

Polycomb-repressed genes are marked by H3K9me3 and H3K27me3 in pluripotent ES cells, but the effects of this combination are altered by H3S10 phosphorylation in differentiated cells. Acquisition of H3K9me3/S10ph at Polycomb-target genes during differentiation reduces binding of Ezh1 and paused RNA Pol II and affects poising of repressed genes.
Abstract
Methylated histones H3K9 and H3K27 are canonical epigenetic silencing modifications in metazoan organisms, but the relationship between the two modifications has not been well characterized. H3K9me3 coexists with H3K27me3 in pluripotent and differentiated cells. However, we find that the functioning of H3K9me3 is altered by H3S10 phosphorylation in differentiated postmitotic osteoblasts and cycling B cells. Deposition of H3K9me3/S10ph at silent genes is partially mediated by the mitogen- and stress-activated kinases (MSK1/2) and the Aurora B kinase. Acquisition of H3K9me3/S10ph during differentiation correlates with loss of paused S5 phosphorylated RNA polymerase II, which is present on Polycomb-regulated genes in embryonic stem cells. Reduction of the levels of H3K9me3/S10ph by kinase inhibition results in increased binding of RNAPIIS5ph and the H3K27 methyltransferase Ezh1 at silent promoters. Our results provide evidence of a novel developmentally regulated methyl-phospho switch that modulates Polycomb regulation in differentiated cells and stabilizes repressed states.
INTRODUCTION
Differentiation of pluripotent stem cells into specialized lineages requires activation of specific gene expression programs and long-term silencing of genes that specify alternative cell fates. Epigenetic modifications of the core histones form complex combinations on nucleosomes that are believed to reinforce activating and silencing effects of transcription factors and participate in the maintenance of cellular memory of transcription states (Jenuwein and Allis, 2001; Turner, 2002). Methylations of lysines 9 and 27 (K9 and K27) of histone H3 are key modifications that have been associated mainly with transcriptional repression. Trimethyl H3K9 (H3K9me3) is well established as a marker for heterochromatin and acts as a binding site for heterochromatin protein 1 (HP1). Trimethyl H3K27 (H3K27me3) is associated with silencing by the Polycomb group (PcG) proteins. Domains of enrichment for H3K27me3 are widely distributed across developmentally regulated genes in pluripotent embryonic stem cells, and many of these regions coexist with the activating H3K4me3 modification to form bivalent domains, which are believed to be involved in poising genes for activation (Azuara et al., 2006; Bernstein et al., 2006a).
The PcG proteins form a number of different complexes in the nucleus, of which the best characterized are Polycomb repressor complex 1 and 2 (PRC1 and PRC2; reviewed by Sawarkar and Paro, 2010). The PRC2 proteins Ezh2 and Ezh1 are both able to methylate H3K27 to generate H3K27me3 and functionally substitute for one another at Polycomb-regulated genes (Shen et al., 2008; Ezhkova et al., 2011). Ezh2 is highly expressed early in development and in proliferating cells, with expression of Ezh1 increasing at later stages of differentiation. Recent studies also implicate Ezh1 and Ezh2 in gene activation. Ezh1 is required for transcription factor recruitment and transcriptional elongation in a myoblast differentiation model (Mousavi et al., 2011; Stojic et al., 2011), and the S21 phosphorylated (S21ph) form of Ezh2 is implicated in transcriptional activation in prostate cancer cells (Xu et al., 2012).
Components of the PRC1 and PRC2 complexes can bind to H3K27me3. This has the potential to form a forward loop, which would have the effect of amplifying and maintaining Polycomb silencing. A second enzymatic activity, which is associated with the PRC1 complex, is the ubiquitin ligase activity of RING1B. This results in ubiquitination of histone H2AK119, a modification that is associated with Polycomb-mediated gene silencing and has also been linked to the presence of a paused serine 5–phosphorylated RNA polymerase II (RNAPIIS5ph) at bivalently marked genes in embryonic stem (ES) cells (Stock et al., 2007).
Although the interaction of the PcG proteins with H3K27me3 forms a central part of the current paradigm for the mechanism of Polycomb-mediated silencing, there is also evidence of a role for H3K9 methylation in Polycomb binding. Drosophila and mammalian Polycomb proteins have been shown to bind to H3K9me3 (Fischle et al., 2003b; Bernstein et al., 2006b; Margueron et al., 2009; Kaustov et al., 2011). Colocalization of H3K9me3 with H3K27me3 was observed at 48% of Polycomb-binding sites on Drosophila polytene chromosomes, and a H3 peptide containing the K9me3 residue was able to compete efficiently for Polycomb binding at a number of sites (Ringrose et al., 2004). A significant proportion (49%) of bivalent domains in murine ES cells have been shown to be marked by H3K9me3 (Bilodeau et al., 2009). The presence of the H3K9me3 modification at ES cell bivalent domains is dependent on the activity of the histone methyltransferase SetDB1. There is also evidence that the PRC2 protein Suz12 interacts with the H3K9 methyl transferase Suv39h1 and promotes H3K9 methylation in differentiated cells but not in undifferentiated ES cells (de la Cruz et al., 2007).
H3K9me3 and H3K27me3 can also occur in conjunction with the adjacent phosphorylated S10 and S28 residues on the same H3 tail, generating double methyl/phospho modifications. Phosphorylation of H3S10 by the Aurora B kinase, together with trimethylation of H3K9, generates high levels of the H3K9me3/S10ph double modification on heterochromatin during the G2 stage of the cell cycle, which then spreads along the entire chromosome at the onset of mitosis. In vitro and in vivo studies show that the presence of S10ph leads to displacement of HP1β from H3K9me3 during condensation of mitotic chromosomes (Fischle et al., 2005). This observation has formed the basis for the methyl-phospho switch hypothesis, which proposes that serine phosphorylation modulates binding of proteins to adjacent methylated lysine residues on the histone tails (Fischle et al., 2003a). The function of H3S10 phosphorylation seems to be context dependent, as H3S10ph is also associated with transcriptionally active promoters in interphase cells, where it is frequently colocalized with peaks of H3 acetylation. This phosphorylation is mediated by the mitogen-activated protein and mitogen- and stress-activated kinases (MSKs; Edmunds and Mahadevan, 2004).
