

Abstract
Background
Androgen receptor (AR) plays a critical role in the progression of both androgen-dependent and androgen-independent prostate cancer (AIPC). Ligand-independent activation of AR in AIPC or castration resistant prostate cancer (CRPC) is often associated with poor prognosis. Recently, tyrosine kinase Ack1 has been shown to regulate AR activity by phosphorylating it at tyrosine 267 and this event was shown to be critical for AIPC growth. However, whether a small molecule inhibitor that can mitigate Ack1 activation is sufficient to abrogate AR activity on AR regulated promoters in androgen-depleted environment is not known.
Methods
We have generated two key resources, antibodies that specifically recognize pTyr267-AR and synthesized a small molecule inhibitor of Ack1, 4-amino-5,6-biaryl-furo[2,3-d]pyrimidine (named here as AIM-100) to test whether AIM-100 modulates ligand-independent AR activity and inhibits prostate cell growth.
Results
Prostate tissue microarray analysis indicates that Ack1 Tyr284 phosphorylation correlates positively with disease progression and negatively with the survival of prostate cancer patients. Interestingly, neither pTyr267-AR expression nor its transcriptional activation was affected by anti-androgens in activated Ack1 expressing or EGF stimulated prostate cells. However, the Ack1 inhibitor, AIM-100, not only inhibited Ack1 activation but also able to suppress pTyr267-AR phosphorylation, binding of AR to PSA, NKX3.1, and TMPRSS2 promoters, and inhibit AR transcription activity.
Conclusion
Ack1 Tyr284 phosphorylation is prognostic of progression of prostate cancer and inhibitors of Ack1 activity could be novel therapeutic agents to treat AIPC.
Keywords: Ack1, AR, CRPC, AIM-100, TNK2, tyrosine kinase inhibitor, tissue microarray, prostate cancer
Introduction
Activation of androgen receptor (AR) is a critical event in the recurrence of most prostate cancers [1,2]. Androgen deprivation therapy is used to treat prostate cancers by suppressing production of testicular androgen either as a result of surgical castration or of administration of luteinizing hormone-releasing hormone superagonists. Treatment with antiandrogens or AR antagonists (e.g., bicalutamide or casodex and flutamide) is also used. These standard systemic treatments although initially present favorable outcomes, eventually relapse with emergence of a more aggressive form of prostate cancer often termed as androgen-independent prostate cancer (AIPC) or castration resistant prostate cancer (CRPC). Mechanisms of AR activation responsible for CRPC includes AR gene amplification or mutation, overexpression of AR or coactivators, changes in interaction with coregulatory molecules including either coactivators and/ or corepressors and ligand-independent AR activation by tyrosine kinases [1–7]. As AR activity acquires resistance to high doses of the antagonists, AR-regulated genes become overexpressed in CRPC [8,9]. However, the precise mechanism of loss of antiandrogen sensitivity in CRPC is not fully understood.
Increasing evidences indicate that tyrosine kinases play a significant role in progression of prostate cancer by regulating AR activity [1,2,5,6,8,10,11]. Heregulin-stimulated HER2 activation induced Ack1 activation and AR tyrosine phosphorylation primarily at Tyr-267, located within the transactivation domain [6]. Further, Ack1 knockdown inhibited heregulin-dependent AR tyrosine phosphorylation and AR activation. Consistent with this data, AIPC samples exhibited tyrosine-phosphorylated AR protein which correlated with tyrosine-phosphorylated Ack1 [6].
Ack1 is a ∼141 kDa non-receptor tyrosine kinase (NRPTK) expressed ubiquitiously and can be activated by variety of growth factor stimuli, for example, heregulin, EGF, FGF, PDGF, and insulin [6,10,12,13]. Upon ligand binding to receptor tyrosine kinase (RTKs), Ack1 is phosphorylated at Tyr284, resulting in kinase activation [10]. Ack1 gene amplification and overexpression is prevalent in many tumor types (including prostate), correlated with poor prognosis, and is associated with increased cell motility and invasiveness and metastasis [10,14–16]. Knockdown of Ack1 increased apoptosis in transformed cells, suggesting that Ack1 signaling enhanced survival [10,17]. Taken together, targeting Ack1 kinase may be a potential therapeutic strategy in prostate cancer.
Despite the awareness of tyrosine kinases and their implications in prostate cancer, a small molecule inhibitor capable of attenuating AR binding to promoters/enhancers of androgen regulated genes in androgen-lacking environment is not available. Here, we report that Ack1 Tyr284 phosphorylation correlates positively with disease progression. Additionally, we generated two critical experimental resources, pTyr267-AR-specific phospho-antibodies and Ack1 inhibitor, AIM-100. AIM-100 inhibited Ack1 Tyr284-phosphorylation attenuated pTyr267-AR transcriptional activity which in turn resulted in cell cycle arrest. Our data suggests that inhibition of Ack1 activity could be highly effective therapeutic strategy for treatment of prostate cancer.
Materials and Methods
Cell Lines, Antibodies, and Plasmids
293T, LNCaP, and DU145 cells were obtained from ATCC. Ack1 mAb (A11), alpha-tubulin (TU-O2), Actin (I-19), pTyr(PY20)HRP conjugate antibodies (Santacruz); pTyr284-Ack1 (Millipore); Her2 Ab-2 (Clone 9G6.10; Thermo Scientific); EGFR (Epitomics) antibodies, were purchased from the respective companies. Flag-tagged AR, myc-tagged constitutively active Ack1 (caAck), kinase dead Ack1 (kdAck) have been described previously [6,10]. EGF, heregulin, DHT, casodex, and flutamide were purchased from Sigma.
