

Abstract
Genomic technologies are becoming a routine part of human genetic analysis. The exponential growth in DNA sequencing capability has brought an unprecedented understanding of human genetic variation and the identification of thousands of variants that impact human health. In this review, we describe the different types of DNA variation and provide an overview of existing DNA sequencing technologies and their applications. As genomic technologies and knowledge continue to advance, they will become integral in clinical practice. To accomplish the goal of personalized genomic medicine for patients, close collaborations between researchers and clinicians will be essential to develop and curate deep databases of genetic variation and their associated phenotypes.
Introduction
Modern DNA sequencing technologies have opened the door to the large-scale characterization of human genomes.1-3 Application of these new technologies to individuals and populations offers the unprecedented opportunity to identify and characterize functional human DNA variants amid the diverse spectrum of genomic variation. Appreciation of DNA as a complex and dynamic molecular anthology is essential for the study of inherited and acquired biological processes. In this article, we review the fundamentals of DNA variation as well as several common sequencing approaches, with emphasis on the application and trajectory of next-generation DNA sequencing technology.
Review of terminology and DNA sequence variation
DNA is a long double-stranded polymer composed of 4 nucleotides which form complementary base pairs (bp) with each other: adenine (A) with thymine (T), and guanine (G) with cytosine (C). Connected 5′ end to 3′ end (referring to the fifth and third carbons of the sugar), these 4 nucleotides are the building blocks of DNA.
DNA is organized into huge, linear, highly structured molecules which form the chromosomes. Chromatin, the physical organization of DNA and associated proteins, participates in regulating DNA function. Genes are the regions of DNA which encode for proteins. Protein coding regions are defined by the presence of exons, read 5′ to 3′, which are made up of codons, triplets of nucleotides which specify amino acids or signal translation stop. The stretches of nonprotein coding DNA between exons are introns. Splice sites mark the exon-intron boundaries and direct the excision of introns from the RNA message.
Control of gene expression
There are numerous functional noncoding DNA elements which participate in gene expression. Promoters, located immediately upstream of genes, are required for gene transcription. DNA regulatory elements which enhance or repress gene expression are often located near (or within introns of) structural genes, but can also lie at great distance. Some elements can control large genomic regions which contain many genes, such as the globin locus control region.4 Additionally, there are numerous DNA regions which transcribe noncoding functional RNAs, for example, transfer RNAs, ribosomal RNAs, and microRNAs. DNA nucleotides can also be reversibly chemically modified, such as by methylation, to affect elements which influence developmental or tissue-specific gene expression, such as occurs during imprinting or cell-lineage differentiation.5,6
DNA sequence variation
DNA is a living molecule in that it is constantly changing. DNA replicates during every mitosis, and recombines and segregates with every meiosis. Although DNA-replicative processes operate at extremely high fidelity, they are not (and cannot be) perfect.7 Thus, DNA variation is rarely but inevitably introduced during the copying of DNA template or ligation of free ends. DNA errors also arise from misrepair of DNA damaged as a result of routine exposure to cellular and environmental sources or by excess ionizing radiation, UV, or chemical insults. DNA-damage repair processes generally exhibit lower fidelity than DNA replication. This error permissiveness is thought necessary to facilitate restoration of a functional genome from corrupted DNA template without stalling DNA repair entirely, and can result in damage-specific patterns of acquired DNA variation.8,9
DNA accumulates variation as time progresses longitudinally over generations (germline variation) and within a single individual over many cell divisions (somatic variation). The vast majority of DNA variants cause no observable phenotype. However, a small fraction of variants are functional and can alter phenotypes.
Any difference in the DNA sequence as compared with a common reference sequence is considered a DNA variant (Figure 1). The simplest type of DNA variant is a change in a single-nucleotide base, known as a single-nucleotide variant (SNV). An SNV which is common in human populations (>1%) can also be known as a single-nucleotide polymorphism (SNP). Another type of DNA variation results from insertion or deletion (known as an indel) of a stretch of nucleotides. Structural variants (typically affecting >1000 bp) are DNA variants which include large indels as well as more complex DNA sequence rearrangements such as inversions (a block of DNA which has flipped “backwards”) and translocations (joining of distant genomic regions). Copy number variants (CNVs) are a type of structural variation resulting from gain or loss of a copy of an entire DNA region by deletion or duplication.