Methyl-phospho switches on the histone H3 tail are not restricted to mitosis. We previously showed that the H3K9me3/S10ph modification is present on facultative heterochromatin in postmitotic plasma cells and also demonstrated that generation of this binary modification in plasma cells requires the activity of the Aurora B kinase (Sabbattini et al., 2007). HP1β does not bind to facultative heterochromatin in plasma cells, and treatment of the cells with the Aurora B inhibitor hesperadin partially reversed this block. Chromatin immunoprecipitation (ChIP) analysis also showed that the binary H3K9me3/S10ph modification marks silent genes in differentiated mesenchymal stem cells, although the identity of the kinase that generates the phospho component of the double modification in these cells was not determined (Sabbattini et al., 2007). Phosphorylation of H3S28 occurs in conjunction with H3K27 methylation during neural differentiation of human teratocarcinoma cells, where it promotes gene activation by displacement of the Polycomb proteins (Gehani et al., 2010).
Here we investigate the roles of H3 methyl-phospho modifications in developmental gene silencing and show that H3K9me3/S10ph marks Polycomb-regulated genes in conjunction with H3K27me3 in differentiated B cells and mesenchymal cells but not in ES cells. We also show that the presence of the H3K9me3/S10ph mark reduces the level of paused RNAPIIS5ph and Ezh1 at Polycomb-regulated promoters. Our results provide evidence for the occurrence of a methyl-phospho switch at Polycomb-regulated genes during differentiation that fine-tunes silencing and contributes to the establishment of an epigenetic profile that is antagonistic to the presence of paused RNAPII.
RESULTS
To characterize the contributions of H3 methyl-phospho modifications to the epigenetic landscape of differentiated cells, we used three different cell systems. Mouse embryonic stem cells provide a well-defined cell culture model representing the undifferentiated pluripotent cells of the inner cell mass in preimplantation blastocysts. Cultured C3H10T1/2 mesenchymal stem cells were induced to differentiate into postmitotic osteoblasts (see Materials and Methods), and primary resting B cells were isolated from mouse spleen and activated for 3 d with lipopolysaccharide (LPS) to provide an example of a cycling differentiated cell type. The double H3K9me3/S10ph modification was analyzed using a commercially available antibody, and the H3K27me3/S28ph modification was detected using an antibody that was raised by injection of a modified peptide into rabbits (described in Materials and Methods). Characterization of the specificities of both antibodies is detailed in Supplemental Figure S1.
The H3K9me3/S10ph double modification is preferentially enriched on Polycomb-regulated genes
We showed previously that the H3K9me3/S10ph double modification is associated with repressed genes in differentiated C3H10T1/2 cells (Sabbattini et al., 2007). To further characterize the distribution of H3K9me3/S10ph in these cells and establish whether it colocalizes with other silencing marks, we carried out ChIP using an antibody against H3K27me3 and compared it with results obtained for H3K9me3/S10ph and H3K9me3 alone (Sabbattini et al., 2007). The precipitated DNA was hybridized to a NimbleGen tiling microarray covering a 2-Mb gene-dense region of mouse chromosome 3. The probed region contains 68 annotated genes and includes cell-type-specific genes with different tissue specificities interspersed with constitutively expressed housekeeping genes. The pattern of enrichment for H3K9me3/S10ph showed striking colocalization with regions of repressed chromatin that were enriched for H3K27me3 (Figure 1A), suggesting that the H3K9me3/S10ph double modification is associated with gene silencing by the Polycomb proteins. The same regions have very low levels of the single H3K9me3 modification (Figure 1A) and show no enrichment for the H3K27me3/S28ph double modification (Figure 1B). These results confirm the presence of a specific and novel combination of H3K9me3/S10ph and H3K27me3 without accompanying S28 phosphorylation at Polycomb-target genes in differentiated mesenchymal cells. They also strongly suggest that there is cross-talk between H3K9me3 and H3K27me3 and that this interaction could be modulated by S10 phosphorylation.
FIGURE 1:

The H3K9me3/S10ph double modification colocalizes with H3K27me3 at Polycomb-regulated genes in differentiated C3H10T1/2 cells. (A) Tiling microarray analysis spanning 1 Mb of mouse chromosome 3, with repressed genes represented in gray and expressed genes shown in purple. The profile of H3K27me3 in differentiated C3H10T1/2 cells is compared with our previously published data for H3K9me3/S10ph and H3K9me3 (Sabbattini et al., 2007). Domains of enrichment of H3K9m3/S10ph and H3K27me3, corresponding to log2 ratios > 0, are shadowed in yellow. Data are represented as the scaled log2 ratio (pull down/input) of the hybridization signal for the DNA immunoprecipitated with the indicated antibody. (B) ChIP analysis was carried out on chromatin from differentiated C3H10T1/2 mesenchymal cells and resting B cells using anti-H3K27me3/S28ph (black histograms) and anti-H3K9me3/S10ph (purple histograms). Immunoprecipitated chromatin was analyzed with primers for the promoters of a panel of silent and active genes. Differences between the panels of genes selected for analysis for each cell type reflect differences in expression profiles between the two cell types. Bars, mean ± SD. n = 2. (C) Examples of profiles extracted from the top 300 promoter signals for H3K9me3/S10ph obtained using the NimbleGen 24,000 mouse promoter array. The promoters of the Hoxb and Hoxa genes shows peaks of enrichment for both H3K9m3/S10ph and H3K27me3 (red peaks, high probability, false discovery rate [FDR] ≤ 0.05; yellow peaks, intermediate probability, FDR > 0.1 and ≤ 0.2). Peaks were obtained using the NimbleGen default algorithm (www.nimblegen.com/products/lit/nimblescan2.3users-guide.pdf). Gene bodies are represented by colored bars with the 5′ ends shown in white.
Analysis of the link between the H3K9me3/S10ph modification and Polycomb silencing was extended genome wide by hybridizing immunoprecipitated DNA from the differentiated mesenchymal cells to a NimbleGen mouse promoter array that covers 24,000 promoters from the mouse genome. The analysis focused on 1.5-kb regions spanning annotated start sites. Strikingly, of the 300 promoters that showed the highest enrichment for H3K9me3/S10ph (Supplemental Table S1), 84% were also enriched for H3K27me3 and included many promoters that had been previously identified as being Polycomb regulated (Boyer et al., 2006; see examples in Figure 1C). Gene Ontology analysis showed a strong enrichment of H3K9me3/S10ph on the promoters of genes that are involved in transcriptional and developmental regulation (see Supplemental Figure S2 and accompanying discussion).