Generation and Purification of pTyr267-AR Phospho-Antibody
Two AR peptides coupled to immunogenic carrier proteins were synthesized: (i) the phosphopeptide: Ac-K-Ahx-QLRGDCMpYAPLLGVP-amide and (ii) the non-phospho peptide: Ac-K-Ahx-QLRGDCMYAPLL-GVP-amide.
Two rabbits were immunized twice with phosphopeptide and ELISA was performed to determine the relative titer of sera against phosphorylated and non-phosphorylated peptides. Two antigen-affinity columns were used to purify the phospho-specific antibodies. The first column was the non-phosphopeptide affinity column. Antibodies recognizing the non-phospho residues of the peptide bound to the column. The flow-through fraction was collected and then applied to the second column, the phospho-peptide column. Antibodies recognizing the phospho-residue bound to the column which was eluted as phospho-specific antibodies.
Ack1 Inhibitor AIM-100 Synthesis
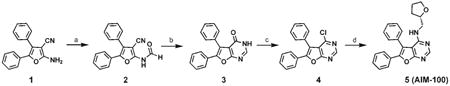
The reported diarylfuropyrimidine Ack1 inhibitor [18], compound 5 shown below was prepared using standard methods [19]. It was named as AIM-100. The synthesis of AIM-100 which starts from commercially available 1 is shown in a scheme below.
Reagents and conditions
(a) Ac2O, HCOOH, 60°C, 6 hr, followed by slow addition of 1 at 0°C then rt 12 hr, 90%; (b) AcOH, microwave heating at 200°C, 60 min, 75%; (c) POCl3, 55°C, 2hr, under argon, 100%; (d) (S)-(+)-Tetrahydrofurfurylamine, EtOH, reflux, 5 hr, 87%.
Flow Cytometry
LNCaP and LAPC4 cells (1 × 105/ml) were treated with AIM-100 (4 μM, 72 hr). Cells were harvested, washed with PBS and were fixed with 80% ethanol. For flow analysis cells were washed with PBS containing 0.2% BSA and treated with propidium iodide and RNAse A (37°C for 1 hr). Cells were analyzed by Becton and Dickenson Flow Cell sorter and quantitated by Modfit software.
Immunoprecipitation, Immunoblotting,Chromatin Immunoprecipitation (ChIP) Analysis, Quantitative RT-PCR, Reporter Assays, and MTT Assays
These assays have been described in our earlier publications [6,10,20].
Tissue Microarray (TMA) Analysis
We generated prostate TMA block with 263 patient samples (Table I). The tissue array slides (4 slides including 2 test duplicate slides, and positive and negative controls) were stained with pTyr284-Ack1 (Milipore) and pTyr267-AR antibodies at 1:100 dilution, overnight. Negative controls were included by omitting pTyr284-Ack1 or pTyr267-AR antibodies during primary antibody incubation or incubating pTyr284-Ack1 antibody with purified activated-Ack1 protein prior to TMA staining. The staining was examined in a blinded fashion by two independent pathologists.
Table I. Prostate TMA Core Distribution.
| Diagnosis | No. of cores | Location |
|---|---|---|
| BPH | 24 | 1–24 |
| PINs (regular) | 25 | 25–49 |
| PINs (high) | 15 | 50–64 |
| Gleason's 6 | 34 | 65–98 |
| Gleason's 7 (3 + 4) | 42 | 99–140 |
| Gleason's 7 (4 + 3) | 40 | 141–180 |
| CRPC | 20 | 181–200 |
| Gleason's 8 & UP | 52 | 201–252 |
| Metastatic cases | 11 | 253–263 |
| Cell line | 4 | 264–267 |
| Total | 267 |
Statistical Analysis
Boxplots were used to summarize the intensity sampling distribution at each progression stage. To assess the association between pTyr284-Ack1 levels and progression stages and groups of prostate cancer, the Spearman's rank correlation coefficient was estimated. Analysis of variance (ANOVA) was performed to examine whether the expression levels of pTyr284-Ack1 differ among different tumor stages and CRPC samples. Tukey–Kramer method was further performed to examine the differences in expression levels between pairs of stages (Table II). The association of the expression levels of pTyr284-Ack1 and the overall survival was assessed using the log-rank test, and then summarized using Kaplan–Meier survival curves.
Table II. P-Values of Tukey–Kramer Multiple Comparisons (Simultaneous Inference) of pTyr284 -Ack1 Intensity Levels Between All Pairs of Stages for Prostate Cancer.
| Normal | BPH | PIN | G6–7 | G8–10 & Met. | AI | |
|---|---|---|---|---|---|---|
| Normal | 1.00 | 0.21 | 0.04* | 0.12 | 0.002* | |
| BPH | 0.03* | 0.0002* | 0.003* | <0.0001* | ||
| PIN | 1.00 | 1.00 | 0.50 | |||
| G6–7 | 1.00 | 0.23 | ||||
| G8–10 & Mets | 0.17 | |||||
| AI |
Significance at 0.05 level.
Results
Tyr284-Phosphorylated-Ack1 Expression Positively Correlates With Disease Progression and Inversely Correlates With the Survival
We generated a TMA of clinically annotated prostate (n=263) tumor samples (Table I). Tyr284 being the primary autophosphorylation site in Ack1 [10,13,21], pTyr284-Ack1 antibodies were used to assess Ack1 activation. To validate pTyr284-Ack1 antibodies for immunohistochemistry, RWPE or mouse embryonic fibroblast (MEFs) cells treated with EGF ligand (or no ligand) and MCF7 cells treated with heregulin (or no ligand) were fixed and used for antibody validation. Ligand treated cells exhibited Tyr284 phosphorylation of Ack1, in contrast untreated cells had significantly low or undetectable levels of pTyr284-Ack1 (data not shown). Subsequently, prostate tumor samples were used for antibody validation; prostate tumors exhibited high levels of pTyr284-Ack1 which was diminished when antibodies were incubated with purified Ack1 protein (Fig. 1A). We have earlier observed that in contrast to pTyr284-Ack1, the total Ack1 levels remained unchanged between normal and tumor samples, as seen by immunobloting and immunohistochemistry [10,13].
