

This study investigated the possibility that CD133 could serve as a potential target for cytotoxic T cells (CTLs) to target glioblastoma cancer stem cells. Two human leukocyte antigen (HLA)-Ap0201-restricted CD133 epitopes capable of inducing cytotoxic responses and of killing CD133 expressing tumor cells were identified. Results support the use of CD133-specific epitope vaccines to target CSCs in glioblastoma and other CD133 expressing tumors.
Keywords: CD133, Immunotherapy, Cancer stem cell, Dendritic cell, Cytotoxic T cell
Abstract
Targeting cancer stem cells (CSCs) with immunotherapy may be an effective means to prevent recurrences in glioblastoma multiforme (GBM). It is well established that CD133 is expressed in the population of GBM tumor cells representing CSCs. This raises a possibility that CD133 could serve as a potential target for cytotoxic T cells (CTLs) to target glioblastoma cancer stem cells. Two potential human leukocyte antigen (HLA)-A*0201-restricted CD133 epitopes, ILSAFSVYV (CD133-405) and YLQWIEFSI (CD133-753), showed strong binding to HLA-A*0201 molecules. In vitro immunogenicity studies generated peptide-specific CD8+ CTLs from normal donors. Autologous monocyte-derived dendritic cells pulsed with the CD133-405 or CD133-753 peptides generated CTLs that efficiently recognized the CD133 epitopes presented in T2 HLA-A*0201 cells and specifically lysed CD133+ HLA-A*0201+ GBM CSCs. These studies demonstrated natural processing and subsequent presentation of these epitopes in GBM CSCs and the ability of CTLs to kill CSCs bearing the antigen. Immunization studies in mice using the mouse homolog CD133 epitopes demonstrated immunogenicity in the absence of autoimmune damage. The results presented in this study support the use of CD133-specific epitope vaccines to target CSCs in glioblastoma and other cancers.
Introduction
Complete eradication of tumor masses requires elimination of cancer stem cells (CSCs), a group of undifferentiated tumor cells that share many properties of normal stem cells and repopulate the tumor by supplying differentiated tumor cells. In general, CSCs are resistant to standard chemotherapy [1, 2] and/or radiotherapy [3], whereas most of differentiated tumor cells are susceptible. Eradication of CSCs therefore requires novel and focused targeting of these cells. Immunotherapy can be a means of targeting these cancer stem cells.
Recently, a small population of cells has been identified in malignant glioblastoma multiforme (GBM) tumors with properties of CSCs [4], and vaccination studies using undefined antigens from these CSCs have shown prolonged survival in an experimental model of the disease [5]. Since most brain tumor patients suffer tumor recurrence due to resistance to traditional therapy [6], an immunotherapy-based method could be used to achieve tumor eradication. This approach would involve the generation of an immune response against the tumor cells by using CSCs antigens that are highly or exclusively expressed in this population.
A highly expressed GBM CSC antigen is CD133. CD133 belongs to a unique family of five transmembrane glycoproteins and is expressed in CSCs of a variety of human tumors [7], as well as some normal tissues and stem cells [8–10]. A recent study [11] suggested a prominent role of CD133 in enhanced DNA repair and apoptosis, thereby providing resistance to radio and chemotherapy. Another study [12] indicated that the presence of the CD133 containing lipid microdomains may be necessary to maintain certain stem cell properties and that their loss might contribute to differentiation. It is therefore possible that the asymmetric distribution of CD133 during cell division results in equal or unequal distribution between nascent daughter cells playing a role in stem cell renewal and commitment to differentiation.
Importantly, a recent and comprehensive study in human tumor samples has demonstrated that higher expression of CD133 is associated with tumor regrowth, malignant progression, and lower survival in glioma patients [13] and the recurrence of glioblastoma [14]. In addition, several studies have recently shown that targeting CD133 results in significant tumor reductions in animal models of head and neck cancer [15], hepatocellular and gastric tumors [16], ovarian cancer [17], and sarcoma [18]. Together, this evidence suggests that CD133 represents an ideal candidate to direct the host cellular immune response to target GBM CSCs.
In the present study we identified potential CD133 human leukocyte antigen (HLA)-A*0201-restricted class I epitopes and evaluated the potential use and safety of such epitopes for a therapeutic vaccine against GBM CSCs. HLA-A*0201 is the most prevalent HLA type in white populations. Our results show for the first time that CD133-derived class I epitopes can induce human-specific cytotoxic CD8+ T cells capable of recognizing and killing CD133 expressing tumor cells. Furthermore, vaccination studies in humanized HLA-A*0201 transgenic mice demonstrated that induction of such immune response did not lead to autoimmunity against nontumorigenic CD133 expressing cells, further supporting the use of these epitopes to treat GBM by CD133 targeted immunotherapy.
Materials and Methods
Prediction of HLA-A*020101-Restricted CD133 Epitopes And Peptides
HLA-A*020101-restricted CD133 epitopes were predicted from the full-length amino acid sequence of human CD133 (GenBank: GI:5174387) using the Immune Epitope Database (http://www.immuneepitope.org). Major histocompatibility complex (MHC) binding and peptide complex stability were determined at ProImmune (Oxford, U.K., http://www.proimmune.com). Binding was analyzed with the Reveal MHC-peptide binding assay (Reveal Biosciences, San Diego, CA, http://www.revealbio.com). Complex stability was determined by calculating the percentage of denaturation of pentamer-MHC complexes over time.
