

It has been observed that embryonic stem cells have the potential to provide unlimited cells and tissues for regenerative medicine. This review suggests potential advantages and limitations of embryonic stem cells derived from parthenogenetic embryos for cell-based therapies.
Keywords: Pluripotent stem cells, Parthenogenesis, Histocompatibility, Imprinting
Abstract
Embryonic stem cells (ESCs) have the potential to provide unlimited cells and tissues for regenerative medicine. ESCs derived from fertilized embryos, however, will most likely be rejected by a patient’s immune system unless appropriately immunomatched. Pluripotent stem cells (PSCs) genetically identical to a patient can now be established by reprogramming of somatic cells. However, practical applications of PSCs for personalized therapies are projected to be unfeasible because of the enormous cost and time required to produce clinical-grade cells for each patient. ESCs derived from parthenogenetic embryos (pESCs) that are homozygous for human leukocyte antigens may serve as an attractive alternative for immunomatched therapies for a large population of patients. In this study, we describe the biology and genetic nature of mammalian parthenogenesis and review potential advantages and limitations of pESCs for cell-based therapies.
Introduction
The potential of pluripotent stem cells (PSCs) for regenerative medicine is virtually unlimited because of their capacity for extensive propagation in an undifferentiated state and their ability to develop into any cell type of the body. During in vitro culture, PSCs rapidly proliferate and self-renew, allowing the generation of sufficient quantities of vital cells that could one day provide treatments for many incurable diseases. Parenthetically, although ongoing clinical trials in the U.S. involve predominately somatic or adult origin stem cells, testing the safety and therapeutic efficacy of PSC-derived cells has begun. Food and Drug Administration-approved clinical trials using embryonic stem cells (ESCs) derived from in vitro fertilized (IVF) embryos currently involve the treatment of macular degeneration [1, 2]. Because ESCs are genetically divergent and most likely different from potential patients, their current uses are restricted to immune privileged areas such as the central nervous system or the eye to minimize rejection of transplanted cells [3]. In addition to ESCs, there are three other PSC types that may provide histocompatible cells for regenerative medicine: ESCs derived by somatic cell nuclear transfer (SCNT), induced pluripotent stem cells (iPSCs), and ESCs derived by parthenogenesis (pESCs). Extensive research in recent years has addressed the biological and therapeutic properties of ESCs and iPSCs with details of their derivation and pluripotent characteristics well covered in numerous reviews [4–7]. Human ESCs produced by SCNT were also recently described, and research defining their genetic, epigenetic, and transcriptional properties is ongoing [8]. Moreover, iPSCs may soon enter the clinical trial arena in Japan [9]. Despite the enormous incentive to find the best PSC type for clinical applications, pESCs derived from parthenogenetic embryos have been largely ignored and relatively little information is available concerning the origin as well as the genetic and epigenetic makeup of this class of PSCs. The aim of this review is to revisit mammalian parthenogenesis, describe the unique genetic and epigenetic features of pESCs, and highlight advantages and disadvantages of pESCs for regenerative medicine.
Mammalian Parthenogenesis: The Understudied Phenomenon
Parthenogenesis (Greek for “virgin birth”) is a form of asexual, uniparental (maternal) reproduction, characteristic of some jawed vertebrate species but not of mammals [10]. Some notable examples include the all-female, obligate parthenogenetic whiptail lizard, Aspidoscelis uniparens [11], and the virgin birth of a hammerhead shark in captivity [10]. Despite the absence of complete parthenogenesis in mammals, in rare but natural instances, or by deliberate manipulation, early parthenote development has been observed.
Normally during maturation, preovulatory mammalian oocytes will reach and remain arrested at metaphase of meiosis II (MII) and subsequent sperm penetration triggers the resumption of meiosis in the oviduct. High levels of maturation-promoting factor (MPF) and a cytostatic factor (CSF) maintain this M-phase arrest. Sperm-induced activation of the MII oocyte is effected through phospholipase C (PLC-ζ) and an increase in intracellular calcium levels [12]. Depending on the species, cytosolic calcium release and reuptake fluctuations will oscillate at a specific amplitude, frequency, and length of time as long as PLC-ζ is present. Calcium/Calmodulin-dependent protein kinase II can decode oscillating calcium signals and activate downstream mechanisms that inhibit MPF and CSF [13]. These events lead to the resumption of meiosis and the segregation of a set of sister chromatids into a second polar body, rendering the oocyte haploid [14] (Fig. 1A). Normal fertilization is completed when sperm and oocyte haploid chromosomes finally join to form a diploid zygote. Unfertilized MII oocytes from certain mammals are sensitive to mechanical or physical stimuli, whereby simple pressure, osmolarity flux, or temperature change can cause spontaneous activation, resulting in meiotic resumption and parthenogenetic development [15, 16].
Figure 1.

Meiosis and zygosity outcomes during parthenogenesis. (A): Normal fertilization with sperm. During the prophase I of meiosis I, the two parental chromosomes (depicted as white and black), each containing sister chromatids, recombine and exchange regions through chromosomal crossover. Meiosis I is resolved by extrusion of one homologous parental chromosome into the first polar body. The remaining homologous chromosome enters into meiosis II but remains arrested at metaphase II until fertilized by sperm. Meiotic progression resumes after fertilization. Sister chromatids segregate, and one chromatid is eliminated in the second PB. The sperm provides the second homologous chromosome to the diploid zygote. (B): Heterozygous parthenogenesis without completion of meiosis. After artificial activation that blocks second PB extrusion, sister chromatids segregate during anaphase II; however, both chromatids are retained within the oocyte, forming a diploid parthenogenetic zygote. Because of earlier meiotic recombination and crossover with the other homologous parental chromosomes, the resulting diploid parthenote exhibits high levels of heterozygosity. (C): Homozygous parthenogenesis after completion of meiosis. Artificial activation methods may not interfere with completion of meiosis and segregation of the second PB. However, the initial haploid genome replicates during mitotic S-phase without undergoing subsequent cell division. Both sister chromatids are retained as a homologous pair resulting in a diploid but homozygous parthenote. (D): Haploid parthenogenesis after completion of meiosis. Parthenogenetic activation renders a haploid genome that is maintained throughout subsequent mitotic divisions. Abbreviations: MII, metaphase II; PB, polar body; PI, prophase I of meiosis I.