Fig. 1.
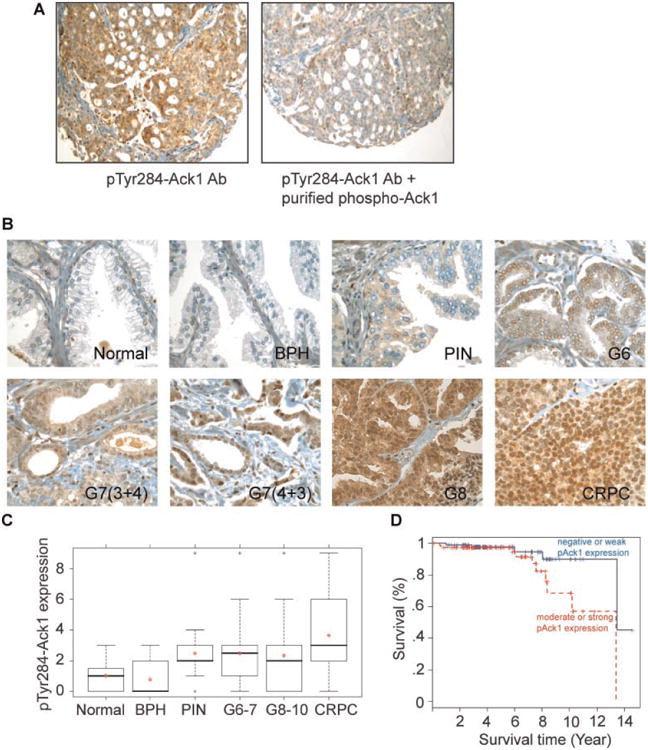
pTyr284-Ack1 expression correlates positively with disease progression and negatively with survival of prostate cancer patients. A: Validation of pTyr284-Ack1antibodies for immunohistochemistry studies. Prostate tumor samples were fixed and sectioned followed by immunohistochemical staining with pTyr284-Ack1 antibodies (left panel). Prostate tumor sample sections were stained with pTyr284 -Ack1 antibodies that were blocked by incubation with purified activated Ack1 protein (right panel). B: TMA sections representing different prostate cancer stages were stained with pTyr284 -Ack1 antibodies. C: Box plots to summarize distributions of staining intensities for pTyr284 -Ack1 in different stages of prostate cancer. The box has lines at the lower quartile (25%), median (50%), and upper quartile values (75%), while the red-cross within the circle marks the mean value. Whiskers extend from each end of the box to the most extreme values within 1.5 times the inter-quartile range from the ends of the box. The data with values beyond the ends of the whiskers, displayed with black circles, are potential outliers. A significant increase in expression of Tyr284-phosphorylated Ack1 (Spearman's rank correlation coefficient = 0.32, P<0.0001) was seen as prostate cancer progressed. D: Kaplan–Meier survival curves shows that individuals with prostate cancer that have moderate to strong staining of pTyr284-Ack1 (>2)have a significantly worse overall survival outcome than those with lower pTyr284-Ack1 levels(log-rank P=0.041).
We analyzed the data from 243 patients samples for correlation. Tyr284-phosphorylated-Ack1 expression is significantly correlated with the severity of disease progression (Spearman's rank correlation coefficient=0.32, P<0.0001; Fig. 1B,C). ANOVA results indicated that Tyr284-phosphorylated Ack1 expression differed significantly among progression stages and CRPC (P<0.0001). The expression levels of pTyr284-Ack1 in CRPC were significantly higher than those in normal, BPH; the expression levels were significantly lower in the BPH samples when compared to almost all of the later stages (Table II). The results from all pairwise comparison using the Tukey–Kramer method for pTyr284-Ack1 between different groups are summarized in Table II. There were 168 individuals with available pTyr284-Ack1 staining and survival information, and these data were used for survival analysis. The expression level of pTyr284-Ack1 in prostate cancer patients was significantly associated with overall survival (log-rank P = 0.041; Fig. 1D). Patients whose tumor expressed lower pTyr284-Ack1 levels have a better survival outcome than those with higher levels.
Generation of Phospho-Antibodies That Specifically Recognize pTyr267-AR
Ack1 has been shown to regulate AR activity by phosphorylating it at tyrosine 267 [6]. To better understand Ack1 function in prostate cancer, we generated antibodies that recognized pTyr267-AR protein. Heregulin treatment of serum and androgen-depleted LNCaP cells resulted in a time-dependent accumulation of endogenous pTyr267-AR (Fig. 2A). Incubation of pTyr267-AR antibodies with AR-phosphopeptide prior to immunoblotting resulted in complete loss of pTyr267-AR recognition (Fig. 2A, 2nd panel). Similarly, LAPC4 cells too displayed time-dependent Tyr267-phosphorylation of endogenous AR (Fig. 2B). Validity of pTyr267-AR antibodies was further confirmed by transfecting 293T cells with kdAck or caAck [10] with AR constructs, followed by immunoblotting with pTyr267-AR antibodies. Coexpression of AR with caAck but not with kdAck resulted in AR Tyr267 phosphorylation, which was detected upon immunoblotting with pTyr267-AR antibodies, while unphosphorylated AR was not recognized (Fig. 2C). Specificity of pTyr267-AR antibodies was further assessed by incubating pTyr267-AR antibodies with AR267-phosphopeptide which resulted in total loss of pTyr267-AR recognition (Fig. 2C, 2nd panel). Serum and androgen-depleted LNCaP cells treated with heregulin ligand exhibited endogenous pTyr267-AR expression which was undetectable in DU145 cells which lack AR, confirming the specificity of the antibodies (Fig. 2D).