Peptides
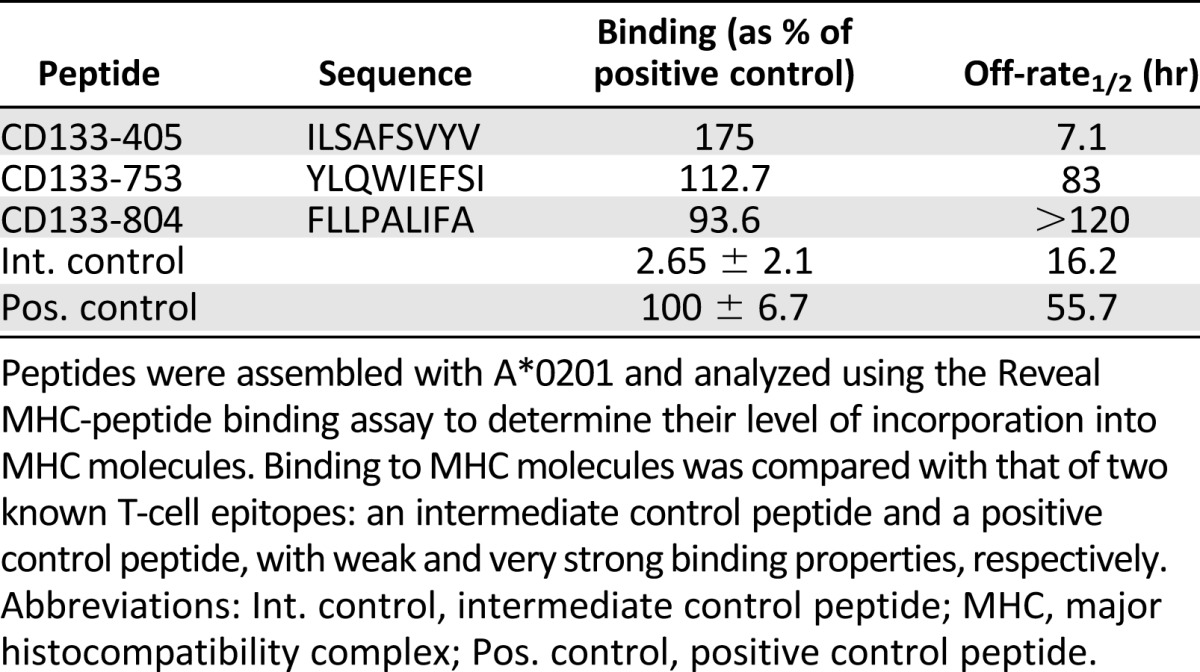
Human CD133-405, CD133-753, and CD133-804 peptides and the mouse homologs were synthesized (Polypeptide Inc., San Diego, CA, http://www.polypeptide.com) with both termini free. The sequences of human CD133 peptides are shown in Table 1. Mouse CD133 epitopes CD133-405 (MLLQVSHYL), CD133-753 (YLQWVLYAI), and CD133-804 (LLLPAVIIA) have predicted binding affinities to HLA-A*0201 that are equal to those of the human sequences (data not shown).
Table 1.
MHC binding of CD133 peptides
Subjects
Peripheral blood mononuclear cells (PBMCs) from HLA-A*0201 healthy donors were obtained from Hemacare Corp. (Van Nuys, CA, http://www.hemacare.com). Each PBMC sample was assigned a different code starting with H for human sample, a number based on the chronological order in which they were obtained, and a T or C based on the laboratory where they were processed and tested (Cedars-Sinai Medical Center or Torrey Pines Institute for Molecular Studies). A total of six different donors were included in this study: H14T, H16T, H17T, H14C, H15C, and H16C.
Generation of Monocyte-Derived DCs From PBMCs
Monocytes were isolated from fresh apheresis obtained from HLA-A*0201 healthy donors (Hemacare). The unwanted cells were labeled and magnetically separated using a Monocyte Enrichment Kit (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com). Enriched monocytes were cultured at 1 × 106 cells per milliliter in Cellgro-DC serum free medium (CellGenix, Freiburg, Germany, http://www.cellgenix.com) supplemented with GM-CSF (1,000 IU/ml) and interleukin (IL)-4 (1,000 IU/ml) (R&D Systems, Minneapolis, MN, http://www.rndsystems.com) in a 75 or 150 cm2 cell culture flask (Fisher Scientific, Pittsburgh, PA, http://www.fisherscientific.com). On day 6, a maturation cytokine cocktail made of monophosphoryl lipid A (2.5 µg/ml) (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) and interferon-γ (IFN-γ) (1,000 IU/ml) (R&D Systems) was added. After an overnight incubation, cells were harvested and the supernatants were saved to determine IL-12 (p70) production (enzyme-linked immunosorbent assay [ELISA]; BD Pharmingen, San Diego, CA, http://www.bdbiosciences.com). Adherent DC were harvested by treatment with a 0.25% trisodium citrate (Sigma-Aldrich) solution for 5 minutes at 37°C. Monocyte-derived dendritic cells (MoDCs) were characterized by expression of CD86, CD11c, CD14, and HLA-DR (BD Pharmingen) molecules by fluorescence-activated cell sorting (FACS) analysis. MoDCs included in all experiments were CD14−CD86+CD11c+HLA-DR+.
Generation of Peptide-Specific Cytotoxic T Cells by In Vitro Stimulation
CD8+ T cells were isolated from frozen apheresis by positive selection using CD8 microbeads and magnetic cell separation columns (Miltenyi Biotec, Auburn, CA, http://www.miltenyibiotec.com) and incubated with autologous MoDCs. Briefly, MoDCs were resuspended in cytotoxic T cell (CTL) media (Iscove’s modified Dulbecco’s medium [IMDM] + 10% HS medium supplemented with l-glutamine [2 mM] and 1× nonessential amino acids) and loaded with peptide (20 µg/ml) for 2 hours at 37°C and 5% CO2. MoDCs were then either γ irradiated at 2,800 rads or treated with mitomycin C (20 μg/ml for 30 minutes at 37°C). MoDCs (3 × 104 cells per well) were cocultured with CD8+ T cells (3 × 105 cells per well) in a 96-well plate at 37°C and 5% CO2 in a final volume of 200 µl of CTL media. Peptide was also added to the culture at a final concentration of 2 μg/ml. After 1 week, cultures were restimulated with newly generated MoDCs loaded with 2µg/ml peptide in the presence of 40 IU/ml IL-2 and 20 ng/ml IL-7. The process was repeated on the following week. On the third or fourth week, IL-2 was replaced with 25 ng/ml IL-15.