Oocyte activation is of relevance to cloning by SCNT, a procedure in which the MII oocyte’s chromosomes are removed mechanically and replaced by a diploid somatic cell nucleus. Because SCNT embryos do not require fertilization by sperm, an artificial means of activation is crucial to release the oocyte cytoplasm from meiotic arrest and initiate cell division. Sophisticated methods of inducing artificial oocyte activation are vital for SCNT research [8] and have consequently yielded successful protocols for artificial parthenogenetic activation that mimic natural, sperm-induced activation and result in efficient preimplantation development [17–19]. To experimentally imitate intracellular calcium oscillations, oocytes are electroporated in calcium-containing medium or exposed to calcium ionophore or ionomycin. Such calcium treatments induce an initial decline in meiotic kinase activities; however, MPF can quickly recover, causing the oocyte to enter another meiotic arrest known as metaphase III [20]. Therefore, to maintain inactivation of meiotic kinases, protocols require additional treatments with either broad protein synthesis or kinase inhibitors such as cycloheximide, 6-dimethylaminopurine (6-DMAP), or roscovitine [18].
During artificial activation of intact MII oocytes, exposure to broad protein synthesis inhibitors, such as 6-DMAP, interferes with the segregation of meiotic chromosomes by blocking cytokinesis and extrusion of the second polar body. As a result, the activated oocyte retains the genetic material of the second polar body, forming a pseudodiploid parthenogenetic embryo. In this scenario, the sister chromatids segregate but remain within the oocyte as a pair of homologous chromosomes (Fig. 1B). Such diploid oocytes then enter mitotic divisions and develop into blastocysts at rates similar to their fertilized counterparts [18]. Initially it was anticipated that such diploid parthenogenetic embryos would exhibit high levels of genomic homozygosity because each pair of homologous chromosomes are, in fact, sister chromatids. However, because of meiotic recombination between parental homologous chromosomes during prophase I, sister chromatids can carry different alleles. Indeed genetic analysis of single-nucleotide polymorphisms (SNPs) and microsatellite loci in pESCs reveals that the frequency of such allelic exchange in the female germline is remarkably high. Heterozygosity levels in nonhuman primate pESCs may reach 64% [17]. The remaining homozygous loci are primarily located in heterochromatic pericentromeric and telomeric regions that experience lower chances of crossover [21, 22]. Thus, parthenogenetic activation that precludes second polar body extrusion results in the development of diploid, heterozygous embryos and pESCs.
Alternatively, activation treatments may not interfere with the completion of meiosis and separation of the second polar body. In the mouse, such embryos can also undergo preimplantation development and support the derivation of haploid pESCs [23, 24] (Fig. 1C). Haploid pESCs can be sorted and maintained for many passages and be used for forward and reverse genetic screening [23, 25] or as a model for the study of germ cells. Surprisingly, epigenetically distinct, haploid androgenetic pESCs (established by removing an oocyte’s MII chromosome and then inserting a single sperm) can be injected into and “fertilize” a MII oocyte to generate genetically modified mice [26, 27].
More often, initially haploid parthenogenetic zygotes develop into diploid embryos in which diploid pESCs can later be derived. Although the mechanism remains largely unknown, there is a point during early mitotic divisions in which the haploid genome duplicates without undergoing cell division in a process referred to as endoreplication [25] or endoreduplication [28] (Fig. 1D). In this scenario the resulting diploid parthenogenetic embryo exhibits a completely homozygous genome [29]. In some cases during natural conception or in vitro fertilization, sperm activates a MII oocyte but fails to contribute genetic material. Such oocytes undergo normal completion of meiosis and form a haploid zygote that subsequently develops into a diploid parthenogenetic embryo [29, 30].
A simplified diagram demonstrating these different genetic outcomes of parthenogenesis is depicted in Figure 1.
Developmental Potential of Parthenogenetic Embryos and pESCs
Parthenotes from most mammalian species are capable of developing into blastocysts at rates comparable to fertilized embryos. Following transfer into recipients, mouse parthenotes can also implant and reach day E9.5 of development, the forelimb stage [31–34]. However, parthenogenetic fetuses cease development because of abnormal placental formation and other anomalies [35]. To further evaluate the developmental capacity of parthenogenetic cells, normal mouse embryos have been aggregated with either parthenogenetic embryos or cultured pESCs to generate chimeras.
When parthenogenetic embryos were combined with normal embryos, the resultant chimeras exhibited fetal growth abnormalities and a selection against parthenogenetic cells in certain tissues, notably in the mesoderm and endoderm lineages, with a particular loss in skeletal muscle [36, 37]. In earlier studies, pESCs contributed to developmentally normal, germline chimeras with low distribution in the skeletal muscle, testes, and hypothalamus but high presence in the cortex, striatum, and hippocampus of the brain [36, 38]. However, recent improvements in ESC derivation, culture, and chimera assays have allowed the production of developmentally normal pESC chimeras displaying an even distribution of pESC progeny throughout the body [25, 28, 39]. Notably, maintaining chimera-competent mouse ESCs and pESCs can be significantly aided by culture with mitogen-activated kinase and glycogen synthase kinase 3 inhibitors [28, 39].
Moreover, an entirely pESC-derived live pup has been born through tetraploid embryo complementation [39], a chimera technique in which a host tetraploid blastocyst only supplies the extraembryonic tissues. In such chimeras, pESCs are forced to form an entire body of offspring. Tetraploid complementation is considered the most stringent test for pluripotency as it demonstrates the ability of pESCs alone to produce an entire mouse [40].
In humans, a clinical case of a male child carrying parthenogenetic cells has been reported [41]. The patient was diagnosed with mild developmental abnormalities, hemifacial microsomy, and signs of sex reversal. Genetic analysis detected chimerism in skin fibroblasts and peripheral blood leukocytes consisting of normal biparental (46,XY) and parthenogenetic (46,XX) cells. The possible origin of such chimerism is unclear but could result from incomplete meiosis after fertilization and retention of the genetic material from the second polar body. In this scenario, a zygote may form three pronuclei. During the first mitotic division, a male and one female pronucleus partition into one blastomere, whereas the second female pronucleus segregates into the other blastomere. The diploid blastomere gives rise to normal biparental (46,XY) cells, whereas the haploid blastomere undergoes endoreduplication during early cleavage to produce diploid parthenogenetic (46,XX) cells. It is likely that the presence of parthenogenetic cells was responsible for the growth abnormalities observed in the patient. More often cases of human parthenogenesis are uncovered when spontaneously activated oocytes give rise to benign ovarian teratoma tumors consisting of disorganized tissues from the three germ layers [42, 43]. In a few rare cases, there has been documentation of fetiform ovarian teratomas (homunculus) of parthenogenetic origin [44–46], with one report describing a highly differentiated solid mass that resembled a “doll-like” structure [47].