Fig. 2.
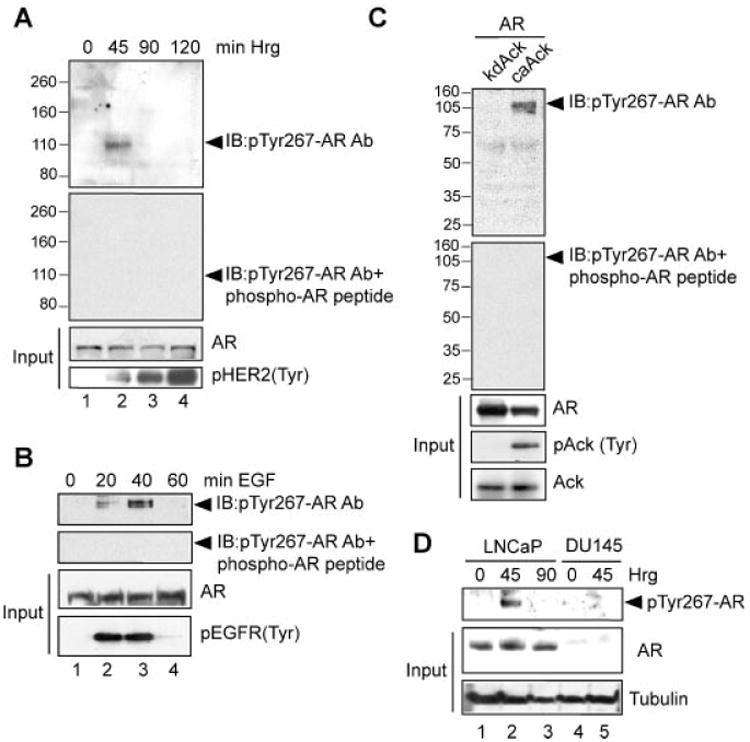
Generation of phospho-antibodies that specifically recognize pTyr267-AR. A: Serum and androgen-depleted LNCaP cells were treated with heregulin (10 ng/ml) for different time intervals and lysates were subjected to immunoblotting with pTyr267-AR antibodies (top panel) or with pTyr267-AR antibodies that were incubated with AR phospho267-peptide (second panel). B: Serumandandrogen-depleted LAPC4cells treated with EGF(10 ng/ml) for different time intervals. Lysates were immunoprecipitated with pTyr-antibodies, followed by immunoblotting with pTyr267-AR antibodies (top panel) or with pTyr267-AR antibodies that were incubated with AR phospho-peptide (second panel). C: HEK293 cells were transfected with the AR expression construct (2 μg) along with the caAck or kdAck expression construct (2 μg). Forty-eight hours after transfection lysates were immunoblotted with pTyr267-AR antibodies (top panel) or with pTyr267-AR antibodies that were incubated with AR phospho-peptide (second panel). D: Serum and androgen-depleted LNCaP and DU145 cells were treated with heregulin (10 ng/ml) for different time intervals. Lysates were immunoprecipitated with pTyr-antibodies, followed by immunoblotting with pTyr267-AR antibodies (top panel).
We have performed TMA staining with pTyr267 AR antibody, representative data is shown in Supplementary Figure 1. It demonstrates significant AR 267-phosphorylation staining in different stages of prostate cancer progression, which correlates well with pTyr284-Ack1 staining (Supplementary Fig. 1).
AR Tyr267-Phosphorylation Is Unaffected by Anti-Androgens
To assess the role Tyr267-phosphorylation of AR in determining sensitivity to antiandrogens, serum and androgen-depleted LNCaP and LAPC4 cells were treated with heregulin or EGF ligands and bicalutamide or flutamide. EGF or heregulin ligand treatment resulted in significant increase in pTyr284-Ack1 and pTyr267-AR expression which was unaffected by bicalutamide or flutamide (Fig. 3A,B, top panels).
Fig. 3.