Analysis of CD133-Specific CTL Immunogenicity by IFN-γ ELISA
After 4 weeks of in vitro stimulation, 10,000 CTLs were removed from each individual well and mixed with an equal number of T2 cells pulsed with the corresponding specific peptides. Cells were cocultured in 96-well plates with 200 μl of CTL culture media at 37°C, 5% CO2 overnight. IFN-γ production was determined in the supernatants by a human IFN-γ ELISA kit (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com).
Analysis of CD133-Specific CTL Immunogenicity by Elispot Assay
Five thousand CTLs from pooled IFN-γ ELISA-positive wells were mixed with 100,000 T2 cells with and without the addition of 10ug/ml of peptides and seeded into 96-well plates. CTLs without T2 cells and CTLs plus 5 μg/ml phytohemagglutinin (PHA) were set as negative and positive controls, respectively. Cells were cultured at 37°C, 5% CO2 overnight. Results were measured using the IFN-γ Elispot kit (BD Biosciences) following the manufacturer’s instructions. Wells were scanned and counted by image analysis at ZellNet Consulting (Fort Lee, NJ, http://zellnet.com). Unpaired t test analysis was used to evaluate the significance of the response between peptide and nonstimulated cultures.
Cytolytic Activity by Chromium Release Assay
Evaluation of the cytolytic activity by 51Cr release was performed at an effector to target (E:T) ratio of 30:1 (2,000 targets cells per well) in a 4-hour standard assay. T2 cells (American Type Culture Collection [ATCC], Manassas, VA, http://www.atcc.org) labeled with chromium-51 (1 mCi) (MP Biomedicals, Solon, OH, http://www.mpbio.com) for 1 hour in a 37°C were used as target cells. The target cells were loaded with peptide and added to the T cell lines with a final peptide concentration of 10 µg/ml. Wells with only labeled target cells were used to determine spontaneous 51Cr release. Wells containing 2% Igepal CA-630 (Sigma-Aldrich) and target cells were used to determine the maximum release. Each CTL was seeded with T2 cells with or without peptide. After 4 hours at 37°C and 5% CO2 in IMDM + 10% human serum (HS) 75 µl of supernatant was transferred to a plate containing 200 µl of scintillation fluid. Plates were counted using a Trilux 1450 Microbeta counter (PerkinElmer, Waltham, MA, http://www.perkinelmer.com). The specific percentage of lysis was calculated as follows: (Experimental release − Spontaneous release)/(Maximum release − Spontaneous release) × 100. The analysis included both the subtraction of the values obtained in the absence of peptide from those in the presence of peptide and the calculation of the fold increase of cytotoxic activity by dividing the values in the presence of peptide by the values in the absence of peptide. Wells that showed fold increases greater than 2 and peptide-specific lysis greater than 10 were considered positive.
Detection of CD133-Specific CTL by HLA-A*0201/Peptide Tetramers
Following analysis at a single-well level by IFN-γ ELISA or chromium release assay, cells from positive wells were pooled together and stained with the specific APC-conjugated HLA-A*0201 tetramer (Beckman Coulter, Brea, CA, http://www.beckmancoulter.com). Briefly, cells (0.1 × 106 to 0.5 × 106) were stained with the tetramer at 5 ng/ml for 20 minutes at room temperature, washed once, and stained with anti-human CD8-Pacific Blue-labeled antibody (BD Biosciences) for 30 minutes at 4°C and analyzed by flow cytometry.
Cytolytic Activity by Caspase 3-Based Killing Assay
Evaluation of the cytolytic activity by caspase 3 assay was developed following the manufacturer protocol instructions (catalog no. 550480; BD Biosciences). In order to specifically gate and analyze the expression of caspase 3 in the target cells, these cells were labeled with DDAO-SE (catalog no. c34553; Invitrogen, Carlsbad, CA, http://www.invitrogen.com) before the coculture with the specific CTLs.
BTSC5 Line, Bone Marrow-Derived Normal Stem Cells, T2 HLA-A*0201, and Caco2 Cells
The brain tumor stem cell line 5 (BTSC5) was generated from a patient tumor sample in our laboratory and maintained in culture as described before [4]. BTSC5 CD133-high cells were enriched from total BTSC5 cells by FACS using the CD133/2 (293C) antibody (130-090-854; Miltenyi Biotec). Healthy donor CD133-positive and -negative bone marrow cells were purchased directly from AllCells Inc. (Alameda, CA, http://www.allcells.com). The CD133+ colorectal adenocarcinoma cell line, Caco-2, and the T2 HLA-A*0201 cell lines were purchased from ATCC and maintained in vitro following ATCC recommendations. BTSC5 and Caco2 cell lines were confirmed to be HLA-A*0201-positive by flow cytometry using fluorescein isothiocyanate (FITC)- or phycoerythrin-labeled anti-human HLA-A2 antibody (551285 and 558570, respectively; BD Pharmingen).
Mouse Immunizations
The mouse 9CB6F1-Tg (HLA-A*0201/H2-Kb) A*0201 transgenic line obtained from Taconic (Germantown, NY, http://www.taconic.com) were immunized three times with Mart1a/Ha-Flu or CD133 peptides emulsified in incomplete Freund’s adjuvant (IFA) and hepatitis B virus (HBV)-peptide every 2 weeks. Depending on the treatment group, mice were immunized with a phosphate-buffered saline (PBS)/ IFA (catalog no. F5506; lot no. 098K8724; Sigma-Aldrich) emulsion consisting of 50 μg of Mart1a and 100 μg of HA-Flu-positive HLA-A*0201 peptides or 50 μg of each CD133 mouse homolog peptide (CD133-21-753, CD133-22-804 and CD133-24-405) and 140 μg of TH-HBV peptide per injection of 100 μl. All the peptide stocks were dissolved in dimethyl sulfoxide at 20 mg/ml. After the peptide PBS mixture was prepared, the PBS mixture was then mixed at a 1:1 ratio with IFA using a Luer-lock syringe and stopcock. Each mouse was injected with one dose of 100 μl of PBS/IFA emulsion s.c. at the base of the tail using a 25-gauge needle. PBS control mice were injected with 100 μl of PBS.
