performed in Excel (Microsoft, Redmond, WA).
Supplementary Material
Acknowledgments
We gratefully acknowledge Roberta B. Nowak and Christine Vu for preparing purified recombinant Tmods. This work was supported by Development Grant 234106 from the Muscular Dystrophy Association (to D.S.G.), Research Grant 200845 from the Muscular Dystrophy Association (to A.S.), National Institutes of Health Grant R01-HL083464 (to V.M.F), National Institutes of Health Grant P30-AR061303 (to V.M.F. and A.S.), and Sanford-Burnham Medical Research Institute startup funds (to A.S.).
Abbreviations used:
- DDecon
distributed deconvolution
- DMD
Duchenne muscular dystrophy
- EDL
extensor digitorum longus
- γcyto-actin
cytoplasmic γ-actin
- SR
sarcoplasmic reticulum
- TA
tibialis anterior
- Tmod
tropomodulin
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0608) on January 15, 2014.
REFERENCES
- Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- Almenar-Queralt A, Lee A, Conley CA, Ribas de Pouplana L, Fowler VM. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J Biol Chem. 1999;274:28466–28475. doi: 10.1074/jbc.274.40.28466. [DOI] [PubMed] [Google Scholar]
- Anderson JE, Bressler BH, Ovalle WK. Functional regeneration in the hindlimb skeletal muscle of the mdx mouse. J Muscle Res Cell Motil. 1988;9:499–515. doi: 10.1007/BF01738755. [DOI] [PubMed] [Google Scholar]
- Bai J, Hartwig JH, Perrimon N. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev Cell. 2007;13:828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Briguet A, et al. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 2008;22:4190–4200. doi: 10.1096/fj.07-099036. [DOI] [PubMed] [Google Scholar]
- Burkholder TJ, Fingado B, Baron S, Lieber RL. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J Morphol. 1994;221:177–190. doi: 10.1002/jmor.1052210207. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Molinari M. Calpain: a protease in search of a function. Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- Castillo A, Nowak R, Littlefield KP, Fowler VM, Littlefield RS. A nebulin ruler does not dictate thin filament lengths. Biophys J. 2009;96:1856–1865. doi: 10.1016/j.bpj.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers MK, Bogan JR, Bogan DJ, Greiner H, Holder M, Grange RW, Kornegay JN. Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of Duchenne muscular dystrophy. Front Pharmacol. 2011;2:89. doi: 10.3389/fphar.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combaret L, Taillandier D, Voisin L, Samuels SE, Boespflug-Tanguy O, Attaix D. No alteration in gene expression of components of the ubiquitin-proteasome proteolytic pathway in dystrophin-deficient muscles. FEBS Lett. 1996;393:292–296. doi: 10.1016/0014-5793(96)00910-6. [DOI] [PubMed] [Google Scholar]
- Coulton GR, Curtin NA, Morgan JE, Partridge TA. The mdx mouse skeletal muscle myopathy: II. contractile properties. Neuropathol Appl Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- Courdier-Fruh I, Briguet A. Utrophin is a calpain substrate in muscle cells. Muscle Nerve. 2006;33:753–759. doi: 10.1002/mus.20549. [DOI] [PubMed] [Google Scholar]
- Czogalla A, Sikorski AF. Spectrin and calpain: a “target” and a “sniper” in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Fowler VM, Sussmann MA, Miller PG, Flucher BE, Daniels MP. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J Cell Biol. 1993;120:411–420. doi: 10.1083/jcb.120.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly P, De Backer F, Van Schoor M, Gillis JM. In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J Physiol. 2007;582:1261–1275. doi: 10.1113/jphysiol.2007.132191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, VanLoock MS, Rybakova IN, Ervasti JM, Egelman EH. The utrophin actin-binding domain binds F-actin in two different modes: implications for the spectrin superfamily of proteins. J Cell Biol. 2002;157:243–251. doi: 10.1083/jcb.200111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Bang ML, Zhang J, Chen J, Lieber RL. Reduced thin filament length in nebulin-knockout skeletal muscle alters isometric contractile properties. Am J Physiol Cell Physiol. 2009;296:C1123–C1132. doi: 10.1152/ajpcell.00503.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Fowler VM. Cytoplasmic gamma-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. J Cell Biol. 2011a;194:105–120. doi: 10.1083/jcb.201011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Fowler VM. Tropomodulin capping of actin filaments in striated muscle development and physiology. J Biomed Biotechnol. 