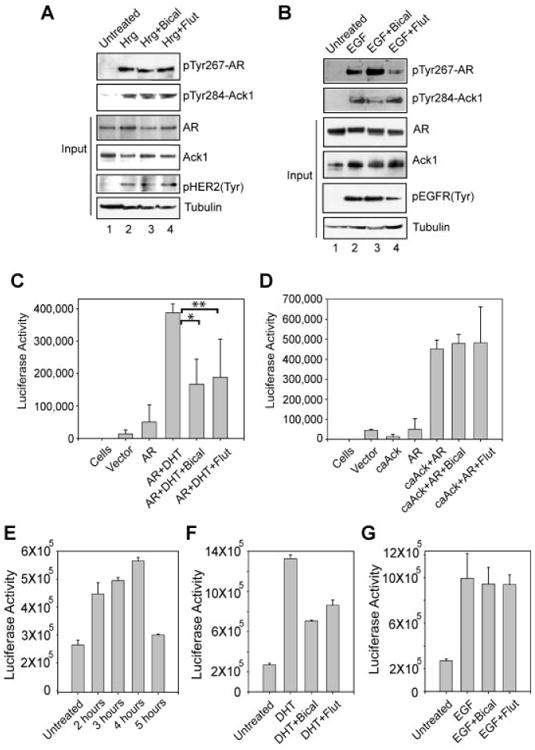
Ack1 targeted ARTyr267-phosphorylation is resistant to anti-androgens. A: Serum and androgen-depleted LNCaP cells were treated with heregulin (10 ng/ml, 45 min) and bicalutamide (1 μM, 16 hr) or flutamide (10 μM for 16 hr). Cell lysates were immunoprecipitated using pTyr267-AR antibodies, followed by immunoblotting with AR antibodies (top panel). B: Serum and androgen-depleted LAPC4 cells were treated with EGF (10 ng/ml, 40 min) and bicalutamide (1 μM, 16 hr) or flutamide (10 μM for 16 hr). Cell lysates were immunoprecipitated with pTyr-antibodies, followed by immunoblotting with pTyr267-AR antibodies (top panel). C: HEK293 cells were transfected with the ARR2PB-luciferase reporter (500 ng) and FLAG-tagged AR vector (500 ng). Twenty-four hours after transfection, cells were treated with DHT (2.5 nM, 16hr) and bicalutamide (1 μM, 16 hr) or flutamide (10 μM, 16 hr) and luciferase activity was determined. Equal expression of AR was confirmed by immunoblotting the lysates with anti-FLAG antibodies. Data are representative of three independent experiments. *P≤0.001; **P≤0.04. D: HEK293 cells were transfected with the ARR2PB-luciferase reporter (250 ng), AR vector (250 ng), and caAck1 (250 ng). Twenty-four hours after transfection, cells were treated with bicalutamide (1 μM, 16 hr) or flutamide (10 μM, 16 hr), and luciferase activity was determined. Equal expression of AR was confirmed by immunoblotting the lysates with anti-FLAG antibodies. Data are representative of three independent experiments. E: LAPC4 cells were transfected with ARR2PB-luciferase reporter construct, serum and androgen-depleted, treated with EGF (10 ng/ml) for various time points and luciferase activity was determined. F: LAPC4 cells were transfected with ARR2PB-luciferase reporter construct, serum and androgen-depleted, treated with DHT (2.5 nM, 16 hr), bicalutamide (1 μM, 16 hr), or flutamide (10 μM, 16 hr), and luciferase activity was determined. G: LAPC4 cells were transfected with ARR2PB-luciferase reporter construct, serum and androgen-depleted, treated with EGF (10 ng/ml, 4 hr), bicalutamide (1 μM, 16 hr), or flutamide (10 μM, 16 hr), and luciferase activity was determined.
To assess whether pTyr267-AR transcriptional activity is resistant to antiandrogens, luciferase assay was performed. AR-dependent reporter construct, ARR2PB-luciferase was transfected with or without AR in HEK293 cells. HEK293 cells have undetectable levels of endogenous AR expression and thus exhibit no AR activity (Fig. 3C). DHT treatment of AR expressing cells exhibited significant upregulation of AR transcription activity, which was significantly inhibited by bicalutamide or flutamide (Fig. 3C). Coexpression of AR with caAck resulted in phosphorylation of AR at pTyr267 (Fig. 2C) and transcriptional activation in the absence of DHT (Fig. 3D). In contrast to inhibition of AR transcription activity by anti-androgens, pTyr267-AR mediated transcription of AR-reporter was unaffected (Fig. 3D).
To determine optimal pTyr267-AR transcription activity, LAPC4 cells were electroporated with ARR2PB-Luc construct, treated with EGF ligand for various time points and luciferase activity was assessed. Serum and androgen-depleted LAPC4 cells treated with EGF ligand for 4 hr exhibited optimal pTyr267-AR transcriptional activity (Fig. 3E). DHT treatment of androgen-depleted LAPC4 cells lead to AR transcriptional activation and bicalutamide or flutamide treatment exhibited 47% and 35%, decrease in AR transcriptional activity, respectively (Fig. 3F). In contrast, EGF treatment of LAPC4 cells lead to pTyr267-AR transcriptional activation and bicalutamide or flutamide treatment exhibited 4.8% and 5.3%, decrease in pTyr267-AR transcriptional activity, respectively (Fig. 3G), indicating that transcriptional activity of endogenous pTyr267-AR was relatively resistant to AR antagonists.
A Small Molecule Inhibitor of Ack1 Suppressed pTyr284-Ack1 and pTyr267-AR Levels
The 4-amino-5,6-biaryl-furo[2,3-d]pyrimidine derivative 5 has recently been reported to be an ATP mimic that inhibits Ack1 kinase activity significantly [18]. The IC50 were calculated to be 0.024 μM (by in vitro kinase assay). Selectivity was further assessed by in vitro kinase assay for related non-RTK Lck, IC50 of which was calculated to be five times higher (0.122 μM) than that of Ack1. However, whether this inhibitor suppresses Ack1 kinase and its physiological substrate's activity in vivo has not yet been demonstrated. We synthesized 5,6-biaryl-furo[2,3-d]pyrimidine using standard methods (compound 5) and named it as AIM-100. To assess the potential inhibition of Ack1 activity, MEFs were treated with AIM-100 overnight. Serum-depleted MEFs were treated with EGF ligand followed by immunoblotting with pTyr284-Ack1 antibody. EGF treatment of MEFs resulted in significant increase in Ack1 activation, as seen by increase in pTyr284-Ack1 levels (Fig. 4A, lane #2, top panel). AIM-100 treatment exhibited significant decrease in Ack1 activation (Fig. 4A, lane #3, top panel). Similar downregulation of Ack1 activation by AIM-100 was seen in EGF-treated LAPC4 cells (Fig. 4B, 2nd panel) and heregulin-treated MCF-7 cells (unpublished data).