Multiple kinase are involved in generating the H3K9me3/S10ph modification in differentiated cells
Kinases known to be capable of phosphorylating H3S10 include Aurora B, MSK1 and MSK2, PKA, RSK2, and PIM1. To determine which kinase or kinases generate the H3K9me3/S10ph modification at Polycomb-repressed genes, we treated differentiated C3H cells with a panel of kinase inhibitors. Incubation with H89, which is a potent inhibitor of MSK1/2 (Edmunds and Mahadevan, 2004; Vermeulen et al., 2009), resulted in reductions of around fourfold in enrichment for H3K9me3/S10ph at a number of silent genes (Figure 2A). Because H89 is known to inhibit other kinases in addition to MSK1/2, we isolated primary mouse embryonic fibroblasts (MEFs) from MSK1/2 double-knockout (MSK1/2-dko) mice (Wiggin et al., 2002). MSK1/2-dko and isogenic wild-type (wt) MEFs were induced to differentiate into osteoblasts (Mukherjee et al., 2010), and the levels of H3K9me3/S10ph were analyzed at Polycomb-repressed genes. The analysis of H3K9me3/S10ph in differentiated MSK1/2-dko MEFs showed reduction of ∼25% in the levels of H3K9me3/S10ph with respect to wt MEFs (Figure 2B).
FIGURE 2:
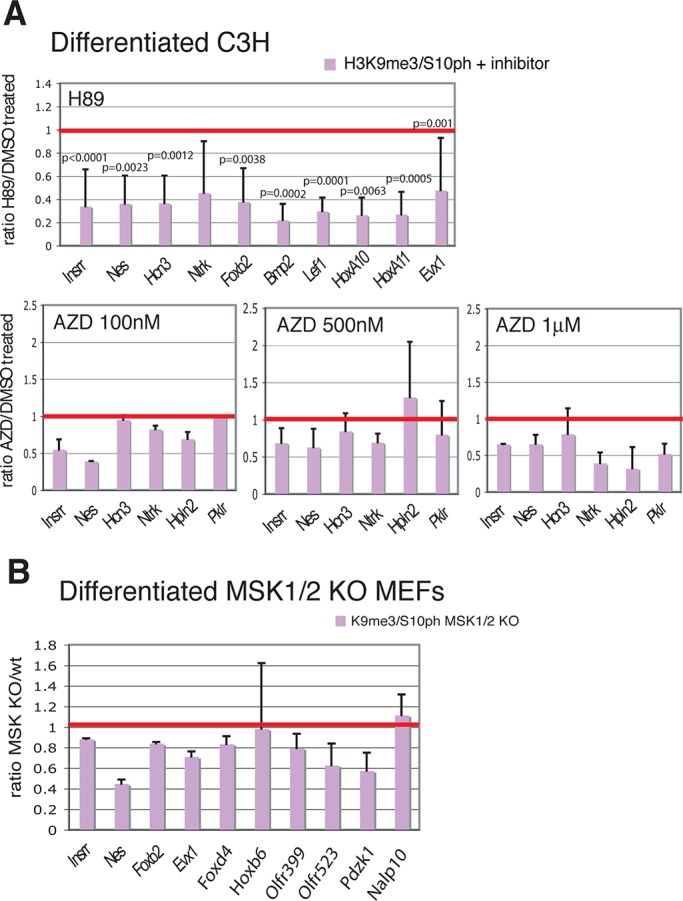
Inhibition of MSK1/2 and Aurora B reduces the levels of H3K9m3/S10ph on repressed genes. (A) ChIP analysis of differentiated C3H cells treated with H89 (top) or AZD1152 (bottom). Results are expressed as ratio of the percentages of input chromatin immunoprecipitated by H3K9m3/S10ph from treated cells and from control cells treated with vehicle. n = 5 biological replicates for the H89-treated cells, n = 2 for cells treated with 100 nM and 1 μM AZD1152, and n = 4 for cells treated with 500 nM AZD1152. The red line corresponds to a ratio of 1, which represents the value for the control cells. Statistical significance of the reductions observed after H89 treatment was assessed by paired t test. (B) Analysis of the levels of H3K9me3/S10ph at selected genes in differentiated MEFs derived from MSK1 and MSK2 double-knockout (MSK1/2 dKO) mice relative to wt cells. n = 2. The silent genes used in this study were selected because they are representative of the following categories: tissue-specific repressed genes located in a gene-dense, 2-Mb region of chromosome 3, chosen as a model of chromosomal organization (Fcrl1, Fcrl5, Insrr, Nes, Hcn3, Ntrk, Hpln2, Bcan, Pklr); genes previously been shown to be silenced by Polycomb complexes (Foxb2, Bmp2, Six1, Lef1, HoxA10, HoxA11; Bernstein et al., 2006b); genes identified by the mouse gene promoter array described in this study in Supplemental Table S1 as having highest H3K9me3/S10ph levels in differentiated C3HT101/2 cells (Foxd4, Evx1, Hoxb6, HoxA10); and repressed genes that are marked by H3K9me3/S10ph and have very low levels of poised RNApolIIS5ph in differentiated C3H101/2 cells (Olfr399, Olfr523, Pdzk1, Nalp10).
The reduction of H3K9me3/S10ph in MSK1/2-dko cells shows that the MSK1/2 kinases are involved in generating the double modification. However, the fact that the effect is smaller than the reduction observed with H89 treatment indicates that other kinases are also involved. Inhibitors of PKA (RpcAMP), RSK2 (BID1870), and PIM1 (Ly294002) were tested singly or in combination, but none of them gave a significant reduction in the level of H3K9me3/S10ph (Supplemental Figure S3). Treatment with AZD1152, which inhibits the Aurora B kinase with high specificity and does not inhibit MSK1/2, did give a reduction of ∼25% (Figure 2A). These results provide evidence that MSK1/2 and Aurora B contribute to generating the H3K9me3/S10ph modification, but they also indicate that other, unidentified kinases can generate the modification and raise the possibility that there is significant redundancy among the kinases that phosphorylate H3S10 at silent promoters.