2011b;2011:103069. doi: 10.1155/2011/103069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Fowler VM. A two-segment model for thin filament architecture in skeletal muscle. Nat Rev Mol Cell Biol. 2013;14:113–119. doi: 10.1038/nrm3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Kim NE, Lewis SA, Hoenecke HR, D'Lima DD, Fowler VM. Thin-filament length correlates with fiber type in human skeletal muscle. Am J Physiol Cell Physiol. 2012;302:C555–C565. doi: 10.1152/ajpcell.00299.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Lewis RA, McKeown CR, Nowak RB, Kim NE, Littlefield RS, Lieber RL, Fowler VM. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J Cell Biol. 2010;189:95–109. doi: 10.1083/jcb.201001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Granzier HL, Akster HA, Ter Keurs HE. Effect of thin filament length on the force-sarcomere length relation of skeletal muscle. Am J Physiol. 1991;260:C1060–1070. doi: 10.1152/ajpcell.1991.260.5.C1060. [DOI] [PubMed] [Google Scholar]
- Gregorio CC, Weber A, Bondad M, Pennise CR, Fowler VM. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature. 1995;377:83–86. doi: 10.1038/377083a0. [DOI] [PubMed] [Google Scholar]
- Hanft LM, Bogan DJ, Mayer U, Kaufman SJ, Kornegay JN, Ervasti JM. Cytoplasmic gamma-actin expression in diverse animal models of muscular dystrophy. Neuromuscul Disord. 2007;17:569–574. doi: 10.1016/j.nmd.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanft LM, Rybakova IN, Patel JR, Rafael-Fortney JA, Ervasti JM. Cytoplasmic gamma-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc Natl Acad Sci USA. 2006;103:5385–5390. doi: 10.1073/pnas.0600980103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger MA, Sonnemann KJ, Fitzsimons DP, Prins KW, Ervasti JM. Context-dependent functional substitution of alpha-skeletal actin by gamma-cytoplasmic actin. FASEB J. 2009;23:2205–2214. doi: 10.1096/fj.09-129783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight PJ, Trinick JA. Preparation of myofibrils. Methods Enzymol. 1982;85(B):9–12. doi: 10.1016/0076-6879(82)85004-0. [DOI] [PubMed] [Google Scholar]
- Law DJ, Allen DL, Tidball JG. Talin, vinculin and DRP (utrophin) concentrations are increased at mdx myotendinous junctions following onset of necrosis. J Cell Sci. 1994;107:1477–1483. doi: 10.1242/jcs.107.6.1477. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Almenar-Queralt A, Fowler VM. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat Cell Biol. 2001;3:544–551. doi: 10.1038/35078517. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Fowler VM. Measurement of thin filament lengths by distributed deconvolution analysis of fluorescence images. Biophys J. 2002;82:2548–2564. doi: 10.1016/S0006-3495(02)75598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield RS, Fowler VM. Thin filament length regulation in striated muscle sarcomeres: pointed-end dynamics go beyond a nebulin ruler. Semin Cell Dev Biol. 2008;19:511–519. doi: 10.1016/j.semcdb.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardahl-Dumesnil M, Fowler VM. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J Cell Biol. 2001;155:1043–1053. doi: 10.1083/jcb.200108026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter GC, Steinhardt RA. Increased activity of calcium leak channels caused by proteolysis near sarcolemmal ruptures. J Membr Biol. 2000;176:169–174. doi: 10.1007/s00232001086. [DOI] [PubMed] [Google Scholar]
- Miciak JJ, Warsing LC, Tibbs ME, Jasper JR, Jampel SB, Malik FI, Tankersley C, Wagner KR. Fast skeletal muscle troponin activator in the dy2J muscular dystrophy model. Muscle Nerve. 2013;48:279–285. doi: 10.1002/mus.23848. [DOI] [PubMed] [Google Scholar]
- Miyauchi-Nomura S, Obinata T, Sato N. Cofilin is required for organization of sarcomeric actin filaments in chicken skeletal muscle cells. Cytoskeleton (Hoboken) 2012;69:290–302. doi: 10.1002/cm.21025. [DOI] [PubMed] [Google Scholar]
- Mourkioti F, et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol. 2013;15:895–904. doi: 10.1038/ncb2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudry RE, Perry CN, Richards M, Fowler VM, Gregorio CC. The interaction of tropomodulin with tropomyosin stabilizes thin filaments in cardiac myocytes. J Cell Biol. 2003;162:1057–1068. doi: 10.1083/jcb.200305031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka R, Kusano K, Abe H, Obinata T. Effects of cofilin on actin filamentous structures in cultured muscle cells. Intracellular regulation of cofilin action. J Cell Sci. 1995;108(Pt 2):581–593. doi: 10.1242/jcs.108.2.581. [DOI] [PubMed] [Google Scholar]