Evaluation of Peptide-Specific CD8+ Responses (Immunogenicity) In Mice
Mice from the PBS control group (n = 10), Mart1a/Ha-Flu-positive control group (n = 10), and CD133 group (n = 20) were sacrificed 2 weeks after the last immunization. An extra group of 10 mice immunized with CD133 peptides was sacrificed 6 weeks after the last immunization. At the time of sacrifice, single cell suspensions were prepared from each mouse spleen and stimulated (25 × 106) in vitro in the presence of peptide and irradiated (2,500 rads) lipopolysaccharide (LPS) blasts (10 × 106) as antigen presenting cells (APCs) in upright 25 cm2 flasks. LPS blasts were prepared by stimulating spleen cells from untreated 9CB6F1-Tg A*0201 mice in vitro with 5 μg/ml LPS (Sigma L2387, Lot#: 099K4024) for 3 days at 37°C. Cultures from PBS immunized mice were stimulated with 5 μg/ml of Mart1a, CD133-21-753, CD133-22-804, and CD133-24-405. Cultures from Mart1a/ Flu-HA immunized mice were stimulated with 5 μg/ml of Mart1a and Flu-HA peptides. Cultures from CD133 immunized mice were stimulated with 5μg/ml of CD133-21-753, CD133-22-804, and CD133-24-405 peptides.
Peptide-stimulated splenocyte cultures were incubated for 7 days at 37°C in 5% CO2 and each culture was then evaluated for peptide reactivity in an IFN-γ Elispot assay (Mabtech, Knockmore, Ireland, http://www.mabtech.ie; antibodies AN18 [catalog no. 3321-3-100] and R4-6A2 Biotin [catalog no. 3321-6-1000]). On day 7, CTL effector cells were harvested and tested at 200,000 and 50,000 effector cells per well in a 96-well flat-bottom plate precoated with a capture anti-IFN-γ monoclonal antibody in RPMI1640 with 10% FBS. Irradiated (2,500 rads) spleen cells from untreated 9CB6F1-Tg A*0201 mice were added as APCs (200,000 cells per well). Peptides were added at 10 μg/ml to duplicate wells and incubated overnight at 37°C,5% CO2. Four wells with no peptide were used for background determination, and duplicate wells were plated with T cells plus Concanavalin A (ConA) (2.5 μg/m; catalog no. 5275; Sigma-Aldrich) as positive controls. Wells were scanned and counted by image analysis at ZellNet Consulting. Unpaired t test analysis was used to evaluate the significance of the response between peptide and nonstimulated cultures.
Results
Identification of CD133 Peptide Candidates and Evaluation of Their Immunogenicity
CD133-40, CD133-804, and CD133-753 peptides were selected from candidate HLA-A*0201-restricted CD133 epitopes generated using an epitope binding prediction service provided by ProImmune. Three candidate epitopes, CD133-405, CD133-753, and CD133-804, showed significant binding affinity to HLA-A*0201, as well as slow off-rates compared with other potential CD133 epitopes (Table 1). The immunogenicity of these peptides was then assessed by evaluating their in vitro capacity to expand naïve CD8+ T cells using PBMCs from healthy human donors. Autologous MoDCs loaded with the CD133-405, CD133-753, or CD133-804 were used to prime purified CD8+ T cells from HLA-A*0201 donors in multiple wells (96-well plates). After four rounds of weekly expansion, the presence of peptide-specific CD8+ T cells was determined by IFN-γ production in the supernatants. Wells showing positive responses were pooled and the frequency of epitope-specific CD8+ T cells was further determined by IFN-γ Elispot. Since epitope CD133-804 did not induce clear CD8+ T cell responses in most of the donors tested (data not shown), it was not considered for further analysis. As shown in Figure 1, CD133-405 and CD133-753 CTLs treated with 5 μg/ml PHA (positive control wells) showed the highest frequency of IFN-γ producing cells. The number of IFN-γ producing cells in response to CD133-405 or CD133-753 CTLs was significantly higher than baseline control wells (CTL plus T2 cells). The results show that CD133-405 and CD133-753 peptides are immunogenic as demonstrated by their capacity to produce peptide-specific IFN-γ producing CD8+ T cells. Since successful immunotherapy would require CD133-specific CTL to kill CD133 tumor expressing cells, we then tested the capacity of CD133-753 and CD133-405-specific CTL to kill target cells by chromium release assay. MoDCs loaded with the CD133-405 or CD133-753 were used to prime purified CD8+ T cells from HLA-A*0201+ donors in multiwell (48 wells in 96-well plates). Mart1a tumor-derived peptide was used as positive control, since there is a high frequency of naïve HLA-A*0201 CD8+ T cells specific to this epitope in the repertoire of healthy donors and these cells are capable of killing peptide-loaded T2 cells [19]. After three rounds of expansion, the killing capacity of peptide-specific CD8+ T cells was evaluated by chromium release assay. Similarly to the evaluation of immunogenicity by IFN-γ production, wells showing positive responses by 51Cr release were pooled and the capacity to kill peptide-loaded T2 cells was further determined by comparing the % lysis at different effector: target (E:T) ratios. Results from two different donors are shown in Figure 2. Together the results presented here indicate not only that CD133-405- and CD133-753-specific CTLs can recognize specific peptide-loaded T2 cells but also that this recognition leads to IFN-γ production and killing of CD133 peptide-loaded target cells.
Figure 1.

In vitro immunogenicity of CD133-405 and CD133-753 peptides evaluated by IFN-γ Elispot. Wells from CD133-753 and CD133-405 CTL cultures showing IFN-γ production were pooled and seeded (5,000 cells per well) with T2 cells (100,000 cells per well) in the presence or absence of the indicated peptide. Results show the number of spots (IFN-γ producing cells) as determined by Elispot. Error bars represent the standard deviation of triplicate wells. Significant differences between peptide stimulated cultures (CTL + T2 + peptide) and control wells (CTL + T2) are indicated as follows: ∗, p = .02; ∗∗, p < .0001. Abbreviations: CTL, cytotoxic T cell; IFN-γ, interferon-γ.