The H3K9me3/S10ph double modification is absent from Polycomb-regulated genes in pluripotent ES cells
The data obtained with differentiated mesenchymal cells demonstrate that the H3K9me3/S10ph double modification marks Polycomb-regulated genes. To test whether the mark is present in pluripotent stem cells, we measured the distribution of the double modification at silent and active genes in mouse ES cells. Analyzing the H3K9me3/S10ph modification in cycling cells presents a problem because the modification is very highly enriched and widely distributed on mitotic chromosomes. This generates a background that makes it difficult to use whole-genome approaches, including microarray analysis, to identify regions that show localized enrichment for the double modification in interphase. To circumvent this problem, we used centrifugal elutriation to purify G1-enriched cell populations from ES cell cultures. We also used elutriation to purify cycling G1 cells from primary splenic B cell cultures that had been activated by treatment with LPS. Examples of these purifications are shown in Supplemental Figure S4. Staining of unsynchronized cell populations with the anti-H3K9me3/S10ph antibody shows a clear distinction between the relatively low levels of staining observed in G1/S and the strongly stained G2/M cells generated by enrichment of the modification on pericentric heterochromatin and across mitotic chromosomes. The high-staining G2/M cells were almost entirely absent from the elutriated G1-enriched cell populations (Supplemental Figure S4).
The chromatin precipitated from the G1-enriched cells was hybridized to an Agilent oligonucleotide tiling microarray covering the 2-Mb gene-dense region of mouse chromosome 3. ChIP profiles across part of the region that contains both active and silent genes are shown in Figure 3. Inspection of the enrichment profiles shows that domains of H3K27me3 are present in ES cells at the promoters of the neurotrophic tyrosine kinase receptor 1 (Ntrk1) gene, the insulin receptor–related receptor (Insrr) gene, the cellular retinoic acid–binding protein II (Crabp2) gene, and the Nestin gene. Several of these genes (Ntrk, Insrr, and Crabp2) have bivalent histone modification patterns in ES cells and also show evidence of bivalency in B cells. Strong peaks of H3K9me3/S10ph enrichment were found at the H3K27me3-marked genes in activated B cells, confirming the results obtained in the differentiated mesenchymal cells. Re-ChIP analysis confirmed that the colocalizing H3K27me3 and H3K9me3/S10ph modifications in activated B cells are located on the same allele (Supplemental Figure S5). The strong enrichment obtained for H3K9me3/S10ph at H3K27me3-modified genes in B cells contrasts with the profile obtained in ES cells, where little or no enrichment was observed for the H3K9me3/S10ph modification at any of these loci (Figure 3).
FIGURE 3:

H3K9me3/S10ph is absent from Polycomb-regulated genes in pluripotent ES cells and correlates with loss of paused RNAPIIS5ph in activated B cells. Enrichment profiles of part of the 2-Mb, gene-dense region of mouse chromosome 3 in ES cells and activated B cells. The analysis was performed using tiled oligonucleotide microarray chips from Agilent (see the Supplemental Methods for details). Hybridization data were analyzed and visualized using DNA Analytics 4.0.85. For reasons of clarity, the alignment line between the traces was removed. Peaks of enrichment (orange squares) were obtained with the Whitehead per-array neighborhood model with the following settings: custom defined f-value, 0.2; maximum distance for two probes to be considered as neighbor, 1000 base pairs; a probe is considered bound if P(X)bar < 0.05; central probe has P(X) = 0.05; and at least one of the neighboring probe has P(X) = 0.1 or at least one neighbor has P(X) < 0.05. For H3K9me3/S10ph the hybridization was carried out with ChIP DNA from elutriated cells in the G1 phase of the cell cycle. Gene bodies are represented by colored bars with the 5′ ends shown in white. Repressed and active genes are colored gray and purple, respectively. Crabp2 is shown in green to indicate that it is expressed at low levels in ES cells. Blue boxes above the genes represent alternative transcripts. Regions containing repressed genes are indicated by dashed boxes.
ChIP analysis of the H3K27me3/S28ph double modification gave quite different results compared with the H3K9me3/S10ph modification. H3K27me3/S28ph was absent from Polycomb-regulated genes in G1 activated B cells (Supplemental Figure S6A), whereas Polycomb-regulated genes were found to be enriched for both modifications in quiescent resting B cells (Figure 1B). Immunofluorescence (IF) analysis further confirmed the difference in distribution of H3K9me3/S10ph and H3K27me3/S28ph. Strong IF signals were obtained for both double modifications in resting B cells and on metaphase chromosomes in activated B cells, whereas IF detected only the H3K9me3/S10ph modification in G1/S/G2 activated B cells and in postmitotic plasma cells (Supplemental Figure S6B). These different patterns suggest that the H3K9me3/S10ph and H3K27me3/S28ph modifications have different regulatory roles during cell differentiation.
Evidence for a developmentally regulated methyl-phospho switch at Polycomb-regulated genes
Bilodeau et al. (2009) reported that a significant proportion (47%) of bivalent domains in mouse ES cells are also marked by H3K9me3. We compared their data set with the cohort of genes from this study that showed high levels of enrichment for H3K9me3/S10ph and H3K27me3 in differentiated C3H mesenchymal cells. Of the 667 annotated genes described by Bilodeau et al. (2009) as being enriched for both H3K9me3 and H3K27me3 in ES cells, 42% are marked by the H3K9me3/S10ph-H3K27me3 combination in differentiated C3H cells (Figure 4). In contrast, H3K9me3/H3K27me3 without the accompanying H3S10ph modification is present in C3H cells at only 2.6% of the H3K9me3-marked bivalent domains that Bilodeau et al. (2009) identified in ES cells (Figure 4). Comparison of our results with their data set provides evidence that a high proportion of the genes that are marked by H3K27me3 and H3K9me3 in ES cells are subject to additional modification through a methyl-phospho switch in differentiated mesenchymal cells. Such a switch has the potential to affect protein binding to H3K9me3. However, the fact that a significant proportion of genes that lack H3K9me3 in ES cells acquire H3K9me3/S10ph during differentiation indicates that H3K9me3/S10ph is also generated through de novo methylation of H3K9 as differentiation progresses.