Figure 1.

Types of DNA sequence variation. (A) SNVs result from the substitution of 1 base, while insertion or deletion (indel) affects a string of nucleotides. (B) Structural variants (typically affecting >1000 bp) include large indels, inversions, duplications, and CNVs.
All types of DNA variation hold the potential to alter the expression or function of genes. SNVs can work directly by misspelling a codon’s amino acid translation (missense), creating a STOP codon (nonsense), or altering splice sites. SNVs can affect gene function by varying the sequence of promoters, regulatory elements, or noncoding RNAs. Indels can also create frameshift variants which shift codon registers to create new amino acid sequences downstream. Large indels can similarly disrupt genes as well as impact entire genomic regions or alter chromatin structure. Inversions and translocations not only disrupt their genomic sites of origin, but can also bring together new combinations of genes and/or regulatory elements. Additionally, CNVs which result in gain or loss of whole copies of functional DNA can affect phenotype via a differential gene dose effect. Thus, any type of DNA variant can affect function, and all categories of DNA variation have been implicated in disease.
DNA sequencing technologies
In the pregenomic era, various technologies were used to localize disease susceptibility genes (cytogenetics, fluorescence in situ hybridization) or to identify susceptibility alleles using DNA sequence variation in linkage analysis in family-based studies (microsatellite markers) or in candidate gene genotyping in unrelated individuals (restriction fragment length polymorphism [RFLP] analysis, allele-specific polymerase chain reaction [PCR]). The completion of the Human Genome Project1,2 and development of dense, genome-wide SNP marker genotyping arrays resulted in dramatic improvements in the design of genetic association studies for complex traits10-12 (Figure 2). These technologic advancements made it possible to efficiently screen the human genome for common polymorphisms associated with clinically relevant traits and ushered in the era of genome-wide association studies (GWAS). In the GWAS design, a large fraction of the commonly varying sites across the human genome are assessed either directly or indirectly (through linkage disequilibrium) for association with quantitative or qualitative phenotypes. While some of the genetic variants associated with complex hematologic traits are located within or near genes known to be involved in disease etiology or trait physiology, the genome-wide approach of GWAS led to discovery of previously unknown loci that provided new insights into disease biology.13,14 Similarly, comparative genomic hybridization (CGH) arrays based on the comparative cohybridization of fluorescently labeled sample and control DNAs have found CNVs to be common and sometimes associated with disease.15,16
Figure 2.

First-generation sequencing and genome-wide association technologies. (A) Stylized schematic of fluorescence-based (Sanger) sequencing chromatogram result showing heterozygosity for T/C at position Y. (B) Cartoon of genome-wide SNP marker genotyping array (SNP array) showing detection of differential hybridization (green or red if homozygous, yellow if heterozygous) of fluorescently labeled DNA representing common SNPs to the chip. (C) Cartoons of results from genetic array data. In GWAS, a “Manhattan plot” is typically used to summarize the large number of P values obtained, as represented by genomic coordinates displayed along the x-axis, with the negative logarithm of the association P value for each SNP displayed on the y-axis. A −log P value, such as indicated by the dashed line, is generated which is considered to meet the threshold for statistical significance. SNPs with significant values would appear above the line (shown in red). (D) For array CGH, gains and losses of DNA are given as a ratio and plotted against genomic position. This example shows loss of a region compared with the reference.
GWAS have exhaustively tested common, usually noncoding, DNA sequence variants and identified many new loci related to hematologic traits. However, rare DNA sequence variants, particularly those within protein-coding sequence, likely also contribute to interindividual variability in the population for hematologic traits or locus heterogeneity for monogenic hematologic syndromes. Recent advances in next-generation DNA sequencing technology allow comprehensive detection of rare DNA sequence variants.