Figure 2.

Cytotoxic activity of peptide-specific CTLs. CD133-405, CD133-753, and Mart1a CTL were generated from donors H14T and H17T. After the third stimulation, each culture well was evaluated by 51Cr release. Wells showing positive responses were pooled after the fourth stimulation and seeded with 51Cr-labeled T2 cells as targets (2 × 103 cells per well) in the presence or absence of peptide. The cytotoxicity activity is expressed as the mean of the percent of specific lysis triggered by the CTL after 4 hours at the indicated CTL:T2 ratios. Error bars indicate the SD of triplicate wells.
Detection of CD133 Peptide-Specific CTL With Tetramers
We next evaluated the immunogenicity of the CD133-405 and CD133-753 peptides using tetramer staining technology. CD133 peptide-specific CTL were generated from six healthy human donors as described in materials and methods. Mart1a peptide was included as a positive control. After three rounds of expansion, individual wells were screened for the presence of epitope-specific CD8+ T cells (positive wells) by killing of peptide-loaded target T2 cells (HT donors) or IFN-γ production (HC donors). In both cases, cells from positive wells were pooled and analyzed by peptide-specific tetramer staining. As shown in Figure 3, all donors showed tetramer-specific cells in the Mart1a-specific CTL. Importantly, CD133-405 and CD133-753 CD8+ T cells could also be efficiently detected using tetramer staining. In particular, among all donors tested, the CD133-405-specific CTLs showed a higher frequency of peptide-specific CD8+ T cells (0.243%–5.940% CD8+ tetramer+ population) than CD133-753 CTL (0.053%–0.418% CD8+ tetramer+ population). As shown in Figure 3, CD8+ T cells from a representative donor before stimulation showed lower tetramer-positive cells than in stimulated T cell lines, suggesting that the increase in the detection of CD133-405- and CD133-753-specific cells by tetramer staining is due to the in vitro immunogenicity of the CD133 peptides.
Figure 3.

In vitro immunogenicity of CD133-405 and CD133-753 peptides evaluated by tetramer staining. CD133-405, CD133-753, and Mart1a (positive control) CTLs were generated from six healthy donors. After the last stimulation, IFN-γ enzyme-linked immunosorbent assay (donors H14C, H15C, and H16C) or chromium release assay (donors H14T, H16T, and H17T) was performed to screen for the presence of peptide-specific T cells in each well. Wells showing positive responses were pooled and stained with anti-CD8 and the specific tetramer. The last panel shows representative ex-vivo staining of peripheral blood mononuclear cells with the tetramers.
CD133 Peptide-Specific CTLs Recognize CD133+ GBM Tumor Cells but Not CD133+ Normal Bone Marrow Cells
We showed that CD133-405 and CD133-753 are immunogenic. In addition, we showed that CD133 peptide-specific CTLs can efficiently recognize peptide-loaded T2 cells and that this recognition leads to IFN-γ production and killing of target cells. However, a more clinically relevant question would be whether these CTLs could kill naturally processed CD133 peptides on patient cancer stem cells. To address this question, the GBM-derived neurosphere cell line (BTSC5) was used as target in a caspase 3 assay. This assay was used to assess cytotoxicity instead of chromium release to avoid biased responses due to the different capacity of the target cells to efficiently incorporate chromium. Since BTSC5 cells are approximately 5% CD133+ (data not shown), CD133+ cells were enriched (>90%) from the original population in order to specifically evaluate the killing of CD133+ expressing cells. Target cells were seeded at a 20:1 E:T ratio and cultured overnight. The Caco2 (CD133+) colon cancer cell line and bone marrow (BM) cells (CD133+ and CD133- fractions; AllCells) from a healthy donor were included as controls. As shown in Figure 4A, both CD133-405 and CD133-753 CTLs specifically lysed BTSC5 line (approximately 30%). Importantly, this killing effect was enhanced on the enriched BTSC5 CD133+ population (approximately 55%). The killing effect on CD133+ BM and CD133− BM populations was shown to be significantly lower (approximately 8%).
Figure 4.

Killing and characterization of CD133 expressing cells. (A): Specific killing of target CD133+ cells. CD133-753 and CD133-405 CTLs from donor H16C were cocultured with the brain tumor-derived stem cell line BTSC5 or CD133+ enriched BTSC5 cells as targets at a 20:1 ratio. The CD133+ colon cancer cell line Caco2 and BM CD133+ and CD133− cells were used as controls. After overnight incubation, caspase 3 staining was analyzed by intracellular staining. Results show the percentage of each target cell population expressing caspase 3. (B): Comparative expression of HLA and CD133 protein. HLA and CD133 expression was assessed by flow cytometry on the indicated cell populations. BM CD133+ cells were enriched from human bone marrow. BTSC5 cells were sorted by fluorescence-activated cell sorting based on CD133 expression (high or low) and cultured for 2 weeks previous to staining. Abbreviations: BM, bone marrow; HLA, human leukocyte antigen.
These observations are particularly important in at least two ways: (a) it strongly implies that CD133-405 and CD133-753 epitopes could be naturally processed and presented by GBM CSC tumor cells, and (b) those processed and presented CD133 epitopes on CSCs can be recognized and killed by both CD133-405- and CD133-753-specific CTLs. Furthermore, these experiments indicated that both CD133-positive and CD133-negative bone marrow cells are less sensitive to recognition and killing by CD133-405 and CD133-753 CTLs. Interestingly, flow cytometry analysis of the CD133 expression on the target populations revealed that the level of CD133 on GBM BTSC5 CD133+ enriched population was higher than in BM CD133+ cells (Fig. 4B), probably explaining the higher sensitivity to killing. In addition, MHC class I expression was also higher on the GBM BTSC5 CD133+ enriched population compared with the bone marrow CD133+ cells from this (Fig. 4B). Together, these results suggest that CD133 and HLA expression on glioma tumor cells leads to a preferential targeted killing of this population in comparison with nontumorigenic CD133+ expressing tissues such as healthy bone marrow-derived cells.