FIGURE 4:
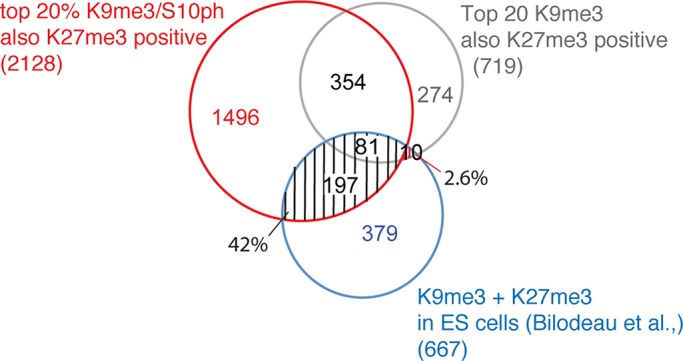
Promoters that are marked by H3K9me3-H3K27me3 in ES cells convert into H3K9me3/S10ph-H3K27me3 in differentiated mesenchymal cells. Among the top 20% promoters enriched for H3K9me3/S10ph in two biological duplicates of differentiated C3H cells, the promoters that were also positive for H3K27me3 were identified and searched for overlap with the promoters identified by Bilodeau et al. (2009) as carrying H3K9me3-H3K27me3 in ES cells. The search was carried out using VENNY (http://bioinfogp.cnb.csic.es/tools/venny/). Of promoters that were identified as being enriched for the H3K9me3-H3K27me3 combination in ES cells, 42% have H3K9me3/S10ph-H3K27me3 in differentiated C3H cells, with only 2.6% maintaining the H3K9me3-H3K27me3 configuration.
Marking by H3K9me3/S10ph is associated with reduced levels of RNAPIIS5Ph at Polycomb-regulated genes in differentiated cells
Stock et al. (2007) described the presence of RNA polymerase II phosphorylated at serine 5 (RNAPIIS5ph) at the promoters and within the coding regions of bivalently marked repressed genes in ES cells. The unusual modification profile and distribution of the RNAPIIS5ph on these genes led to the hypothesis that the polymerase is paused in readiness for transcriptional activation in response to appropriate signals. We compared the distribution of RNAPIIS5ph with enrichment patterns for H3K27me3 and the double H3K9me3/S10ph modification in ES cells, activated B cells, and differentiated mesenchymal cells (Figures 3 and 5A). Genes that are enriched for H3K27me3 in ES cells (Ntrk1, Crabp2, Nestin, Bcan, Hpln2) showed strong positive signals for RNAPIIS5ph close their 5′ ends. In contrast, the same genes, which are marked by the H3K27me3-H3K9me3/S10ph combination in B cells, showed a near absence of RNAPIIS5ph in these cells (Figure 3). Several of the genes that lacked RNAPIIS5ph in B cells were bivalent in these cells (see, e.g., the H3K4me3 and H3K27me3 profiles for Crabp2, Nestin, and Brevican). Analysis using the 8WG16 antibody, which detects hypophosphorylated and intermediately phosphorylated RNAPII (Stock et al., 2007), gave very low signals, indicating a general absence of RNAPII from these genes (Supplemental Figure S7). ChIP analysis of differentiated C3H cells also showed that RNAPIIS5ph was absent from repressed genes that were enriched for H3K27me3 in these cells (Figure 5A). These results show that the presence of the H3K9me3/S10ph double modification correlates with a substantial reduction in the level of the paused RNAPIIS5ph at bivalent and nonbivalent promoters that are marked by H3K27me3 in differentiated cell types.
FIGURE 5:

Reduction of H3K9me3/S10ph is associated with in increased level of paused RNAPIIS5ph at repressed genes in differentiated C3H cells. (A) Chromatin from differentiated C3H cells was precipitated with antibodies that recognize H3K9me3/S10ph, H3K27me3, Ezh1, Ezh2, and RNAPIIS5ph and hybridized to an Agilent tiling oligonucleotide microarray covering the 2-Mb region of mouse chromosome 3 described in Figure 4. Peaks of enrichment are indicated by orange squares. Blue lines are alignment lines. Regions containing repressed genes are indicted by dashed boxes. (B) Effect of H89-induced reduction of H3K9me3/S10ph at repressed genes on binding of RNAPIIS5ph in differentiated C3H mesenchymal cells. The y-axis shows the ratio of enrichment of RNAPIIS5ph at the promoters of the indicated genes in H89- treated cells relative to enrichment cells treated with vehicle (dimethyl sulfoxide). Bars show mean ± SD. n = 5 biological replicates. Statistical significance was assessed by paired t test.
The ability of H89 to substantially reduce the level of K9me3/S10ph allowed us to test whether the presence of the double modification is causally linked to the low levels of the paused RNAPIIS5ph at Polycomb-regulated promoters in differentiated C3H cells. ChIP analysis showed that reduced enrichment for H3K9me3/S10ph after inhibition with H89 is accompanied by a significant increase in the binding of RNAPIIS5ph (Figure 5B). In line with this observation, the decrease in H3K9me3/S10ph observed in MSK1/2 KO MEFs also correlates with increased binding of RNAPIIS5ph on repressed genes (Supplemental Figure S8). These findings provide evidence that the H3K9me3/S10ph modification is involved in displacing poised RNA Pol II from Polycomb-regulated promoters during differentiation.
Binding of Polycomb proteins to H3K9me3 is blocked by H3S10 phosphorylation
Polycomb proteins have been shown to bind to H3K9me3 in addition to their canonical binding to H3K27me3 (Fischle et al., 2003b; Bernstein et al., 2006b; Margueron et al., 2009; Kaustov et al., 2011). It has been suggested that the binding to H3K9me3 synergizes with binding to H3K27me3 to promote Polycomb recruitment to the H3 tail (Ringrose et al., 2004). Our observations raise the possibility that phosphorylation of H3S10 could modulate this synergistic effect. To investigate this possibility, we analyzed binding of Polycomb proteins to H3 peptides containing the H3K9me3 and H3K9me3/S10ph modifications by surface plasmon resonance (SPR). The SPR assay was used to investigate the binding efficiency of recombinant Cbx4, Cbx7, and Cbx8 to H3 peptides containing K9me3, either alone or in conjunction with S10ph. Our results show that phosphorylation of S10 drastically reduced the interaction of the Cbx proteins with the K9me3 (Figure 6A).