First-generation DNA sequencing
DNA sequencing has always been at the core of human genetic analysis because sequencing is the only method that can provide the genotype at every position.1 In fact, capillary-based, fluorescence-based sequencing, known as Sanger sequencing, continues to be a mainstay technology to rapidly analyze any small region across a handful of samples.17 For fluorescence-based (Sanger) sequencing, the region of interest is first amplified from the genome by PCR. The amplified target is added to standard nucleotides (A, C, G, T) containing a mix of terminators, which are modified nucleotides each labeled with a different fluorophore. A DNA polymerase copies the target starting from an oligonucleotide primer, and as the DNA is synthesized (extended) the incorporation of fluorescently labeled terminators randomly stops synthesis such that a ladder of differently sized products is generated ending at each base in the target sequence. This cycle is repeated similar to PCR, generating many copies of the laddered products and enhancing the detection of each modified base that terminates a fragment. By subjecting the resulting ladder to single-base-resolution capillary electrophoresis, the fluorescence of the terminator in each fragment (from shortest to longest) is detected (Figure 2). The resulting sequence can exceed 600 bp. Fluorescence-based (Sanger) sequencing is still considered the gold standard, particularly in diagnostic situations. Therefore, this technology is still in common use, although higher-throughput technologies are rapidly being integrated into clinical laboratories.
Next-generation DNA sequencing
The development of next-generation sequencing (NGS) has changed the comprehensiveness of human genetic analysis and significantly reduced the costs associated with sequencing a genome.18-21 Today, for clinical evaluations, whole-exome sequencing (coding regions only) or whole-genome sequencing (coding and noncoding) of the individual22-26 are thought to be appropriate and practical testing modalities. The choice between exome or genome ultimately depends on cost and the need for noncoding data for clinical assessment. Exome sequencing is currently cheaper than whole-genome sequencing, although that may change in the future. In addition to exome and whole-genome sequencing, specific target gene panels can be optimized for NGS.
Sample preparation and library construction
There are several basic steps that are common to all massively parallel sequencing approaches (Figure 3).27,28 The first step is the generation of an in vitro library from the sample (DNA, or RNA converted to complementary DNA [cDNA]). The quality of the library is critical in determining sequencing efficiency. Originally, to prepare the sample, DNA fragments were physically sheared. Now, a number of enzymatic approaches have been developed29,30 which greatly simplify the process and increase the uniformity of library production. The throughput of NGS has increased to the point where samples from different individuals can often be sequenced together by uniquely bar-coding individual samples during library construction and then pooling samples prior to amplification. After sequencing, the barcodes permit the sequences to be separated or deconvoluted.
Figure 3.
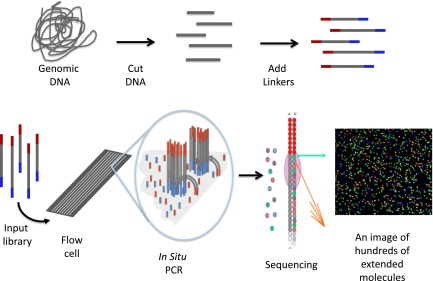
Schematic of 1 form of NGS. The process starts by randomly cutting genomic DNA (or cDNA) into short fragments (a few hundred base pairs in length). Oligonucleotide linkers are added to the fragments to generate a library in vitro. Libraries are introduced into a microscope slide with flow channels containing complementary oligonucleotides on the surfaces of the channel to ones on the libraries, thus allowing hybridization to attach millions of individual molecules to discrete locations on the slide. In situ PCR is performed to copy the individual fragments of the library to enhance sequencing detection. Single-base extension by a DNA polymerase with all 4 dye terminators extends the sequence 1 base. The image of the base extension is captured. This cycle and is repeated a 100 times from 1 end of the molecule and 100 times from the other.