CD133-Derived Homolog Mouse Peptides Are Immunogenic in the HLA-A*0201 Transgenic Mice Model Without Inducing Autoimmunity
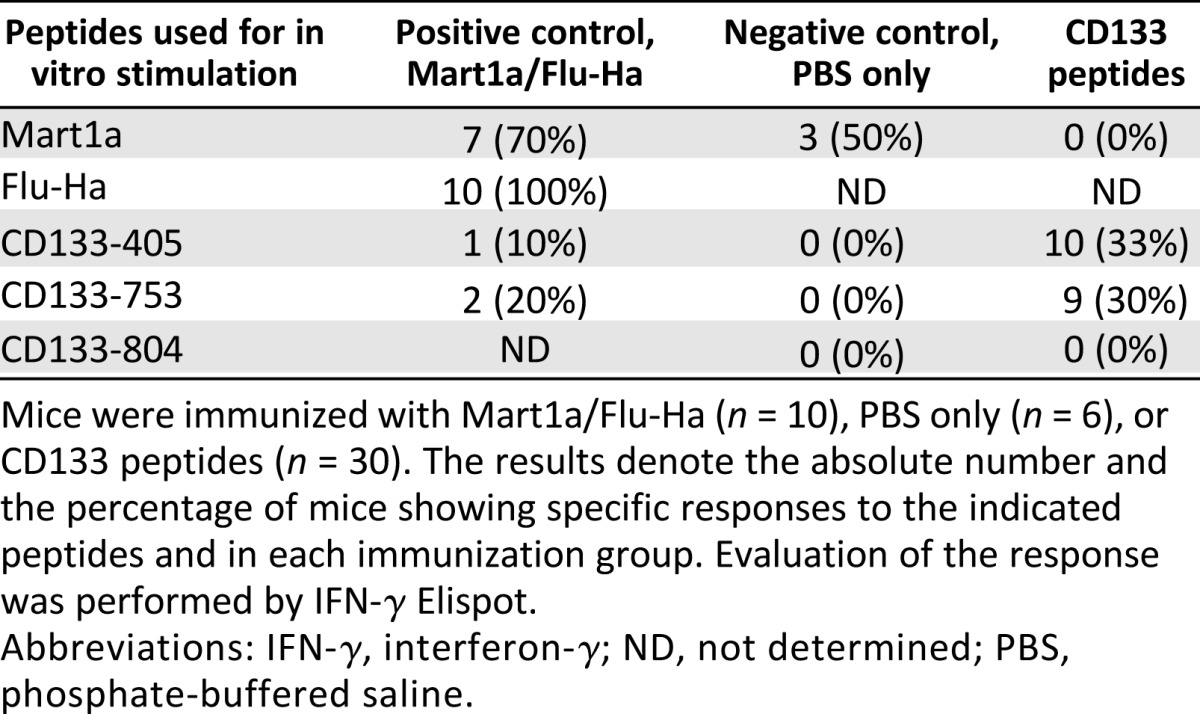
CD133 is expressed on different stem and progenitor cells from a variety of tissues (reviewed in [20]), raising the possibility that undesired autoimmunity might be a side effect triggered by CD133 peptide vaccine therapy. To evaluate the immunogenicity and the potential for autoimmunity, mouse homolog peptides of the CD133-405 and CD133-753 epitopes were used to immunize HLA-A*0201 transgenic mice. These homolog peptides were shown to strongly bind to HLA-A*0201 (data not shown). Mice were immunized three times at 3-week intervals and sacrificed 2 weeks after the last immunization. An additional group was sacrificed 6 weeks after the last immunization. Immunogenicity was evaluated by quantifying the number of IFN-γ producing cells by Elispot in the spleen after in vitro culture for 1 week with peptide-pulsed LPS-stimulated APCs. Figure 5 shows the results for representative mice. Table 2 shows the percentage of mice showing positive responses to each of the CD133 peptides and a mixture of Mart1a/Flu peptides. Of the mice immunized with the positive control antigens (Mart1a and Flu-Ha peptides), 100% showed responses to Flu and 70% to Mart1a. Interestingly, some level of response to CD133 peptides was observed in this positive control group. These responses may be a consequence of a low level of activation of unrelated (heterologous) T cells as a result of the inflammatory responses elicited by the adjuvant used in the immunization. This highlights the value of using dendritic cells loaded with peptide as a means of specifically activating the desired antitumoral T cell populations and avoiding high level of bystander activation due to adjuvant stimulation. Mice in the negative control group were negative for CD133 peptides, whereas in the CD133 treatment group, 33% mice responded to CD133-405, 30% to CD133-753, and none to CD133-804, confirming the results obtained in vitro with PBMCs from healthy donors. Together, these studies demonstrated the immunogenicity of mouse homolog CD133-753 and -405 peptides in HLA-A*0201 transgenic mice. To evaluate the autoimmune effect, toxicology studies were performed on 10 organs (heart, lung, liver, kidney, stomach, intestine, brain, bone marrow, gonads, and eyes) from mice with immune responses (Histo-Scientific Research Laboratories, Frederick, MD, http://hsrl.org). Results from these studies (data not shown) showed no toxicity or lymphocytic infiltration in any of these organs, supporting a lack of autoimmunity related to the immune response to these peptides. Together these studies support the safety and immunogenicity of these peptides as a potential vaccine to target CD133 cancer stem cells.
Figure 5.