FIGURE 6:

Binding of Polycomb proteins to H3K9me3 is affected by the H3S10ph modification. (A) SPR was used to measure binding of purified histidine (His)-tagged Cbx4, 7, and 8 to peptides corresponding to amino acids 1–15-Y of histone H3 containing the K9me3 or the K9me3/S10ph modifications. Peptides were injected over the indicated His-Cbx proteins immobilized on a Biacore flow cell (see Materials and Methods). Red and blue lines indicate binding of K9me3 and K9me3/S10ph peptides, respectively. (B) Binding of Ezh1- and Ezh2-containing PRC2 complexes to histone H3 peptides was assessed by peptide capture. Nuclear extracts from NIH3T3 fibroblasts were used for peptide capture assays as described in the Supplemental Methods. Pull downs were performed with biotinylated peptides derived from the N-terminus of histone H3, corresponding to amino acids 1–20. The peptides were unmodified or contained K9me3 or combined K9me3/S10ph. Captured proteins were identified by Western blot. (C) Reduction of the H3K9me3/S10ph modification by treatment with H89 increases recruitment of Ezh1 to Polycomb-repressed genes but has little effect on Ezh2 binding. Binding of Ezh1 and Ezh2 to promoters of repressed genes was analyzed after treatment with H89 or vehicle (dimethyl sulfoxide). The effect of H89 on the levels of Ezh1 binding across a panel of 13 genes was statistically significant (p = 0.0007, analysis of variance). Bars show mean ± SD. n = 7 biological replicates. No significant effect of H89 treatment was observed on binding of Ezh2 to repressed genes, consistent with its absence from silent genes in these cells.
We also investigated the effect of S10ph on the binding of the PRC2 proteins Ezh1 and Ezh2 to H3K9me3. Plasmon resonance was unsuitable for this purpose, as binding of these proteins to histone H3 outside the context of the PRC2 complex is very inefficient. We therefore used a peptide capture assay to compare binding of Ezh1- and Ezh2-containing PRC2 complexes to H3K9me3. The results show that phosphorylation of H3S10 strongly reduces binding of both proteins to H3K9me3 (Figure 6B). These results provide support for the hypothesis that S10 phosphorylation forms part of a binary switch that modulates binding of Polycomb proteins to chromatin.
Dynamic switches in binding of PRC2 components at silent and active promoters during mesenchymal differentiation
We used quantitative PCR-ChIP and ChIP-on-chip analysis to characterize the in vivo interaction of PRC2 proteins with genes that are marked by the H3K9me3/S10ph-H3K27me3 combination in differentiated C3H mesenchymal cells. The results show that Ezh1 is the major PRC2 H3K27 methyltransferase that is bound to repressed genes in differentiated C3H cells (Figure 5 and Supplemental Figure S9). In contrast, little or no binding of Ezh2 is observed at repressed genes.
To test whether phosphorylation of H3S10 functions in vivo as a modulator of Ezh1 and Ezh2 binding to repressed genes, we treated differentiated C3H cells with H89. We then used ChIP to compare binding of Ezh1 and Ezh2 at selected Polycomb-repressed genes in the H89-treated cells with the binding levels in untreated cells (Figure 6C). Binding of Ezh1 showed a mean increase after H89 treatment that ranged from 1.5- to 3-fold and was statistically significant (analysis of variance, p = 0.0007). Binding of Ezh2 to the repressed genes remained low and was not altered by H89 treatment (Figure 6C). These data led us to conclude that Ezh1 is the major H3K27 methyltransferase that binds to repressed genes in differentiated mesenchymal cells. They also indicate that binding of Ezh1 to Polycomb-repressed genes is reduced by the presence of the S10ph modification in differentiated cells.
DISCUSSION
The Polycomb proteins play key roles in maintaining transcriptional repression at various stages of development and cell differentiation. In the early embryo, this involves poising of genes for activation, whereas the later stages are believed to involve fixing of transcriptional states in differentiated cells. The results described in this article identify significant changes in the epigenetic landscape of Polycomb-regulated genes that occur during differentiation of mammalian cells. In particular, we describe a novel combination of the binary histone H3K9me3/S10ph modification with Polycomb-associated H3K27me3. In differentiated mesenchymal cells and B cells, these three histone modifications combine to mark Polycomb-regulated genes, whereas in pluripotent ES cells, the H3K9me3/S10ph double modification is largely absent from genes that are marked by H3K27me3. It is intriguing that H3S10ph in conjunction with acetyl-H3K9 is associated with gene activation through binding of 14-3-3 proteins (Winter et al., 2008), whereas our results indicate that the same modification in combination with H3K9 methylation acts to reinforce silencing. This finding provides further evidence of a combinatorial histone code in which a given modification can participate in a number of different combinations that give rise to radically different functional outcomes.
Our results show that repressed genes that encode developmental transcriptional regulators are enriched for the H3K9me3/S10ph-H3K27me3 combination in differentiated cells. This observation suggests that acquisition of the H3K9me3/S10ph modification could be involved in fixing cellular identity during the later stages of differentiation. Use of inhibitors and knockout cells indicates that the phosphorylation of H3S10 that generates the H3K9me3/S10ph modification is partly mediated by MSK1/2 and Aurora B. However, our data also suggest that there is a degree of redundancy with other, unidentified kinases also able to generate the double modification in differentiated cells. This raises interesting questions about the nature of the signaling pathways that lead to generation of the double modification and suggests that the presence of the H3K9me3/S10ph modification at repressed genes could represent a shared endpoint for different signaling pathways that are involved in fixation of repressed states in a number of differentiated cell types.
Our results and those of Bilodeau et al. (2009) demonstrate that a proportion of the genes that acquire the double H3K9me3/S10ph modification in differentiated mesenchymal cells are already marked by H3K9me3 in ES cells. This suggests that the double modification arises from a bona fide methyl-phospho switch at these genes during differentiation and cell commitment. However, our data also suggest that de novo methylation of H3K9 and phosphorylation of H3S10 are responsible for generating the H3K9me3/S10ph modification at some Polycomb-regulated genes during development. This is likely to be linked to shifts in gene expression patterns as cells differentiate, an explanation that is supported by the observation that bivalent domains are lost and acquired at developmentally regulated genes during differentiation of neural cells (Mohn et al., 2008).