Amplification
After a library is constructed, the molecules within the library are amplified to generate additional copies, ensuring robust detection.28 Amplification is one of the steps in the process that introduces biases as it decreases sequence coverage for some regions, that is, GC-rich regions such as some promoters and first exons, and introduces errors prior to sequencing. Errors that arise during the copying process are random, and their presence makes each individual sequence read less accurate. These errors are not usually miscalled as variants because the sequence is ultimately determined by a consensus of multiple unique reads, which are reads distinguished by their unique genomic positions, sequences, and/or lengths. However, when low-level detection is desired, such as in cancer sequencing, errors introduced during amplification must be considered. Low-level tumor variants are usually called only when they are seen more than twice, giving greater confidence that the observed variant is truly present and not an artifact of the process. Thus, greater depth (usually 300 times or more) among the unique reads is desirable in explorations of heterogeneous samples, as occurs in malignancy.26,31
Sequencing
There are a number of formats and chemistries used in NGS.27,28 Many use fluorescent dyes in a manner similar to fluorescence-based (Sanger) sequencing. Sequence detection for NGS is performed in channels, chambers, nanowells, or on assembled nanoballs. What varies substantially among the platforms is the approach used to obtain the sequence. One of the most widely applied technologies (available from Illumina) uses reversible dye terminator sequencing.20 In this system, the molecular library is captured in a channel and then amplified to generate a small cluster from each captured molecule. Next, DNA polymerase and all 4 dye terminators are flowed through the channel, resulting in fluorescent base extension for each cluster. Fluorescence is then read for the hundreds of millions of clusters found in the channel simultaneously. The dye terminators are then reversed by flowing reagents through the channel to clip off the fluorophore and repair the nucleotide, readying the base to be extended again. This whole process is known as a cycle, which is then repeated. Typically, 100 bp sequence reads are obtained from each end of the cluster, although read lengths from 50 to >200 bp are possible. An entire run from multiple channels can generate ∼600 gigabases (Gb) of sequence in an 11-day period. With a new upgrade, 120 Gb can be generated in ∼27 hours, or an entire whole genome every day at >30-fold coverage. This is truly massively parallel sequencing, and these approaches continue to improve and evolve.
Other sequence systems routinely generate whole-genome sequence data, such as those from Complete Genomics and Life Technologies. Complete Genomics does not sell an instrument, but provides a service in which a DNA sample is sent to the company and the sequence returned. In their approach, library construction leads to the production of a massive array of nanoballs, which are sequenced by a combination of hybridization and DNA ligation.32 Life Technologies’ SOLiD platform also uses DNA ligation, rather than DNA polymerization, to sequence on a massive scale.33
Other next- and third-generation sequencing platforms
Many other NGS platforms are available, and more are under development. Some have sufficient throughput to sequence the human exome, and all are capable of handling targeted gene panels or RNA sequencing applications. The Ion Torrent (Life Technologies) is unique in detecting the slight change in pH that takes place when each base is added 1 nucleotide position at a time34 and is similar in concept to 454 (Roche) which detects a base addition by the generation of a pyrophosphate.18 Another unique platform from Pacific Biosciences measures fluorescent base incorporation in single molecules in real time.35 Although throughput is not as high as other NGS platforms, it can produce long reads up to 25 kilobases (kb) in length and directly detect DNA methylation.36
Long read length can be advantageous when sequencing more complex regions of the human genome. Single-molecule sequencing holds the promise of long read length capabilities and is an active area of technology development. For example, in nanopore sequencing37 single DNA molecules are moved through a narrow pore and each base is detected in real time,38,39 which offers the potential of being able to generate molecule long reads. While this and other promising platforms are possible in the future, existing NGS technologies are also working toward longer reads to improve assembly of individual genome sequences and detection of difficult to call variants, such as larger indels and structural variants.
Sequence assembly
After sequence reads are generated by an NGS platform, they are typically aligned and assembled on a human reference sequence. The human reference sequence serves as a scaffold for read placement using a rapid indexing approach that finds the best match taking into account errors and variants in the reads. Although this form of assembly is not perfect, it is effective and fast at assembling the vast amounts of NGS data. In fact, the majority of the genome (∼90%) can be reliably mapped with this approach.