Fig. 4.
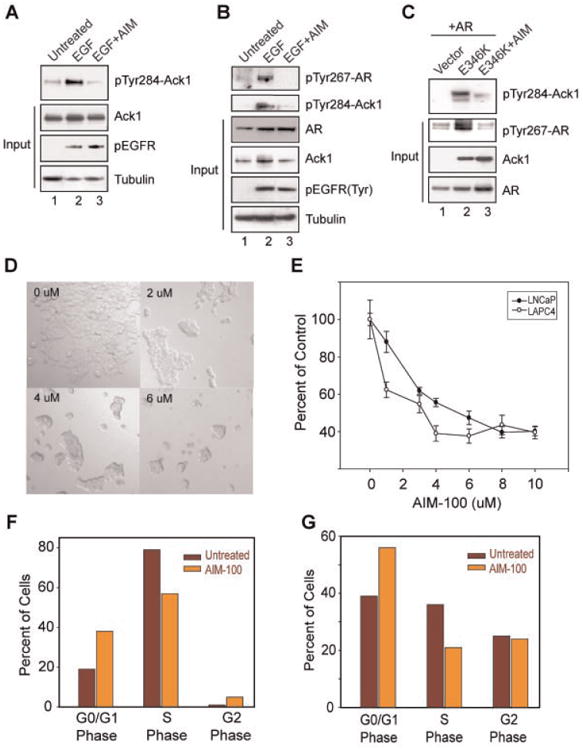
Ack1 small molecule inhibitor AIM-100 inhibited pTyr284 -Ack1 expression. A: Serum-depleted MEFs were treated (or untreated) with EGF (10 ng/ml, 10 min) and AIM-100 (0.8 μM, 16hr), and lysates were immunoblotted with pTyr284-Ack1 antibodies (top panel). B: Serum and androgen-depleted LAPC4 cells were treated (or untreated) with EGF (10ng/ml, 40 min) and AIM-100 (0.8 μM, 16 hr). Cell lysates were immunoprecipitated using pTyr antibodies, followed by immunoblotting with pTyr267-AR (top panel) and pTyr284-Ack1 antibodies (second panel). C: HEK293 cells were transfected with empty vector or Ack1 mutant E346K and Flag-tagged AR construct, treated with AIM-100 (1.6 μM, 16 hr) and whole cell lysates were immunoblotted with pTyr284-Ack1 antibodies (top panel). Cell lysates were also immunoprecipitated using pTyr antibodies, followed by immunoblotting with pTyr267-AR antibodies (second panel). D: LNCaP cells were untreated or treated with AIM-100 (2–6 μM) for 48hr and cells photographed using differential interference contrast imaging. AIM-100 treatment significantly inhibited the growth of cells. E: LNCaP and LAPC4 cells were untreated or treated with AIM-100 (1–10 μM) for 72 and 108 hr, respectively, and MTTassay was performed. Experiment was performed twice with eight replicates, a representative data set is shown. F: Cell cycle analysis by flow cytometry. LNCaP cells were untreated or treated with AIM-100 (3 μM) for 72hr. Cells were stained with propidium iodide and DNA content was measured. Experiment was performed thrice, and representative data set is shown. G: LAPC4 cells were untreated or treated with AIM-100 for 72 hr. Cells were stained with propidium iodide and DNA content was measured by flow cytometry. Experiment was performed thrice, a representative data set is shown (with 10,000 cells counted). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Recently, we identified a somatic autoactivating mutation E346K in Ack1 [13]. We generated HA-tagged E346K mutants of Ack1 and determined the effect of AIM-100 on Ack1 autoactivation. HEK293 cells transfected with E346K mutant exhibited high level of Tyr284-phosphorylated Ack1 (Fig. 4C, top panel). However, AIM-100 treatment resulted in significant downregulation of activated Ack1 (Fig. 4C, top panel), suggesting that AIM-100 is a physiological Ack1 inhibitor.
To test the effect of Ack1 inhibition on AR Tyr267-phosphorylation, LAPC4 cells were either untreated or treated with EGF and AIM-100. EGF treatment resulted in increased endogenous pTyr267-AR levels, however, AIM-100 treatment exhibited significant decrease in AR Tyr267-phosphorylation (Fig. 4B, top panel). Further, autoactivated Ack1 (E346K mutant) mediated AR Tyr267-phosphorylation was also inhibited upon AIM-100 treatment (Fig. 4C, panel 2). Taken together with earlier data, it indicates that AIM-100 is effective in repressing oncogene induced or ligand modulated AR Tyr267-phosphorylation.
AIM-100 Inhibits Prostate Cancer Cell Proliferation
To determine whether Ack1 inhibition affects cell proliferation, LNCaP cells were treated with AIM-100 for 48 hr. Differential interference contrast imaging revealed that AIM-100 treatment significantly inhibited the growth of cells (Fig. 4D). Further, MTT assay of LNCaP and LAPC4 cells exhibited significant decrease in cell growth with increasing concentrations of AIM-100 (Fig. 4E).
To determine whether this decrease in cell proliferation is due to cell cycle arrest, we performed cell cycle analysis of LNCaP and LAPC4 cells that were untreated or treated with AIM-100. Cells were stained with propidium iodide and DNA content was measured by flow cytometry. In contrast to untreated cells, AIM-100 treatment increased the proportion of cells in G0/G1 phase of cell cycle by 19% and concurrently decreased cells in S phase by 23% (Fig. 4F,G), suggesting that AIM-100 treatment results in cell cycle arrest in G0/G1 phase.