Imprinting and Parthenogenesis
As described above, mammalian parthenogenetic embryos do not support full-term development, leading to the pivotal question of why they are developmentally incompetent whereas other female vertebrates produce healthy parthenogenetic offspring. The general consensus implicates mammalian-specific, genomic imprinting: the deviation from Mendelian inheritance in which parent-of-origin epigenetic marks result in strictly paternal or maternal expression of some genes [48, 49].
Since the early discovery of a few imprinted genes in the mouse [50–53], more than 100 imprinted genes have been revealed (see Geneimprint [http://www.geneimprint.org] and MouseBook [http://www.mousebook.org]) [54]. Because parthenotes lack sperm alleles, paternally expressed imprinted genes are functionally absent. Conversely, maternally expressed imprinted genes are transcribed from both alleles, leading to overexpression and associated developmental abnormalities. Because many imprinted genes are directly involved in fetal growth pathways, the genome-wide dosage imbalance serves as a barrier to normal fetal development in parthenotes [48]. Studies have determined that monoallelic gene expression from the paternal genome is critical for extraembryonic tissue formation and function [32, 34]. For example, paternally expressed Igf2 and its maternal regulators, Igfr2, Grb10, and H19, are critical for placental growth and nutrient diffusion [51, 55–60]. In the mouse, parthenogenetic fetuses display poor development of the extraembryonic compartment [32]. Given the strong evidence linking proper imprinting to normal extraembryonic tissue formation, it is likely that placental growth defects are the main barrier to full-term mammalian parthenogenesis.
Several theories have been proposed to explain the evolution of mammalian imprinting, including its role in complex placentation, maternal behavior and lactation [61], neonatal feeding [62], and the coevolution of viviparity and genomic imprinting [63, 64]. When comparing eutherian, marsupial, and monotreme mammals, there is a phylogenetic connection between the length of gestation, the complexity of placentation, and the level of genomic imprinting. Imprinted genes are extensive among eutherians, but fewer in marsupials, and absent in the egg-laying monotremes [64]. The parent-offspring conflict hypothesis articulates the need for balance between the fecundity of the mother and the father’s genetic agenda to increase the fitness of his offspring. This hypothesis reasons that genomic imprinting is a parental tug of war, in which paternally expressed imprinted genes have evolved to extract as many resources as possible from the mother to ensure healthy development of the father’s offspring. In contrast, maternally expressed imprinted genes will regulate fetal growth to conserve the reproductive fitness of the mother [65]. Because the gestational period and placental dependence of eutherian mammals are higher than that of marsupials and monotremes, parent-offspring conflict may have been the initial evolutionary driver for the selection of genomic imprinting [64].
In the mouse, deliberate genetic manipulations can circumvent the developmental barriers of full-term parthenogenesis. Viable bimaternal mice have been generated by combining the genomes of two unrelated oocytes. The trick to this elaborate technique was using a nongrowing oocyte from a newborn mouse that had undergone complete imprint erasure but had not yet re-established maternal imprint signatures. To further mimic normal expression levels of paternally silenced genes, the paternal Igf2-H19 and Dlk1-Dio3 differentially methylated regions were deleted in the imprint-free oocyte [66]. Subsequently, the nuclei from these double-knockout imprint-free oocytes were reconstructed by serial nuclear transfer and then fused with mature imprinted oocytes. Following artificial activation, such bimaternal parthenogenetic embryos developed into live offspring [66]. This impressive study showed that the correction of just a few imprinted domains was sufficient to rescue parthenogenetic development. Interestingly, such bimaternal mice had reduced weights but lived approximately 30% longer than control females, suggesting that paternal imprints could be regulating longevity and energy-conserving genes [67]. Although the removal of two paternally imprinted regions allowed for normal development in the bimaternal mouse, mammalian parthenotes may still harbor other epigenetic abnormalities that are detrimental for offspring. Children born with uniparental imprinting diseases such as Beckman-Wiedemann, Silver-Russell, Prader-Willi, or Angleman syndromes display acute developmental defects ranging from mental retardation to various physical abnormalities [68].
Parthenogenetic ESCs for Autologous and Allogeneic Therapies
Despite the fact that genomic imprinting prevents the full-term development of parthenogenetic embryos, therapeutic applications of cells and tissues derived from pESCs are still merited. Mammalian pESCs have been established from several species (reviewed by Cibelli et al. [69]), including humans. The literature refers to parthenote-derived stem cells as “embryonic,” and, although indeed they are derived from parthenogenetic embryos, it should be noted that the oocytes involved are not fertilized, so some ethical issues associated with human embryos would thereby be avoided. Notably, parthenogenetic embryo development and ESC derivation efficiencies are similar to that of sperm-fertilized counterparts [17, 19, 70]. In addition, pESCs are morphologically indistinguishable from biparental controls, with similar growth and culture characteristics. Although parthenogenetic embryos maintain aberrant imprints and thus cannot develop to term, established pESCs can correct some of these defects and display normal gene expression [17, 71]. Global transcriptional profiling suggested that pESCs are similar to ESCs derived from fertilized embryos with strong expression of genes implicated in the maintenance of pluripotency, self-renewal, genome surveillance, and cell fate determination [29]. As expected, expression of several paternally imprinted genes was downregulated in pESCs compared with biparental ESCs. However, there were no differences noted in expression levels of maternally expressed imprinted genes in parthenotes [29].