Immunogenicity of peptides in vivo. Cytotoxic CD8+ T cell lines were generated from splenocytes from mice immunized with CD133 peptides, Mart1a/Flu-HA, or PBS only. After 7 days of in vitro stimulation, the reactivity of the CTLs was tested by IFN-γ Elispot. Representative mice showing positive responses from each immunization group are shown. Imm-CD133-1 and Imm-CD133-2 are two different mice that were immunized with all CD133 peptide analogs (CD133-21-753, CD133-22-804, and CD133-24-405) as described in Materials and Methods. The unpaired t test was used for statistical analysis, and significant differences (p < .005) between peptide and control wells (no peptide) are denoted with an asterisk. Abbreviations: ConA, Concanavalin A; CTL, cytotoxic T cell; IFN-γ, interferon-γ; Imm, immunized; PBS, phosphate-buffered saline; Pept., peptide.
Table 2.
Immunogenicity of CD133-derived peptides in vivo
Discussion
GBM comprises the most malignant of primary brain tumors and remains the most prevalent tumor in the CNS [21]. Despite recent advances in therapeutic modalities, morbidity and mortality resulting from GBM have not significantly improved [22]. Relapse in GBM, even after total resection, is likely due to residual CSCs in the brain parenchyma that can resist traditional therapies, including surgery, radiotherapy, and chemotherapy. Possibly in combination with conventional cancer therapies, immunotherapy targeting GBM CSCs, offers a future modality for otherwise incurable GBM. In this study, our aim was to evaluate CD133, known to be expressed in GBM CSCs, as a target for vaccine immunotherapy. We identified two HLA-A*0201-restricted class I CTL epitopes, CD133-405 (ILSAFSVYV) and CD133-753 (YLQWIEFSI), from the amino acid sequence of the full-length CD133.
CD133 is one of several proteins that are known to be overexpressed in GBM CSCs. The exact cellular function(s) of CD133 is not well known; however, its association with resistance to radiation and chemically induced cell death has been well established. As a large protein with five transmembrane domains CD133 may trigger a cascade of intracellular signals to activate a host of genes that are involved in cellular survival and DNA repair. This appears to be the case, at least in the mouse model, where CD133 in radioresistant cells triggers phosphorylated ataxia telangiectasia mutated (ATM) pathways to activate BCL-2 [11], although the nature of the signal generated by CD133 remains elusive. These properties of CD133 make it an ideal target for T cell-mediated immunotherapy. Because of its large size, there should be a larger number of potential epitopes to be discovered. Also, because of its expression in radio- and chemoresistant GBM CSCs in adult brain, immunotherapy can specifically target those cells that survive radiation and chemotherapy. Other approaches have also been used to deplete CD133+ cells. A study using a drug-conjugated antibody showed that the selective elimination of CD133+ cells can inhibit tumor cell proliferation in vitro and effectively delay tumor growth in vivo in a mouse model [16]. However, antibody therapy has the problem that only a small percentage of antibody can penetrate into tumor tissue, making antibody therapy less efficacious in vivo. A recent study used oncolytic measles virus to specifically eliminate CD133+ cells [23]; however, therapy using oncolytic viruses is still in the early stages of development, and the safety of the approach is under evaluation.
The results presented here clearly show that CD133-405 and CD133-753 peptides are defined CD133 CTL epitopes restricted by HLA-A*0201. They bind strongly to HLA-A*0201 and form a stable peptide-MHC complex compared with other candidate CD133 epitopes and other known HLA-A*0201 immunodominant epitopes. Interestingly, among other CD133 CTL epitopes that were screened, several showed strong binding capacity but did not form stable MHC-peptide complexes as CD133-405 and CD133-753 did, which may have rendered these epitopes less immunogenic. Interestingly, CD133-804 showed strong binding and a significantly long off-rate and still was shown to be nonimmunogenic. It is then possible to suggest that a low level of CD133-804 precursor T cells exist in the normal human repertoire, precluding a substantial expansion using peptide stimulation. Alternatively, there are a comparable number of precursors for CD133-804 and the immunogenic epitopes CD133-405 and CD133-753, but since the affinity of the peptide for the HLA molecule is so high, the T cells became tolerant or deleted because of specific peptide activation.
Immunogenicity of CD133-405 and CD133-753 was demonstrated by in vitro induction of CTLs specific for CD133-405 or CD133-753 using peptide-loaded autologous MoDCs from healthy HLA-A*0201 donors. In vitro priming and expansion of these two CD133 peptide-specific CTLs are clearly shown by tetramer staining and by IFN-γ Elispot. These studies also showed that the CD133-405 epitope triggered a stronger immune response than CD133-753 epitope.
Killing studies by coculturing CD133-405- or CD133-753-specific CTLs with peptide-loaded HLA-A*0201 target T2 cells showed recognition and lysis of epitope bearing target cells. In addition, these CTLs recognized and lysed GBM tumor cells enriched for cancer stem cells. This specific recognition and lysis was dose-dependent and correlated with the CD133 expression level on the target cells. Indeed, lower levels of recognition and killing was observed with normal bone marrow CD133+ cells consistent with lower expression of CD133 and MHC class I on these cells (Fig. 4B). Others have reported the low expression of MHC class I on normal neural stem cells [24], and low or negative expression on embryonic stem cells from humans [24, 25] or mouse [26], as well as spermatogonial stem cells [27].
Since CD133 is also expressed in normal stem cells and tissues, studies aimed to evaluate the potential for autoimmunity were performed in transgenic HLA-A*0201 mice that were immunized with mouse homologs of the CD133-405 and 753 epitopes. Immune responses were observed in these mice without any apparent autoimmunity. However, it is important to highlight here that it is unclear at this point whether these responses would be strong enough to mediate immune rejection of a tumor. Future studies using tumor grafts would help to determine the lack of autoimmunity in the presence of antitumor activity.
Conclusion
This study identified HLA-A*0201-restricted CD133 epitopes, CD133-405 and CD133-753. Unlike other potential CD133 epitopes examined, these two epitopes were strongly immunogenic in PBMCs from healthy donors and also naturally presented by GBM CSCs lines. Furthermore, studies in HLA-A*0201 expressing mice showed that these epitopes are indeed immunogenic in vivo but do not lead to autoimmune responses. Taken together, these results support CD133 as a suitable CTL target for GBM CSC immunotherapy. Based on these findings, a phase I trial of a dendritic cell vaccine using these CTL epitopes in recurrent glioblastoma has been initiated.