AIM-100 Suppressed pTyr267-AR Recruitment to Androgen-Regulated Gene Regulatory Sequences
To assess whether AIM-100 blocks recruitment of AR to promoters/enhancers of AR-target genes, ChIP analysis followed by qPCR of the PSA, TMPRSS2, and NKX3.1 androgen responsive enhancers (AREs) was performed. In LAPC4 cells treated with DHT, significant increase in AR binding to the PSA ARE was seen (Fig. 5A). In contrast, EGF-treated LAPC4 cells exhibited marginal increase in AR binding to the PSA ARE. However, when ChIP analysis was performed using pTyr267-AR antibodies, substantial increase in binding pTyr267-AR to PSA ARE was seen upon EGF treatment (Fig. 5B). Control ChIP experiment performed using pTyr267-AR or AR antibodies in DU145 cells that lack AR, did not exhibit any amplification of PSA ARE (data not shown). Interestingly, when cells were treated with Ack1 inhibitor AIM-100, pTyr267-AR binding to PSA ARE was significantly compromised (Fig. 5B). ChIP analysis on the GAPDH gene was performed as a negative control, which showed no recruitment of AR and pTyr267-AR to the GAPDH gene promoter (data not shown). Similar to PSA, ChIP performed using AR and pTyr267-AR antibodies followed by qPCR for TMPRSS2 and NKX3.1 AREs too exhibited pTyr267-AR binding to TMPRSS2 and NKX3.1 AREs, which was abolished upon AIM-100 treatment (Fig. 5C–F).
Fig. 5.
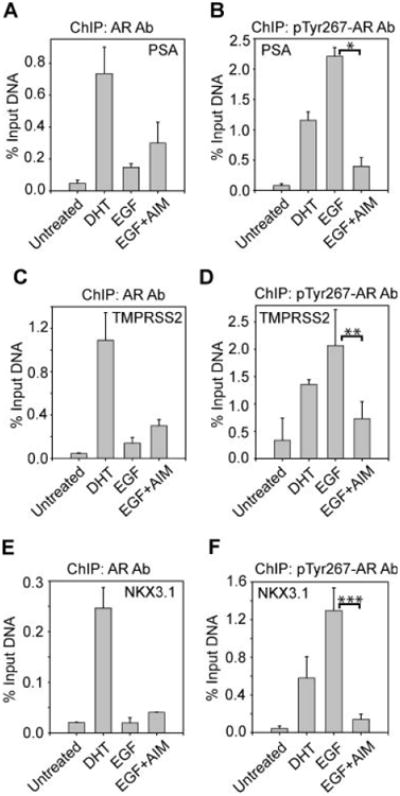
AIM-100 inhibits pTyr267-AR binding to AREs. A,C,E: Serum and androgen-depleted LAPC4 cells were treated (or untreated) with DHT (5 nM, 16 hr) or EGF (10 ng/ml, 1 hr), and AIM-100 (0.8 μM, 16 hr) and ChIP analysis for total AR binding to the PSA(A), TMPRSS2 (C), and NKX3.1 (E) AREs was performed followed by quantitative PCR. Data shown are representative of three independent experiments. B,D,F: Serum and androgen-depleted LAPC4 cells were treated with DHT (5 nM, 16 hr) or EGF (10 ng/ml, 1 hr), and AIM-100 (0.8 μM, 16 hr) and ChIP analysis for pTyr267-AR binding to the PSA (B), TMPRSS2 (D) and NKX3.1 (F) AREs was performed followed by quantitative PCR. Data shown are representative of three independent experiments. *P≤ 0.001; **P≤ 0.006; ***P≤0.014.
AIM-100 Suppressed pTyr267-AR Transcriptional Activity
Further, to probe whether Ack1-mediated AR Tyr267-phosphorylation in the absence of androgen was sensitive to AIM-100 treatment, luciferase assay was performed. DHT treatment of LAPC4 cells resulted in significant upregulation of AR luciferase activity that was unaffected by AIM-100 (Fig. 6A). In contrast, EGF treatment resulted in upregulation of AR luciferase activity that was attenuated by AIM-100 (Fig. 6B). Similarly, significant reduction in pTyr267-AR activity was observed when HEK293 cells were transfected with caAck and AR and luciferase activity was measured in presence of AIM-100 (Fig. 6C).
Fig. 6.