The potential of human pESCs for regenerative medicine is evident through studies that demonstrate similar in vitro and in vivo differentiation potential to ESCs. For example, when injected into immunodeficient mice, both human and nonhuman primate pESCs readily form teratomas that are indistinguishable in composition from biparental controls [17, 19, 29, 30, 70, 72]. Upon in vitro differentiation, human pESCs have provided β cells, fibroblasts, cardiomyocytes, and neurons [19, 73], and monkey pESC have been differentiated into cardiomyocytes, smooth muscle, dopaminergic neurons, adipocytes, and ciliated epithelium [17, 29, 72, 74, 75]. When implanted into live rodents, pESC-derived cells show similar tissue engraftment potential as ESCs [22, 75–77]. Therapeutically, pESC-derived neurons can relieve Parkinson’s symptoms in rats [75]. In the mouse, cardiomyocytes derived from pESCs enhanced myogenesis and repair after cardiac infarction [76, 77], and pESC-derived hematopoietic progenitors have supported long-term hematopoiesis [78]. Although more transplantation studies are needed to draw accurate conclusions, it is encouraging that tumors were not observed with engrafted differentiated pESCs [75–78].
Parthenogenetic ESCs are endowed with several unique features that could prove clinically useful. Parthenogenetic activation protocols involve rather simple procedures requiring little manipulation expertise compared with SCNT. They can be derived with high efficiency, so as little as one stimulation cycle could provide a reproductively normal young woman with a sufficient number of oocytes to derive several genetically matched stem cell lines. As an example of pESC derivation efficiencies, in the monkey model, one ovarian stimulation supported derivation of 3 pESC lines [17], and, in humans, 21 oocytes rendered 4 pESC lines [19]. When comparing pESC derivation rates with other PSCs, they are far higher than iPSCs (∼0.001%–4.4%) [6] and are similar in efficiency to SCNT-ESCs (2.3%–20%) [8] and intracytoplasmic sperm injection-ESCs (27%) [15].
Of particular advantage, heterozygous pESCs should support autologous transplantations as a result of immune compatibility. When three rhesus macaque pESCs derived from the same female were analyzed for heterozygosity using polymorphic short tandem repeats (STR), pESC-1, -2, and -3 were identical to the oocyte donor at 87.2%, 69.2%, and 71.8% of loci tested, respectively (Table 1) [17]. Such high heterozygosity levels are explained by the extensive shuffling of alleles between parental homologous chromosomes at prophase I, and, for this reason, most alleles are afforded equal opportunity for germline transmission. Maintaining heterozygosity within major histocompatibility complex (MHC) [human leukocyte antigen (HLA) for humans] regions is most critical for pESCs that would avoid immune recognition of missing alleles [22]. For example, the pESC-1 cell line was a complete match for both alleles in this region (rhesus monkey MHC region located on the chromosome 6), whereas pESC-2 was homozygous (Table 1). The absence of a second allele may provoke an immune response by cytotoxic natural killer (NK) cells (see below). Therefore, human pESC lines should be analyzed for heterozygosity, in particular HLA loci, to ensure the best match for future autologous histocompatible transplantations. In addition, heterozygous pESCs could be a valuable research tool to study human recombination hotspots in the female germline. Through genome-wide SNP or STR genotyping, areas of sister chromatid exchange can be pinpointed by identifying zygosity changes along the chromosome [22]. A map of meiotic recombination hotspots in the female germline would most certainly be a valuable resource for the study of human diseases.
Table 1.
Zygosity levels of Rhesus macaque ESC and pESC lines by microsatellite parentage analysis
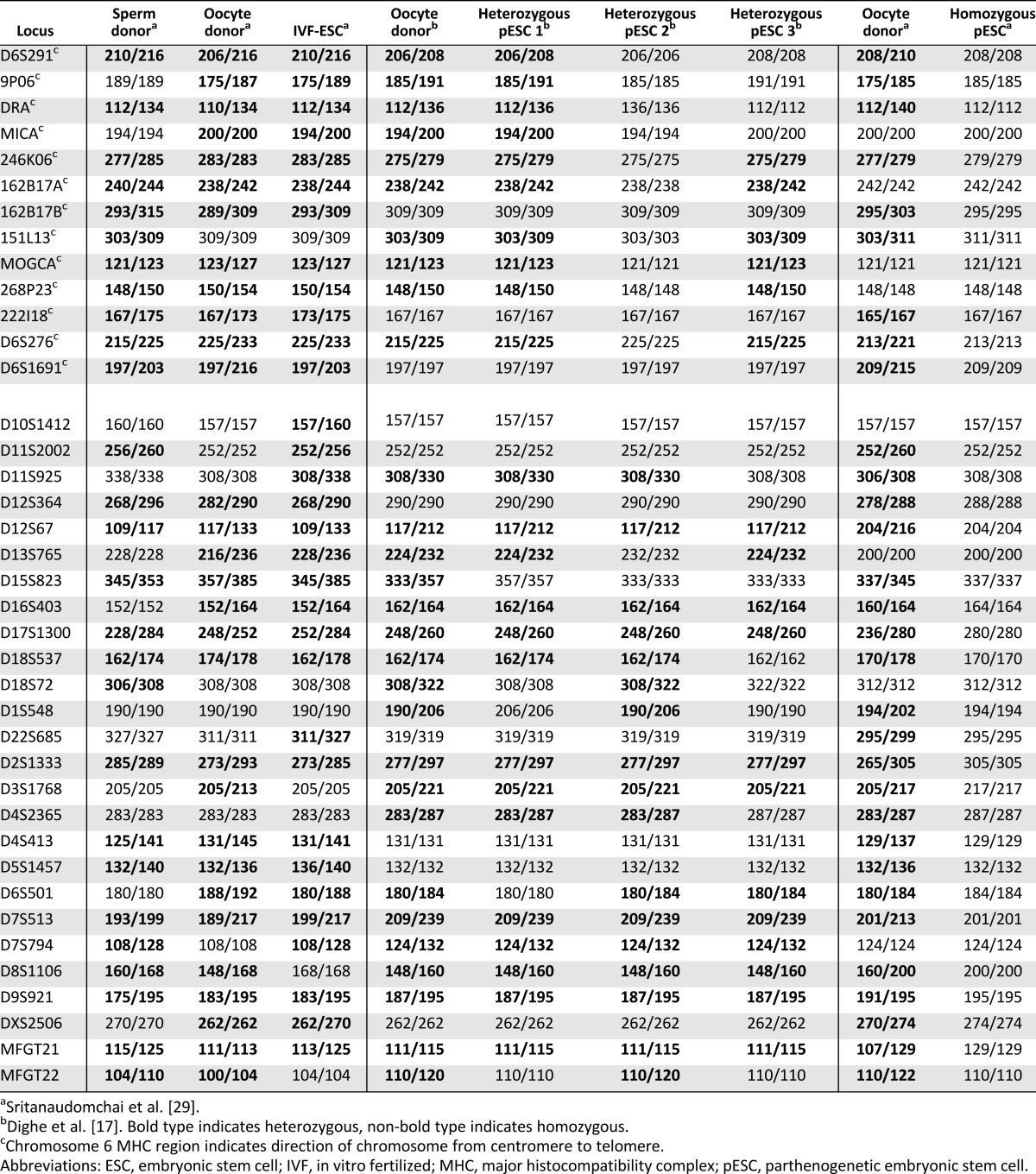
Unrelated patients could also benefit from allogeneic homozygous pESCs. As described above, a slight change in the oocyte activation protocol supports derivation of homozygous pESCs. With the projected, and seemingly impractical, high cost of iPSCs or SCNT-ESCs for personalized cell therapy, a viable alternative for providing PSC-derived cells to patients would involve banking stem cell lines. The immense diversity of HLA alleles in the human population is projected to be more than 1,500 at 12 different loci [79], presenting an arduous challenge for banking an adequate spectrum of lines for intended patients. However, to avoid transplant rejection and the need for immunosuppressive therapy, it is essential that banked human PSC lines match critical HLAs within four loci: HLA-A, HLA-B, HLA-C, and HLA-DRB1 [80].