However, not all variants in the sequenced genomes are represented in the reference sequence, and short NGS read length can pose problems in assembly. Particularly challenging are large indels and other structural variations which cannot be assembled simply by aligning reads to the reference scaffold. Emerging tools to identify and characterize these variants include the addition of other sequences into the assembly and/or use of alternative algorithms.40,41 Genes with high sequence similarity (homology) also pose dilemmas by generating highly similar short sequence reads originating from different genes. Obtaining longer paired-end NGS reads will solve many of these issues. The use of hybrid approaches to genome sequence and assembly also holds promise for improving sequence assembly by combining short- and long-read NGS technologies or assembling short reads into longer ones molecularly.42 Additionally, the selected use of de novo sequence assembly, which assembles sequences without a reference scaffold43,44 and/or the application of new approaches to resolve human sequence haplotypes,45,46 could also improve our ability to analyze these challenging regions.
Variant calling
Once assembled, sequence variants are called in the dataset. In the early days of NGS, variants were identified by counting the number of times they appeared in unique reads to a set threshold. For example, if a region had 30 overlapping reads where 15 were called C at 1 position and 15 were called T at that same position, a heterozygous C/T variant would be called because each haplotype should be equally represented if the data were obtained in an unbiased fashion. Of course, in reality, sequencing reads obtained by any method are not unbiased, and these biases must be considered in analysis of the data.
The accuracy of SNV calling is high for most NGS platforms, although there is still significant variation among platforms because the error profiles and decoding schemes are not the same. Indels (small and large) and CNVs are more problematic both in specificity and sensitivity. Statistical and machine learning approaches are being applied to find variants and accurately and reproducibly call genotypes,47 and many new algorithms can now handle different types of variants, that is, indels or CNVs.48-50 Target gene panels are already in use for cancer sequencing, where the deeper sequence coverage obtained for each base increases the sensitivity and specificity of identifying variants.26 New approaches are also being developed to identify somatic mutations present at lower levels in samples.51
Thus, variant-calling capabilities are quite good with some variant types, such as SNVs, but face challenges for calling other, particularly larger, variants. The rapid pace of method development promises even higher sensitivity and specificity in the identification of complex variation in NGS data in the near future.
Applications of NGS to human genetics
Many lessons are emerging from the large-scale application of NGS in human genetics. Sequencing of the human exome and other large, targeted gene panels reveals that the level of rare variation in the human genome is far greater than expected.52,53 NGS data have also confirmed that the rarest variants in the human population are the youngest ancestrally and predicted to be the most deleterious.54 These findings have stimulated the development of high-throughput approaches to resequence genes in thousands of samples using the same capture methods successfully applied for exome sequencing. Simplifying and increasing the cost effectiveness is a key focus area of technology development. Critical to this will be optimizing approaches for multiplex PCR to capture hundreds or thousands of genomic targets in a single reaction.55 Combined with extensive sample bar coding, hundreds of samples could be sequenced with ease across thousands of regions.56 Already, systems such as molecular inversion probes are offering new ways to advance NGS capabilities and facilitate sequencing of the thousands of individuals required to pursue rare variant discoveries in human genetics.57
The scale of massively parallel sequencing opens new avenues for all forms of biological analysis, including analysis of sequence variants (shown in Table 158-63).64 Variant discovery and RNA sequencing are the principal applications today for NGS.65 Exome sequencing and genome sequencing have been successful in discovering causal variants in individuals with rare, highly penetrant monogenic disorders.66 The application of exome sequencing and exome-based genotyping arrays to more complex phenotypes in large population samples is under way through consortia such as the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP). Early results suggest exome sequencing and newer statistical approaches to analyzing rare variants can be used to further characterize genetically heterogeneous traits in large population-based studies.67,68
Table 1.