Our data clearly show that the two double methyl-phospho modifications H3K9me3/S10ph and H3K27me3/S28ph are separately regulated in differentiated C3H cells and cycling activated B cells. The only cells in which we have detected colocalization of the two double modifications are quiescent resting B cells. H3K9me3/S10ph and H3K27me3/S28ph also show different kinetics in mitosis, with H3K9me3/S10ph present during G2, whereas H3K27me3/S28ph is detected only at the onset of chromosome condensation in metaphase. Phosphorylation of H3S28 by MSK1/2 blocks binding of PRC2 proteins to H3K27me3 and has been associated with displacement of Polycomb and accompanying gene activation during ES cell differentiation and in response to stress (Gehani et al., 2010). Our results indicate that acquisition of the H3K9me3/S10ph modification has the opposite effect on gene regulation, as it is associated with loss of gene poising and substantial changes in the binding profile of Polycomb proteins in terminally differentiated cells.
A key feature of the poised state of gene expression observed in ES cells is the presence of the serine 5–phosphorylated form of RNAPII at the 5′ ends of developmentally regulated genes (Stock et al., 2007). We show that reducing the level of H3K9me3/S10ph at repressed genes in differentiated mesenchymal cells by treatment with H89 increases the level of RNAPIIS5ph at these genes. This provides further evidence that the H3K9me3/S10ph modification is associated with the transition from a poised to a fully repressed state.
The mechanism behind the effect of the H3K9me3/S10ph modification on RNAPIIS5ph binding is unclear. However, phosphorylation of H3S10 has been shown to displace binding of a wide range of proteins from H3K9me3 (Fischle et al., 2005; Vermeulen et al., 2010). Our results show that the presence of the double H3K9me3/S10ph modification reduces binding of Ezh1 to repressed genes. In vitro binding studies and in vivo peptide competition studies suggest that Polycomb proteins can bind efficiently to H3K9me3 (Ringrose et al., 2004; Margueron et al., 2009; Xu et al., 2010), suggesting a model in which synergy between H3K9me3 and H3K27me3 enhances the relatively low binding affinity of Polycomb proteins for both modifications (Figure 7). The increase in Ezh1 binding after reduction of the levels of the H3K9me3/S10ph by treatment with the kinase inhibitor H89 supports the idea that part of the function of S10 phosphorylation is to modulate the level of Polycomb binding. Ezh1 has been shown to interact with RNA Pol II at active genes in differentiating myoblasts, raising the possibility that the level of Ezh1 binding modulates the level of paused RNAPIIS5ph at repressed genes in differentiated cells (Figure 7).
FIGURE 7:

Model describing the role of the H3K9me3 and H3K9me3/S10ph modifications in polycomb recruitment and gene silencing. In pluripotent ES cells (top), many bivalent genes are also marked by H3K9me3, which binds PRC2 with higher affinity than H3K27me3. The synergy between H3K9me3 and H3K27me3 enhances polycomb binding. In differentiated cells (bottom), phosphorylation of H3S10 blocks binding of PRC2 to H3K9me3 and reduces overall levels of Polycomb binding to genes that are marked by H3K27me3. This contributes to reducing the levels of paused RNAPIIS5ph at these genes. The presence of the H3K9me3 modification also blocks acetylation of H3K9 and formation of the H3K9ac/S10ph modification, which recruits 14-3-3 proteins to active promoters.
Although an association between reduced binding of a Polycomb protein and more stable silencing might seem counterintuitive, it is becoming increasingly clear that developmental silencing of gene expression by Polycomb proteins is often associated with maintenance of gene plasticity at the pluripotent and multipotent stages of cell differentiation. Our results suggest that changes in histone modifications have a role in modulating Polycomb binding and converting the relatively plastic silencing that is observed early in development into a more stable repressed state in differentiated cells. These observations highlight a growing shift in the Polycomb paradigm from a purely repressive function for Polycomb proteins toward a scenario in which they are highly versatile interactors with the transcriptional machinery that can mediate silencing, gene poising, or transcriptional activation, depending on the context.
MATERIALS AND METHODS
Cell culture
Isolation of resting B cells from mouse spleen and activation with LPS were carried out as previously described (Sabbattini et al., 2007). C3H10T1/2 mesenchymal stem cells were expanded in DMEM medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM GlutaMAX, 2 mM nonessential amino acids, and 2 mM sodium pyruvate. Differentiation into osteoblasts was induced as described in Sabbattini et al. (2007), except that β-glycerol phosphate was omitted from the medium. For kinase inhibition, 500 nM AZD1152 (Astra Zenica, Alderley Park, Macclesfield, Cheshire, UK) and H89 (Sigma-Aldrich, St. Louis, MO) were added to the differentiation medium for 7 h. Different H89 preparations showed variable potency in inhibiting MSK1/2. Therefore several concentrations of inhibitor were tested for each experiment. Concentrations of H89 found to be effective in reducing H9K9me3/S10ph levels were between 3 and 20 μM.
Centrifugal elutriation
G1 cell populations of mouse embryonic stem cells and LPS-activated B cells were obtained by means of centrifugal elutriation in a JE-5.0 elutriation system equipped with a 5-ml Sanderson chamber (356943; Beckman Instruments, Fullerton, CA) and a MasterFlex peristaltic pump (Banfalvi, 2008). A detailed description of the elutriation conditions is given in the Supplemental Methods. G1 cells were resuspended in culture media and processed immediately for ChIP using the antibodies H3K9me3/S10ph (ab5819; Abcam, Cambridge, UK), H3K27me3/S28ph (this article), and H3K27me3 (07-449; Upstate-Millipore, Billerica, MA) as a positive control and anti-rabbit immunoglobulin G (sc2027; Santa Cruz Biotechnology, Santa Cruz, CA) as a negative control.
Chromatin immunoprecipitation
Chromatin immunoprecipitation experiments were performed on formaldehyde-fixed cells as described by Szutorisz et al. (2005) with modifications described in the Supplemental Methods. Antibodies used for ChIP are listed in Supplemental Table S2. The antibody against H3K27me3/S28ph was raised by immunizing rabbits against a synthetic peptide (KAARK(me3)S(ph)APATGG-C). To minimize cross-reactivity with the homologous K9me3/S10ph region, the antibody was immunodepleted by incubation with NeutrAvidin agarose beads (Thermo Scientific, Pierce, Rockford, IL) conjugated to a peptide containing the H3K9me3/S10ph epitope (biotin-ARTKQTARK(me3)S(ph)TGGKAP-RKQL). ChIP for RNAPII and H2Aubq were performed as previously described (Stock et al., 2007).