Estimates are available relative to the feasibility of banking IVF-derived ESC lines. For the Japanese population, hypothetically 850 randomly derived ESC lines could match 60% of patients at three important HLA loci (A, B, and DR), whereas only 55 randomly selected homozygous pESC lines would be required to completely match 80% of patients [81]. Likewise in the U.K., 150 random or 100 O-blood-group ESCs would provide for less than 20% of the population, whereas 10 selected HLA-homozygous pESCs could benefit 38% of the people [82]. Because HLA-homozygous individuals are incredibly rare, banking ESCs or iPSCs from these individuals would be a difficult challenge. In contrast, HLA-homozygous pESCs could be easily obtained from any reproductively healthy woman.
Potential Limitations of pESCs
Although homozygous pESCs appear to be tantalizing candidates for allogeneic immunomatching, important questions should be addressed before considering them for future clinical use. Critics argue that cytotoxic NK cells may display hybrid resistance in that they are able to detect and react to the levels of host antigens, including the lack thereof [22, 83]. It is hypothesized that heterozygous individuals treated with homozygous allografts may experience a NK cell immune response because of missing HLA alleles. However, mouse studies have demonstrated that homozygous-MHC pESCs and differentiated embryoid bodies are not rejected when injected into immunomatched heterozygous-MHC mice. In contrast, heterozygous-MHC pESCs and embryoid bodies with partial mismatch to the host are not tolerated well [22, 76]. Simply put, MHCa/a can engraft in an MHCa/b host, but MHCa/b shows poor survival in MHCa/a mice. Although these mouse results are encouraging, humans are far more polymorphic at HLA loci, making immune tolerance outcomes more complicated.
Another potential concern with homozygous pESCs is that loss of heterozygosity (LOH) is considered a detrimental genetic outcome often associated with cancer. According to the Knudson tumorigenesis hypothesis [84], two harmful mutations in both parental alleles are required for complete loss of a functional tumor suppressor gene; thus, heterozygous individuals are insulated by the normal second allele. Inherited retinoblastoma is the classic example for tumorigenic LOH. In this disease, the infant is born with one defective copy of the tumor suppressor Rb1, and the second copy will often mutate during childhood, leading to retinal cancer. It is believed that the genome carries many harmful mutations, but most are masked by a functional second allele. Therefore, loss of heterozygosity through parthenogenesis could result in functional expression of defective genes. For example, knockdown or deficiency of the tumor suppressor p53 can lead to increased DNA damage and escape from apoptosis [85]. In addition, because p53 controls expression of pluripotency genes NANOG and OCT4, its knockdown may lead to persistence of undifferentiated cells [85, 86]. For this reason it would be critical to thoroughly screen homozygous pESC lines, for coding region mutations to address these safety concerns before clinical use. Various high-throughput genome interrogation assays, including next-generation exome sequencing and SNP arrays, are becoming routine for the detection of pathogenic mutations [21, 87, 88]. In parallel, novel genome-editing technologies, such as zinc finger nucleases and transcription activator-like effector nucleases, were recently developed that allow efficient and precise targeting and correction of underlying gene mutations in stem cells [89, 90].
To circumvent the allogeneic complications of unmatched ESCs, patient-specific iPSCs and SCNT-ESCs have been developed for autologous transplantation. When considering the future of PSCs for therapeutic purposes, it is estimated that preparing clinical-grade iPSC and SCNT-ESC products for individualized medicine will be costly and most likely unattainable for most patients. Secondly, the genetic integrity of both iPSCs and SCNT-ESCs remains unclear given that their genetic material originates from a somatic cell and may harbor age-acquired mutations [91, 92]. Additionally, although there are nonintegrating and nonviral methods for deriving iPSCs, reprogramming itself can generate and select for genetic abnormalities [91]. Several reports suggest that iPSCs harbor high levels of copy number variations [93, 94]. In the context of cell therapy, this could be detrimental for the recipient, as cancer-related mutations have also been found in iPSCs [87]. Indeed, when comparing transplanted undifferentiated and differentiated ESCs and iPSCs, immunogenicity [95] and tumor formation [96, 97] have been documented more often with iPSCs. However, recent studies have shown transplantation improvements with differentiated iPSCs and ESCs [98, 99]. In yet another drawback, reprogramming of somatic cells to pluripotency may not always be complete. Notably, several studies have found that some iPSCs retain an epigenetic memory of the parental somatic cell, which can bias their differentiation toward certain fates [100].