Selected applications of NGS
| Sequencing applications | Examples | Selected references |
|---|---|---|
| Genomic variation | Clinical genetics, cancer analysis | 24, 25 |
| Coding and targeted resequencing | Mendelian genetics, rare variant detection, cancer sequencing | 22, 23, 26, 53 |
| RNA profiling | RNA-Seq for expression, alternative splicing and cancer analysis | 58, 65 |
| DNA methylation | Epigenetic profiling | 36, 69 |
| Active regulatory regions | Dnase-seq, FAIRE-Seq | 59, 72 |
| DNA-DNA interactions | Hi-C | 70, 71 |
| Protein-DNA interactions | Chip-Seq, Chia-PET | 60, 73 |
| Mutagenesis profiling | Functional profiling of proteins or DNA elements, ie, (promoters, enhancers) | 64, 78, 79 |
| Immunoprofiling | HLA, T-cell receptor profiling | 61-63 |
The types of digital profiling that can be tackled by NGS are nearly unlimited, including methylation,36,69 chromatin profiling,70 structural DNA interactions,71,72 as well as many others.73,74 Among the alternative applications, RNA sequencing is the most widely applied. In RNA-seq, read counts are used to measure gene expression in the sample.75 Importantly, RNA sequencing also assesses alternative splicing and isoform usage, which, along with high sensitivity, offers major advantage over microarray analysis. RNA-seq brings new challenges for optimizing sample preparation and analyzing the resulting data, which are active areas of development.76 NGS is emerging as the method of choice for analyzing biology and genomic regulatory elements on a large-scale,64,77,78 highlighted by the myriad of new insights from the ENCODE project.79
Promise and challenges in the genomic era
Benefits of genomic-scale DNA sequencing are being realized as NGS is widely applied in research and being piloted for some clinical applications, such as pharmacogenomics, inherited congenital syndromes, inherited cancer risk genes, and tumor profiling.80,81 There are several examples where genome sequencing has been used for diagnosis of rare, Mendelian disorders,23,24 and new opportunities are available through the Centers for Mendelian Genomics.82 Moreover, noninvasive prenatal diagnostic screening by genome sequencing is under way.83 Detailed functional characterization of the consequences of sequence variation in genetic regulatory elements or protein function at single-nucleotide resolution are now possible through massively parallel reporter assay analysis.64,78
Genomic technologies are incredibly powerful but still hindered by the physical limits of the amount of DNA which can be sequenced at a time and the computing power needed to analyze the resulting data. As read lengths become longer and DNA sequencing becomes cheaper, the ability to deeply characterize entire genomes is expected to continue its current path of exponential growth. However, as the cost of storing raw data for long periods of time is offset by the uncertain practical utility of maintaining it,84 there is movement to only maintain analyzed results and, if needed, simply resequence at a future date. The ability to generate comprehensive personal sequence data also increases the likelihood of capturing large numbers of incidental findings; this will require development of consensus on procedures for maintaining and returning incidental results that vary in penetrance, clinical relevance, and medical actionability.85
Large-scale genomic sequencing of well-phenotyped population-based cohort or case-control studies may help to define the role of rare or lower frequency genetic variants, perhaps explaining some of the “missing heritability” of common, complex diseases and quantitative traits. However, extremely large sample sizes (in the hundreds of thousands) may be required for adequate statistical power. Consortia such as the NHLBI ESP have begun to evaluate the association of lower-frequency coding variants with hematologic traits, demonstrating the benefits of sequencing approaches to characterize genetically heterogeneous traits in large population-based studies. Another goal of ESP is to share these datasets with the scientific community, both through National Institutes of Health (NIH) repositories of genetic variants (dbGaP, dbSNP) and through the Exome Variant Server (http://evs.gs.washington.edu/EVS/), a web-based application that can be queried by gene or chromosomal location for a detailed summary of all identified sequence variants (Figure 486). Another outgrowth of large exome sequencing consortia has been the development of the Illumina Infinium Human Exome BeadChip, a lower-cost genotyping array that interrogates lower-frequency nonsynonymous, nonsense, and splice-site variants.
Figure 4.

Screenshot from exome variant server of the MPL gene showing part of a summary table of variants discovered through the NHLBI ESP. The current data release is taken from 6503 unrelated European American and African American samples drawn from multiple ESP cohorts and represents all of the ESP exome variant data. Users can select summary characteristics of interest for display and query sequence variants by gene, rsID (the variant identifier in dbSNP, if known), chromosomal location, or batch. The corresponding attributes (eg, allele counts or frequencies overall or by ethnicity, various evolutionary conservation scores such as GERP, phastCons, functional annotation) can be viewed on the web or downloaded as text-formatted files. Color coding is used to annotate variants according to genomic function (eg, splice/nonsense/frameshift, missense, synonymous, UTR). A functional prediction for each missense variant is shown using the Polyphen2 prediction algorithm.86 GERP, genomic evolutionary rate profiling; UTR, untranslated region.