Microarrays
Microarrays were generated by tiling oligonucleotides of 50 bases (NimbleGen) or 60 bases (Agilent Technologies, Santa Clara, CA) with 100-base resolution. ChIP and input DNA samples were amplified using GenomePlex Whole Genome Amplification system (Sigma). Microarray analysis using NimbleGen microarrays was performed as described in Sabbattini et al. (2007). The NimbleGen promoter microarray covered >24,000 mouse promoters, spanning from −1000 to +500 base pairs with respect to the transcription start site. DNA labeling with Cy3 or Cy5 and hybridization to the microarrays was carried out by (Nimblegen, Madison, WI). For the Agilent microarrays, 3 μg of whole-genome amplified material was labeled with Cy3 (input) and Cy5 (IP DNA) by random priming. Two biological replicate ChIP/microarrays were performed for each antibody for each cell type. Full details of the hybridization protocols for the Agilent arrays and data analysis for both NimbleGen and Agilent arrays can be found in the Supplemental Methods.
Bioinformatic analysis
Functional annotation clustering of the promoter microarray data was carried using the DAVID Gene Ontology tool (Huang et al., 2009).
Immunofluorescence microscopy
Immunofluorescence analysis was carried out as previously described (Sabbattini et al., 2007). Secondary antibodies used for fluorescence detection were Alexa 488–anti-rabbit and Alexa 488–anti-rat (both from Molecular Probes, Eugene, OR). Images were collected by confocal microscopy using a SP5 microscope (Leica Microsystems, Wetzlar, Germany) and LAS-AF software.
Animal procedures
Animals used in the study were maintained and handled according to the guidelines of the Imperial College Subcommittee for Animal Research and the regulations set out by the British Home Office.
Supplementary Material
Acknowledgments
We thank Francisco Ramirez for assistance with the statistical analysis and Marion Leleu for assistance with bioinformatics. This research was funded by the Medical Research Council UK. S.N. was funded by an EU Marie Curie Research Training Fellowship (HPRN-CT 00504228). A.F. was funded by a Gordon Piller Research Studentship from Leukaemia and Lymphoma Research.
Abbreviations used:
- ANOVA
analysis of variance
- C3H
C3H10T1/2 cell line
- ChIP
chromatin immunoprecipitation
- H3K9me3
trimethyl lysine 9 of histone H3
- H3K27me3
trimethyl lysine 27 of histone H3
- H3S10ph
phospho serine 10 of histone H3
- LPS
lipopolysaccaride
- MEFS
mouse embryonic fibroblasts
- MSK1/2-dKO
MSK1 and 2 double-knockout
- PcG
Polycomb group proteins
- PRC1
Polycomb repressor complex 1
- PRC2
Polycomb repressor complex 2
- RNAPIIS5ph
RNA polymerase II phosphorylated at serine 5
- SPR
surface plasmon resonance
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0628) on January 15, 2014.
*These authors contributed equally to this study.
The authors declare that they have no conflict of interest.
REFERENCES
- Azuara V, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- Banfalvi G. Cell cycle synchronization of animal cells and nuclei by centrifugal elutriation. Nat Protoc. 2008;3:663–673. doi: 10.1038/nprot.2008.34. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006a;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse Polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006b;26:2560–2569. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–2489. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- de la Cruz CC, Kirmizis A, Simon MD, Isono K-I, Koseki H, Panning B. The polycomb group protein SUZ12 regulates histone H3 lysine 9 methylation and HP1 alpha distribution. Chromosome Res. 2007;15:299–314. doi: 10.1007/s10577-007-1126-1. [DOI] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J Cell Sci. 2004;117:3715–3723. doi: 10.1242/jcs.01346. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Lien W-H, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011;25:485–498. doi: 10.1101/gad.2019811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003a;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003b;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehani SS, Agrawal-Singh S, Dietrich N, Christophersen NS, Helin K, Hansen K. Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol Cell. 2010;39:886–900. doi: 10.1016/j.molcel.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis C. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kaustov L, et al. Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem. 2011;286:521–529. doi: 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Wang AH, Sartorelli V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol Cell. 2011;45:255–262. doi: 10.1016/j.molcel.2011.11.019. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Wilson EM, Rotwein P. Selective signaling by Akt2 promotes bone morphogenetic protein 2-mediated osteoblast differentiation. Mol Cell Biol. 2010;30:1018–1027. doi: 10.1128/MCB.01401-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L, Ehret H, Paro R. Distinct contributions of histone H3 lysine 9 and 27 methylation to locus-specific stability of polycomb complexes. Mol Cell. 2004;16:641–653. doi: 10.1016/j.molcel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Sabbattini P, Canzonetta C, Sjoberg M, Nikic S, Georgiou A, Kemball-Cook G, Auner HW, Dillon N. A novel role for the Aurora B kinase in epigenetic marking of silent chromatin in differentiated postmitotic cells. EMBO J. 2007;26:4657–4669. doi: 10.1038/sj.emboj.7601875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawarkar R, Paro R. Interpretation of developmental signaling at chromatin: the polycomb perspective. Dev Cell. 2010;19:651–661. doi: 10.1016/j.devcel.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, Pombo A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428–1435. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- Stojic L, et al. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin. 2011;4:16. doi: 10.1186/1756-8935-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szutorisz H, Canzonetta C, Georgiou A, Chow CM, Tora L, Dillon N. Formation of an active tissue-specific chromatin domain initiated by epigenetic marking at the embryonic stem cell stage. Mol Cell Biol. 2005;25:1804–1820. doi: 10.1128/MCB.25.5.1804-1820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Berghe WV, Beck IME, De Bosscher K, Haegeman G. The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci. 2009;34:311–318. doi: 10.1016/j.tibs.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JSC. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S, Simboeck E, Fischle W, Zupkovitz G, Dohnal I, Mechtler K, Ammerer G, Seiser C. 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J. 2008;27:88–99. doi: 10.1038/sj.emboj.7601954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci USA. 2010;107:19266–19271. doi: 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.