In contrast, pESCs are directly derived from oocytes, which are evolutionarily protected from somatic mutations to maintain the integrity of the germline. Parthenogenetic ESCs display robust capacity for differentiation owing to the oocyte’s intrinsic ability to induce pluripotent cells that are similar to ESCs. When considering all limitations, there are distinct obstacles for each PSC type, and it remains to be determined whether these aberrations are formidable barriers to effective cell therapy.
Conclusion
Transplant rejection and the harsh side effects of lifelong immunosuppression are currently major obstacles to the feasibility of cell therapy. HLA-knockout ESCs, HLA-screened ESCs, and patient-specific PSCs have been considered for cell banking. Each approach has its pitfalls. Production of clinical-grade cells and tissues for regenerative medicine could be time consuming and costly, and such cells may still be immunogenic despite controlled efforts. Immunocompatible pESCs present unique and attractive advantages for regenerative medicine. They can be efficiently derived, and estimates suggest that fewer than 100 HLA-homozygous pESC lines could provide compatible cells for greater than 90 million people [76]. Studies suggest that pESCs are indistinguishable from ESCs and iPSCs and perhaps even less tumorigenic [77]. However, abnormal imprinting and high levels of homozygosity may complicate applications of pESCs as well as other PSCs in regenerative medicine. Clearly, each PSC has unique advantages and disadvantages. Research on the utility of human pESCs in the context of cell therapies should be evaluated and outcomes compared between various PSC types.
Acknowledgments
We thank Dr. Don Wolf for helpful discussions and critical reading of the manuscript and past and current members of the Mitalipov laboratory for support in providing research data. This work was supported by grants from the Leducq Foundation and National Institutes of Health (T32HD007133, R01HD063276, R01HD057121, R01HD059946, R01EY021214, and P51OD011092).
Author Contributions
B.D.: conception and design, manuscript writing, collection and/or assembly of data; S.M.: conception and design, financial support, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Schwartz SD, Hubschman JP, Heilwell G, et al. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet. 2012;379:713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- 2.Advanced Cell Technology. Safety and Tolerability of Sub-retinal Transplantation of Human Embryonic Stem Cell Derived Retinal Pigmented Epithelial (hESC-RPE) Cells in Patients With Stargardt's Macular Dystrophy (SMD). Available at http://clinicaltrials.gov/show/NCT01469832 Accessed July 8, 2013.
- 3.Drukker M, Benvenisty N. The immunogenicity of human embryonic stem-derived cells. Trends Biotechnol. 2004;22:136–141. doi: 10.1016/j.tibtech.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tachibana M, Amato P, Sparman M, et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153:1228–1238. doi: 10.1016/j.cell.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyranoski D. Stem cells cruise to clinic. Nature. 2013;494:413. doi: 10.1038/494413a. [DOI] [PubMed] [Google Scholar]
- 10.Chapman DD, Shivji MS, Louis E, et al. Virgin birth in a hammerhead shark. Biol Lett. 2007;3:425–427. doi: 10.1098/rsbl.2007.0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crews D, Grassman M, Lindzey J. Behavioral facilitation of reproduction in sexual and unisexual whiptail lizards. Proc Natl Acad Sci USA. 1986;83:9547–9550. doi: 10.1073/pnas.83.24.9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saunders CM, Larman MG, Parrington J, et al. PLC zeta: A sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development. 2002;129:3533–3544. doi: 10.1242/dev.129.15.3533. [DOI] [PubMed] [Google Scholar]
- 13.Fan HY, Huo LJ, Meng XQ, et al. Involvement of calcium/calmodulin-dependent protein kinase II (CaMKII) in meiotic maturation and activation of pig oocytes. Biol Reprod. 2003;69:1552–1564. doi: 10.1095/biolreprod.103.015685. [DOI] [PubMed] [Google Scholar]
- 14.Ducibella T, Fissore R. The roles of Ca2+, downstream protein kinases, and oscillatory signaling in regulating fertilization and the activation of development. Dev Biol. 2008;315:257–279. doi: 10.1016/j.ydbio.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dighe V, Clepper L, Pedersen D, et al. Heterozygous embryonic stem cell lines derived from nonhuman primate parthenotes. Stem Cells. 2008;26:756–766. doi: 10.1634/stemcells.2007-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitalipov SM, Nusser KD, Wolf DP. Parthenogenetic activation of rhesus monkey oocytes and reconstructed embryos. Biol Reprod. 2001;65:253–259. doi: 10.1095/biolreprod65.1.253. [DOI] [PubMed] [Google Scholar]
- 19.Paull D, Emmanuele V, Weiss KA, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Susko-Parrish JL, Leibfried-Rutledge ML, Northey DL, et al. Inhibition of protein kinases after an induced calcium transient causes transition of bovine oocytes to embryonic cycles without meiotic completion. Dev Biol. 1994;166:729–739. doi: 10.1006/dbio.1994.1351. [DOI] [PubMed] [Google Scholar]
- 21.Kim K, Ng K, Rugg-Gunn PJ, et al. Recombination signatures distinguish embryonic stem cells derived by parthenogenesis and somatic cell nuclear transfer. Cell Stem Cell. 2007;1:346–352. doi: 10.1016/j.stem.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Kim K, Lerou P, Yabuuchi A, et al. Histocompatible embryonic stem cells by parthenogenesis. Science. 2007;315:482–486. doi: 10.1126/science.1133542. [DOI] [PubMed] [Google Scholar]