The prospect of personalized medicine truly seems to be within reach as DNA sequencing technologies continue to become cheaper, faster, and provide more information. However, there remain significant challenges to fulfilling the promise of personalized medicine via deciphering of individual human genomes. Complete reference human genomes and maps of human genetic variation are incomplete, in part due to limitations in detecting structurally complex variants, and in part due to the need for comprehensive DNA sequence data on many individuals from ethnically diverse populations in order to represent human genetic diversity.
In summary, significant expansion of our knowledge of the human genome in conjunction with rapid technological advancements in DNA sequencing technologies has led to the identification of genetic variants responsible for hundreds of diseases. This number will only continue to grow as NGS capabilities are now within reach of most research laboratories and are being developed in clinical settings. Maintenance of the current steep trajectory in the understanding of human genetic diversity and successful application of that knowledge for the benefit of human health requires active collaboration between genomic experts and medical scientists to generate the accessible, deeply characterized, well-annotated, and diverse genomic and biological reference data to realize the potential of the human genome.
Acknowledgments
This work was supported by the National Institutes of Health NHLBI Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls available through the Exome Variant Server: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Authorship
Contribution: D.A.N., J.M.J., and A.P.R. contributed to writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jill M. Johnsen, Research Institute, Puget Sound Blood Center, Seattle, WA 98104; e-mail: JillJ@psbc.org.
References
- 1.Lander ES, Linton LM, Birren B, et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome [published correction appears in Nature. 2001;411(6838):720]. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 4.Palstra RJ, de Laat W, Grosveld F. Beta-globin regulation and long-range interactions. Adv Genet. 2008;61:107–142. doi: 10.1016/S0065-2660(07)00004-1. [DOI] [PubMed] [Google Scholar]
- 5.Ballestar E. An introduction to epigenetics. Adv Exp Med Biol. 2011;711:1–11. doi: 10.1007/978-1-4419-8216-2_1. [DOI] [PubMed] [Google Scholar]
- 6.Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev. 2012;22(2):72–78. doi: 10.1016/j.gde.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 8.Jiricny J. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. 2013;5(4):a012633. doi: 10.1101/cshperspect.a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 10.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6(2):95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 11.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008;17(R2):R156–R165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gieger C, Radhakrishnan A, Cvejic A, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480(7376):201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Harst P, Zhang W, Mateo Leach I, et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012;492(7429):369–375. doi: 10.1038/nature11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14(2):125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 17.Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet. 2006;38(3):375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- 18.Shendure J, Porreca GJ, Reppas NB, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309(5741):1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 19.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors [published correction appears in Nature. 2006;441(7089):120]. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wetterstrand KA. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). www.genome.gov/sequencingcosts. Accessed June 24, 2013.