Acknowledgments
This work was supported by grants from ImmunoCellular Therapeutics Ltd. G.L. is currently affiliated with the Center for Translational Research, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai.
Footnotes
Contributed equally as first authors.
Author Contributions
J.J.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; V.A.J., C.P.: conception and design, data analysis and interpretation, manuscript writing; G.L., H.W., A.B., Z.L., M.X.: collection and assembly of data; J.B.: conception and design, financial support, data analysis and interpretation, manuscript writing; J.S.Y.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
J.B. is a compensated officer and shareholder of Immunocellular Therapeutics; V.A.J. and A.B. have sponsor research agreements with Immunocellular Therapeutics; G.L. has uncompensated intellectual property rights; C.P. has a sponsor research agreement; J.S.Y. is an officer, director, and shareholder of Immunocellular Therapeutics and has intellectual property rights.
References
- 1.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salmaggi A, Boiardi A, Gelati M, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54:850–860. doi: 10.1002/glia.20414. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 5.Pellegatta S, Poliani PL, Corno D, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 6.Cheng JX, Liu BL, Zhang X. How powerful is CD133 as a cancer stem cell marker in brain tumors? Cancer Treat Rev. 2009;35:403–408. doi: 10.1016/j.ctrv.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Mizrak D, Brittan M, Alison MR. CD133: Molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 8.Gallacher L, Murdoch B, Wu DM, et al. Isolation and characterization of human CD34(-)Lin(-) and CD34(+)Lin(-) hematopoietic stem cells using cell surface markers AC133 and CD7. Blood. 2000;95:2813–2820. [PubMed] [Google Scholar]
- 9.Bussolati B, Bruno S, Grange C, et al. Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005;166:545–555. doi: 10.1016/S0002-9440(10)62276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.am Esch JS, Knoefel WT, Klein M, et al. Portal application of autologous CD133+ bone marrow cells to the liver: A novel concept to support hepatic regeneration. Stem Cells. 2005;23:463–470. doi: 10.1634/stemcells.2004-0283. [DOI] [PubMed] [Google Scholar]
- 11.Chiou SH, Kao CL, Chen YW, et al. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE. 2008;3:e2090. doi: 10.1371/journal.pone.0002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer N, Wilsch-Bräuninger M, Karbanová J, et al. Haematopoietic stem cell differentiation promotes the release of prominin-1/CD133-containing membrane vesicles—a role of the endocytic-exocytic pathway. EMBO Mol Med. 2011;3:398–409. doi: 10.1002/emmm.201100147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeppernick F, Ahmadi R, Campos B, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 14.Shibahara I, Sonoda Y, Saito R, et al. The expression status of CD133 is associated with the pattern and timing of primary glioblastoma recurrence. Neuro-oncol. 2013;15:1151–1159. doi: 10.1093/neuonc/not066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldron NN, Kaufman DS, Oh S, et al. Targeting tumor-initiating cancer cells with dCD133KDEL shows impressive tumor reductions in a xenotransplant model of human head and neck cancer. Mol Cancer Ther. 2011;10:1829–1838. doi: 10.1158/1535-7163.MCT-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith LM, Nesterova A, Ryan MC, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer. 2008;99:100–109. doi: 10.1038/sj.bjc.6604437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skubitz AP, Taras EP, Boylan KL, et al. Targeting CD133 in an in vivo ovarian cancer model reduces ovarian cancer progression. Gynecol Oncol. 2013;130:579–587. doi: 10.1016/j.ygyno.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stratford EW, Bostad M, Castro R, et al. Photochemical internalization of CD133-targeting immunotoxins efficiently depletes sarcoma cells with stem-like properties and reduces tumorigenicity. Biochim Biophys Acta. 2013;1830:4235–4243. doi: 10.1016/j.bbagen.2013.04.033. [DOI] [PubMed] [Google Scholar]
- 19.Dutoit V, Rubio-Godoy V, Pittet MJ, et al. Degeneracy of antigen recognition as the molecular basis for the high frequency of naive A2/Melan-a peptide multimer(+) CD8(+) T cells in humans. J Exp Med. 2002;196:207–216. doi: 10.1084/jem.20020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu A, Oh S, Wiesner SM, et al. Persistence of CD133+ cells in human and mouse glioma cell lines: Detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 2008;17:173–184. doi: 10.1089/scd.2007.0133. [DOI] [PubMed] [Google Scholar]
- 21.Ohgaki H. Epidemiology of brain tumors. Methods Mol Biol. 2009;472:323–342. doi: 10.1007/978-1-60327-492-0_14. [DOI] [PubMed] [Google Scholar]
- 22.Preusser M, de Ribaupierre S, Wöhrer A, et al. Current concepts and management of glioblastoma. Ann Neurol. 2011;70:9–21. doi: 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 23.Bach P, Abel T, Hoffmann C, et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res. 2013;73:865–874. doi: 10.1158/0008-5472.CAN-12-2221. [DOI] [PubMed] [Google Scholar]
- 24.McLaren FH, Svendsen CN, Van der Meide P, et al. Analysis of neural stem cells by flow cytometry: Cellular differentiation modifies patterns of MHC expression. J Neuroimmunol. 2001;112:35–46. doi: 10.1016/s0165-5728(00)00410-0. [DOI] [PubMed] [Google Scholar]
- 25.Drukker M, Katz G, Urbach A, et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci USA. 2002;99:9864–9869. doi: 10.1073/pnas.142298299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grusby MJ, Auchincloss H, Jr, Lee R, et al. Mice lacking major histocompatibility complex class I and class II molecules. Proc Natl Acad Sci USA. 1993;90:3913–3917. doi: 10.1073/pnas.90.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubota H, Avarbock MR, Brinster RL. Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc Natl Acad Sci USA. 2003;100:6487–6492. doi: 10.1073/pnas.0631767100. [DOI] [PMC free article] [PubMed] [Google Scholar]