- 23.Leeb M, Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–134. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latham KE, Akutsu H, Patel B, et al. Comparison of gene expression during preimplantation development between diploid and haploid mouse embryos. Biol Reprod. 2002;67:386–392. doi: 10.1095/biolreprod67.2.386. [DOI] [PubMed] [Google Scholar]
- 25.Elling U, Taubenschmid J, Wirnsberger G, et al. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Shi L, Wang BA, et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–617. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Shuai L, Wan H, et al. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature. 2012;490:407–411. doi: 10.1038/nature11435. [DOI] [PubMed] [Google Scholar]
- 28.Leeb M, Walker R, Mansfield B, et al. Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development. 2012;139:3301–3305. doi: 10.1242/dev.083675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sritanaudomchai H, Ma H, Clepper L, et al. Discovery of a novel imprinted gene by transcriptional analysis of parthenogenetic embryonic stem cells. Hum Reprod. 2010;25:1927–1941. doi: 10.1093/humrep/deq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin G, OuYang Q, Zhou X, et al. A highly homozygous and parthenogenetic human embryonic stem cell line derived from a one-pronuclear oocyte following in vitro fertilization procedure. Cell Res. 2007;17:999–1007. doi: 10.1038/cr.2007.97. [DOI] [PubMed] [Google Scholar]
- 31.Kaufman MH, Barton SC, Surani MA. Normal postimplantation development of mouse parthenogenetic embryos to the forelimb bud stage. Nature. 1977;265:53–55. doi: 10.1038/265053a0. [DOI] [PubMed] [Google Scholar]
- 32.Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–550. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- 33.McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37:179–183. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- 34.Surani MA, Barton SC, Norris ML. Nuclear transplantation in the mouse: Heritable differences between parental genomes after activation of the embryonic genome. Cell. 1986;45:127–136. doi: 10.1016/0092-8674(86)90544-1. [DOI] [PubMed] [Google Scholar]
- 35.Tarkowski AK, Witkowska A, Nowicka J. Experimental partheonogenesis in the mouse. Nature. 1970;226:162–165. doi: 10.1038/226162a0. [DOI] [PubMed] [Google Scholar]
- 36.Allen ND, Barton SC, Hilton K, et al. A functional analysis of imprinting in parthenogenetic embryonic stem cells. Development. 1994;120:1473–1482. doi: 10.1242/dev.120.6.1473. [DOI] [PubMed] [Google Scholar]
- 37.Fundele RH, Norris ML, Barton SC, et al. Temporal and spatial selection against parthenogenetic cells during development of fetal chimeras. Development. 1990;108:203–211. doi: 10.1242/dev.108.1.203. [DOI] [PubMed] [Google Scholar]
- 38.Keverne EB. Importance of the matriline for genomic imprinting, brain development and behaviour. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110327. doi: 10.1098/rstb.2011.0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Liu Z, Huang J, et al. Birth of parthenote mice directly from parthenogenetic embryonic stem cells. Stem Cells. 2009;27:2136–2145. doi: 10.1002/stem.158. [DOI] [PubMed] [Google Scholar]
- 40.Kang L, Wang J, Zhang Y, et al. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135–138. doi: 10.1016/j.stem.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Strain L, Warner JP, Johnston T, et al. A human parthenogenetic chimaera. Nat Genet. 1995;11:164–169. doi: 10.1038/ng1095-164. [DOI] [PubMed] [Google Scholar]
- 42.Linder D, McCaw BK, Hecht F. Parthenogenic origin of benign ovarian teratomas. N Engl J Med. 1975;292:63–66. doi: 10.1056/NEJM197501092920202. [DOI] [PubMed] [Google Scholar]
- 43.Oliveira FG, Dozortsev D, Diamond MP, et al. Evidence of parthenogenetic origin of ovarian teratoma: Case report. Hum Reprod. 2004;19:1867–1870. doi: 10.1093/humrep/deh345. [DOI] [PubMed] [Google Scholar]
- 44.Lee YH, Kim SG, Choi SH, et al. Ovarian mature cystic teratoma containing homunculus: A case report. J Korean Med Sci. 2003;18:905–907. doi: 10.3346/jkms.2003.18.6.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiss JR, Burgess JR, Kaplan KJ. Fetiform teratoma (homunculus) Arch Pathol Lab Med. 2006;130:1552–1556. doi: 10.5858/2006-130-1552-FTH. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg JA, Clancy TE. Fetiform teratoma (homunculus) Rev Obstet Gynecol. 2008;1:95–96. [PMC free article] [PubMed] [Google Scholar]
- 47.Kuno N, Kadomatsu K, Nakamura M, et al. Mature ovarian cystic teratoma with a highly differentiated homunculus: A case report. Birth Defects Res A Clin Mol Teratol. 2004;70:40–46. doi: 10.1002/bdra.10133. [DOI] [PubMed] [Google Scholar]
- 48.Kono T. Genomic imprinting is a barrier to parthenogenesis in mammals. Cytogenet Genome Res. 2006;113:31–35. doi: 10.1159/000090812. [DOI] [PubMed] [Google Scholar]
- 49.Kaneko-Ishino T, Kohda T, Ishino F. The regulation and biological significance of genomic imprinting in mammals. J Biochem. 2003;133:699–711. doi: 10.1093/jb/mvg090. [DOI] [PubMed] [Google Scholar]
- 50.Barlow DP, Stöger R, Herrmann BG, et al. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature. 1991;349:84–87. doi: 10.1038/349084a0. [DOI] [PubMed] [Google Scholar]
- 51.DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- 52.Ferguson-Smith AC, Cattanach BM, Barton SC, et al. Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature. 1991;351:667–670. doi: 10.1038/351667a0. [DOI] [PubMed] [Google Scholar]
- 53.Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 54.Geneimprint. Imprinted Genes: by Species. Available at http://www.geneimprint.org/site/genes-by-species Accessed February 14, 2013.