- Ochala J, Gokhin DS, Iwamoto H, Fowler VM. Pointed-end capping by tropomodulin modulates actomyosin crossbridge formation in skeletal muscle fibers. FASEB J. 2014;28:408–415. doi: 10.1096/fj.13-239640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Ono K. Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J Cell Biol. 2002;156:1065–1076. doi: 10.1083/jcb.200110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenheijm CA, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2) J Struct Biol. 2010;170:334–343. doi: 10.1016/j.jsb.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenheijm CA, Witt CC, Stienen GJ, Labeit S, Beggs AH, Granzier H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet. 2009;18:2359–2369. doi: 10.1093/hmg/ddp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–250. doi: 10.1091/mbc.E05-06-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. 2013;201:499–510. doi: 10.1083/jcb.201212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakova IN, Humston JL, Sonnemann KJ, Ervasti JM. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- Rybakova IN, Patel JR, Davies KE, Yurchenco PD, Ervasti JM. Utrophin binds laterally along actin filaments and can couple costameric actin with sarcolemma when overexpressed in dystrophin-deficient muscle. Mol Biol Cell. 2002;13:1512–1521. doi: 10.1091/mbc.01-09-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A, et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010;143:1059–1071. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby J, Morris C, Morris L, Sweeney L. A proteasome inhibitor fails to attenuate dystrophic pathology in mdx mice. PLoS Curr. 2012;4:e4f84a944d8930. doi: 10.1371/4f84a944d8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby J, Pendrak K, Zadel M, Tian Z, Pham J, Carver T, Acosta P, Barton E, Sweeney HL. Leupeptin-based inhibitors do not improve the mdx phenotype. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1192–1201. doi: 10.1152/ajpregu.00586.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Obinata T. Actin concentration and monomer-polymer ratio in developing chicken skeletal muscle. J Biochem. 1986;99:751–759. doi: 10.1093/oxfordjournals.jbchem.a135534. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Tidball JG. Calpain concentration is elevated although net calcium-dependent proteolysis is suppressed in dystrophin-deficient muscle. Exp Cell Res. 1992;203:107–114. doi: 10.1016/0014-4827(92)90045-a. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Baque S, Uhm CS, Daniels MP, Price RL, Simpson D, Terracio L, Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res. 1998;82:94–105. doi: 10.1161/01.res.82.1.94. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Spencer MJ. Calpains and muscular dystrophies. Int J Biochem Cell Biol. 2000;32:1–5. doi: 10.1016/s1357-2725(99)00095-3. [DOI] [PubMed] [Google Scholar]
- Turner PR, Schultz R, Ganguly B, Steinhardt RA. Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle. J Membr Biol. 1993;133:243–251. doi: 10.1007/BF00232023. [DOI] [PubMed] [Google Scholar]
- Wadosky KM, Li L, Rodriguez JE, Min JN, Bogan D, Gonzalez J, Patterson C, Kornegay JN, Willis M. Regulation of the calpain and ubiquitin-proteasome systems in a canine model of muscular dystrophy. Muscle Nerve. 2011;44:553–562. doi: 10.1002/mus.22125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Pennise CR, Babcock GG, Fowler VM. Tropomodulin caps the pointed ends of actin filaments. J Cell Biol. 1994;127:1627–1635. doi: 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Pennise CR, Fowler VM. Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP.P(i)-actin to ADP-actin at all pointed filament ends. J Biol Chem. 1999;274:34637–34645. doi: 10.1074/jbc.274.49.34637. [DOI] [PubMed] [Google Scholar]
- Wencel-Drake JD, Okita JR, Annis DS, Kunicki TJ. Activation of calpain I and hydrolysis of calpain substrates (actin-binding protein, glycoprotein Ib, and talin) are not a function of thrombin-induced platelet aggregation. Arterioscler Thromb. 1991;11:882–891. doi: 10.1161/01.atv.11.4.882. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Gokhin DS, Kimura S, Nowak RB, Fowler VM. Tropomodulins: pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton (Hoboken) 2012;69:337–370. doi: 10.1002/cm.21031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Speicher KD, Speicher DW, Fowler VM. Mammalian tropomodulins nucleate actin polymerization via their actin monomer binding and filament pointed end-capping activities. J Biol Chem. 2010;285:33265–33280. doi: 10.1074/jbc.M110.144873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.