- 22.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 25.Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchard CC, Smith C, Salipante SJ, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14(4):357–366. doi: 10.1016/j.jmoldx.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 28.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 29.Adey A, Morrison HG, Asan, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11(12):R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aird D, Ross MG, Chen WS, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12(2):R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327(5961):78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 33.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19(9):1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 35.Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 36.Flusberg BA, Webster DR, Lee JH, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Branton D, Deamer DW, Marziali A, et al. The potential and challenges of nanopore sequencing. Nat Biotechnol. 2008;26(10):1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manrao EA, Derrington IM, Laszlo AH, et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat Biotechnol. 2012;30(4):349–353. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherf GM, Lieberman KR, Rashid H, Lam CE, Karplus K, Akeson M. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nat Biotechnol. 2012;30(4):344–348. doi: 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8(1):61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7(2):119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Hu Y, Bolund L, Wang J. State of the art de novo assembly of human genomes from massively parallel sequencing data. Hum Genomics. 2010;4(4):271–277. doi: 10.1186/1479-7364-4-4-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28(14):1838–1844. doi: 10.1093/bioinformatics/bts280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitzman JO, Mackenzie AP, Adey A, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29(1):59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters BA, Kermani BG, Sparks AB, et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487(7406):190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karakoc E, Alkan C, O’Roak BJ, et al. Detection of structural variants and indels within exome data. Nat Methods. 2012;9(2):176–178. doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krumm N, Sudmant PH, Ko A, et al. NHLBI Exome Sequencing Project. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Olivier M. Current analysis platforms and methods for detecting copy number variation. Physiol Genomics. 2013;45(1):1–16. doi: 10.1152/physiolgenomics.00082.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tennessen JA, Bigham AW, O’Connor TD, et al. Broad GO; Seattle GO; NHLBI Exome Sequencing Project. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelson MR, Wegmann D, Ehm MG, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337(6090):100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu W, O’Connor TD, Jun G, et al. NHLBI Exome Sequencing Project. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants [published correction appears in Nature. 2013;495(7440):270]. Nature. 2013;493(7431):216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turner EH, Ng SB, Nickerson DA, Shendure J. Methods for genomic partitioning. Annu Rev Genomics Hum Genet. 2009;10:263–284. doi: 10.1146/annurev-genom-082908-150112. [DOI] [PubMed] [Google Scholar]
- 56.Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6(5):315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Roak BJ, Vives L, Fu W, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pickrell JK, Marioni JC, Pai AA, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paul DS, Albers CA, Rendon A, et al. HaemGen Consortium. Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci. Genome Res. 2013;23(7):1130–1141. doi: 10.1101/gr.155127.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148(1-2):84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang C, Krishnakumar S, Wilhelmy J, et al. High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc Natl Acad Sci U S A. 2012;109(22):8676–8681. doi: 10.1073/pnas.1206614109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19(10):1817–1824. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fowler DM, Araya CL, Fleishman SJ, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7(9):741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 66.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 67.Auer PL, Johnsen JM, Johnson AD, et al. Imputation of exome sequence variants into population- based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am J Hum Genet. 2012;91(5):794–808. doi: 10.1016/j.ajhg.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnsen JM, Auer PL, Morrison AC, et al. NHLBI Exome Sequencing Project. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood. 2013;122(4):590–597. doi: 10.1182/blood-2013-02-485094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23(3):555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duan Z, Andronescu M, Schutz K, et al. A three-dimensional model of the yeast genome. Nature. 2010;465(7296):363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mercer TR, Edwards SL, Clark MB, et al. DNase I-hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet. 2013;45(8):852-859. [DOI] [PMC free article] [PubMed]
- 73.Li XY, Biggin MD. Genome-wide in vivo cross-linking of sequence-specific transcription factors. Methods Mol Biol. 2012;809:3–26. doi: 10.1007/978-1-61779-376-9_1. [DOI] [PubMed] [Google Scholar]
- 74.Shendure J, Lieberman Aiden E. The expanding scope of DNA sequencing. Nat Biotechnol. 2012;30(11):1084–1094. doi: 10.1038/nbt.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Melnikov A, Murugan A, Zhang X, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30(3):271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patwardhan RP, Hiatt JB, Witten DM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.ENCODE Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15(4):258–267. doi: 10.1038/gim.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biesecker LG. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: lessons from the ClinSeq project. Genet Med. 2012;14(4):393–398. doi: 10.1038/gim.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bamshad MJ, Shendure JA, Valle D, et al. Centers for Mendelian Genomics. The Centers for Mendelian Genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am J Med Genet A. 2012;158A(7):1523–1525. doi: 10.1002/ajmg.a.35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra4. [DOI] [PMC free article] [PubMed]
- 84.Stein LD. The case for cloud computing in genome informatics. Genome Biol. 2010;11(5):207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jamal SM, Yu JH, Chong JX, et al. Practices and policies of clinical exome sequencing providers: analysis and implications. Am J Med Genet A. 2013;161A(5):935–950. doi: 10.1002/ajmg.a.35942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]