- 55.Ogawa H, Shindo N, Kumagai T, et al. Developmental ability of trophoblast stem cells in uniparental mouse embryos. Placenta. 2009;30:448–456. doi: 10.1016/j.placenta.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 56.Kono T, Sotomaru Y, Katsuzawa Y, et al. Mouse parthenogenetic embryos with monoallelic H19 expression can develop to day 17.5 of gestation. Dev Biol. 2002;243:294–300. doi: 10.1006/dbio.2001.0561. [DOI] [PubMed] [Google Scholar]
- 57.Kono T, Obata Y, Yoshimzu T, et al. Epigenetic modifications during oocyte growth correlates with extended parthenogenetic development in the mouse. Nat Genet. 1996;13:91–94. doi: 10.1038/ng0596-91. [DOI] [PubMed] [Google Scholar]
- 58.Obata Y, Kaneko-Ishino T, Koide T, et al. Disruption of primary imprinting during oocyte growth leads to the modified expression of imprinted genes during embryogenesis. Development. 1998;125:1553–1560. doi: 10.1242/dev.125.8.1553. [DOI] [PubMed] [Google Scholar]
- 59.Constância M, Hemberger M, Hughes J, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 60.Sibley CP, Coan PM, Ferguson-Smith AC, et al. Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc Natl Acad Sci USA. 2004;101:8204–8208. doi: 10.1073/pnas.0402508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li L, Keverne EB, Aparicio SA, et al. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- 62.Plagge A, Gordon E, Dean W, et al. The imprinted signaling protein XL alpha s is required for postnatal adaptation to feeding. Nat Genet. 2004;36:818–826. doi: 10.1038/ng1397. [DOI] [PubMed] [Google Scholar]
- 63.Edwards CA, Rens W, Clarke O, et al. The evolution of imprinting: Chromosomal mapping of orthologues of mammalian imprinted domains in monotreme and marsupial mammals. BMC Evol Biol. 2007;7:157. doi: 10.1186/1471-2148-7-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Renfree MB, Suzuki S, Kaneko-Ishino T. The origin and evolution of genomic imprinting and viviparity in mammals. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120151. doi: 10.1098/rstb.2012.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore T, Haig D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 66.Kawahara M, Wu Q, Takahashi N, et al. High-frequency generation of viable mice from engineered bi-maternal embryos. Nat Biotechnol. 2007;25:1045–1050. doi: 10.1038/nbt1331. [DOI] [PubMed] [Google Scholar]
- 67.Kawahara M, Kono T. Longevity in mice without a father. Hum Reprod. 2010;25:457–461. doi: 10.1093/humrep/dep400. [DOI] [PubMed] [Google Scholar]
- 68.Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- 69.Cibelli JB, Cunniff K, Vrana KE. Embryonic stem cells from parthenotes. Methods Enzymol. 2006;418:117–135. doi: 10.1016/S0076-6879(06)18008-8. [DOI] [PubMed] [Google Scholar]
- 70.Mai Q, Yu Y, Li T, et al. Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Res. 2007;17:1008–1019. doi: 10.1038/cr.2007.102. [DOI] [PubMed] [Google Scholar]
- 71.Horii T, Kimura M, Morita S, et al. Loss of genomic imprinting in mouse parthenogenetic embryonic stem cells. Stem Cells. 2008;26:79–88. doi: 10.1634/stemcells.2006-0635. [DOI] [PubMed] [Google Scholar]
- 72.Cibelli JB, Grant KA, Chapman KB, et al. Parthenogenetic stem cells in nonhuman primates. Science. 2002;295:819. doi: 10.1126/science.1065637. [DOI] [PubMed] [Google Scholar]
- 73.Gonzalez R, Garitaonandia I, Abramihina T, et al. Deriving dopaminergic neurons for clinical use: A practical approach. Sci Rep. 2013;3:1463. doi: 10.1038/srep01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vrana KE, Hipp JD, Goss AM, et al. Nonhuman primate parthenogenetic stem cells. Proc Natl Acad Sci USA. 2003;100(suppl 1):11911–11916. doi: 10.1073/pnas.2034195100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sánchez-Pernaute R, Studer L, Ferrari D, et al. Long-term survival of dopamine neurons derived from parthenogenetic primate embryonic stem cells (cyno-1) after transplantation. Stem Cells. 2005;23:914–922. doi: 10.1634/stemcells.2004-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Didié M, Christalla P, Rubart M, et al. Parthenogenetic stem cells for tissue-engineered heart repair. J Clin Invest. 2013;123:1285–1298. doi: 10.1172/JCI66854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Ye X, Mao L, et al. Transplantation of parthenogenetic embryonic stem cells ameliorates cardiac dysfunction and remodelling after myocardial infarction. Cardiovasc Res. 2013;97:208–218. doi: 10.1093/cvr/cvs314. [DOI] [PubMed] [Google Scholar]
- 78.Eckardt S, Leu NA, Bradley HL, et al. Hematopoietic reconstitution with androgenetic and gynogenetic stem cells. Genes Dev. 2007;21:409–419. doi: 10.1101/gad.1524207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rao MS, Auerbach JM. Estimating human embryonic stem-cell numbers. Lancet. 2006;367:650. doi: 10.1016/S0140-6736(06)68261-5. [DOI] [PubMed] [Google Scholar]
- 80.Bray RA, Hurley CK, Kamani NR, et al. National marrow donor program HLA matching guidelines for unrelated adult donor hematopoietic cell transplants. Biol Blood Marrow Transplant. 2008;14:45–53. doi: 10.1016/j.bbmt.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 81.Nakajima F, Tokunaga K, Nakatsuji N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells. 2007;25:983–985. doi: 10.1634/stemcells.2006-0566. [DOI] [PubMed] [Google Scholar]
- 82.Taylor CJ, Bolton EM, Pocock S, et al. Banking on human embryonic stem cells: Estimating the number of donor cell lines needed for HLA matching. Lancet. 2005;366:2019–2025. doi: 10.1016/S0140-6736(05)67813-0. [DOI] [PubMed] [Google Scholar]
- 83.Höglund P, Sundbäck J, Olsson-Alheim MY, et al. Host MHC class I gene control of NK-cell specificity in the mouse. Immunol Rev. 1997;155:11–28. doi: 10.1111/j.1600-065x.1997.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 84.Knudson AG., Jr Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qin H, Yu T, Qing T, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. doi: 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 86.Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 87.Gore A, Li Z, Fung HL, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laurent LC, Ulitsky I, Slavin I, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Joung JK, Sander JD. TALENs: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Urnov FD, Rebar EJ, Holmes MC, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 91.Young MA, Larson DE, Sun CW, et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10:570–582. doi: 10.1016/j.stem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abyzov A, Mariani J, Palejev D, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martins-Taylor K, Nisler BS, Taapken SM, et al. Recurrent copy number variations in human induced pluripotent stem cells. Nat Biotechnol. 2011;29:488–491. doi: 10.1038/nbt.1890. [DOI] [PubMed] [Google Scholar]
- 94.Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 95.Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 96.Hayashi K, Ohta H, Kurimoto K, et al. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011;146:519–532. doi: 10.1016/j.cell.2011.06.052. [DOI] [PubMed] [Google Scholar]
- 97.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 98.Guha P, Morgan JW, Mostoslavsky G, et al. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell. 2013;12:407–412. doi: 10.1016/j.stem.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 99.Araki R, Uda M, Hoki Y, et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013;494:100–104. doi: 10.1038/nature11807. [DOI] [PubMed] [Google Scholar]
- 100.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]