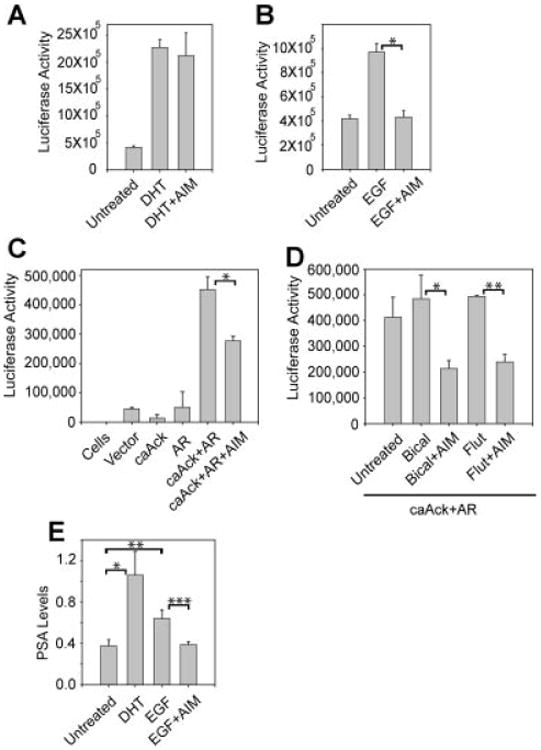
AIM-100 suppressed androgen-independent pTyr267-AR transcriptional activity and PSA gene expression. A: LAPC4 cells were transfected with the ARR2PB-luciferase reporter. Cells were serum and androgen-depleted (24 hr), treated with DHT (10 nM, 16 hr), and AIM-100 (0.8 μM, 16 hr) and luciferase activity was determined. Data are representative of three independent experiments. B: LAPC4 cells were transfected with the ARR2PB-luciferase reporter. Cells were serum and androgen-depleted (24 hr), treated with EGF (10 ng/ml, 3 hr) and AIM-100 (0.8 μM, 16 hr), and luciferase activity was determined. Data are representative of three independent experiments. *P≤0.008. C: HEK293 cells were transfected with the ARR2PB-luciferase reporter (500 ng), AR vector (500 ng) and caAck1 (500 ng). 24 hr after transfection, cells were treated with AIM-100 (1.6 μM, 16 hr) and luciferase activity was determined. Data are representative of three independent experiments. *P≤0.02. D: HEK293 cells were transfected with the ARR2PB-luciferase reporter (500 ng), AR vector (500 ng), and caAck1 (500 ng). Twenty-four hours after transfection, cells were treated with bicalutamide (1 μM, 16 hr) or flutamide (10 μM, 16 hr), and AIM-100 (1.6 μM, 16 hr) and luciferase activity was determined. Data are representative of three independent experiments. *P≤0.07, **P≤0.05. E: Serum and androgen-depleted LAPC-4 cells were treated with DHT (5 nM, 16 hr), EGF (10 ng/ml, 3 hr), and AM-100 (0.8 μM, 16 hr). Quantitative RT-PCR for PSA mRNA was performed. Data are representative of three similar independent experiments. *P≤0.009; **P≤0.06; ***P≤0.01.
We have observed that the coexpression of AR with caAck resulted in transcriptional activation of pTyr267-AR which was unaffected by antiandrogens (Fig. 3). When these cells were treated with AIM-100, significant decrease in pTyr267-AR transcriptional activation was noticed (Fig. 6D). To further determine whether Ack1 inhibition modulates pTyr267-AR target gene expression, PSA mRNA levels were measured by qRT-PCR. LAPC4 cells treated with DHT exhibited significant increase in PSA mRNA levels as compared to androgen-deprived cells (Fig. 6E). Addition of EGF stimulated PSA mRNA levels in the absence of androgens, which was significantly downregulated by AIM-100 treatment, which is comparable to untreated cells (Fig. 6E).
Discussion
Our data suggests that AR Tyr267-phosphorylation and transcriptional activation by pTyr284-Ack1 could be one potential mechanism of acquisition of resistance to antiandrogens by prostate cancer cells. Recently, two compounds RD162 and MDV3100 have been reported to bind AR with five- to eightfold greater affinity than bicalutamide [22]. These second generation antiandrogens were suggested to be better suited for treatment of CRPC because of their ability to inhibit AR binding to AREs. We have observed that AIM-100 is highly efficient in suppressing pTyr267-AR binding to various AREs, suggesting that Ack1 kinase inhibitor/s could also be as effective for treatment of AIPC as RD162 and MDV3100. To our knowledge, this is the first report wherein a non-RTK inhibitor suppressed AR binding to AREs and inhibited transcription of AR-target genes. Whether, Tyr267-phosphorylation of AR results in conformational changes that are sufficient to modulate affinity of AR LBD for antiandrogens remains to be seen.
The data in Figure 5B,D,F show that DHT increases pTyr267-AR occupancy on the PSA, TMPRSS2, and Nkx3.1 promoters. Can androgen ligands regulate pTyr267-AR phosphorylation? ChIP analysis on DHT-treated prostate cells reveals that pTyr267-AR is recruited to the ARE elements in the presence of DHT. Our unpublished data indicates that androgen ligands do not directly phosphorylate AR at Tyr267. However, the basal levels of pTyr267-AR present in LAPC4 cells seem to be efficiently recruited to ARE elements upon androgen addition. However, addition of growth factors such as EGF to LAPC4 cells increases pTyr267-AR's occupancy two- to threefold over DHT-treated cells suggesting that tyrosine phosphorylation promotes significant AR recruitment even at low concentration of androgen ligand.
Doublet bands were seen in immunoblots that were stained for p267Tyr-AR antibodies (Figs. 2B, 3B, 4C). Previously, we have demonstrated that Ack1 phosphorylates AR at 267 and 363 tyrosine residues, with 267 being the major phosphorylation site [6]. Thus, the doublet bands in the immunoblots are likely to be due to AR phosphorylation at tyrosine 267 alone or both tyrosines 267 and 363.
AR has been shown to be phosphorylated at serine residues upon EGF treatment and at Tyr534 by Src [5,23]. Although a selective Ack1 inhibitor, AIM-100 could inhibit other non-RTKs, for example, Src and Lck. Thus, AIM-100 could potentially inhibit AR transcriptional activity due to other Tyr/Ser-phosphorylations. Indeed, for the same reasons, the cell cycle arrest seen by AIM-100 treatment may not be exclusively due to Ack1 inhibition. However, contribution of other tyrosine kinases in pTyr267-ARphosphorylation, binding of pTyr267-AR to AREs and stimulation of AR transcription activity is likely to be minimal. Ack1 is the only tyrosine kinase known so far to cause robust AR Tyr267-phosphorylation, therefore, we believe that the effects of AIM-100 treatment shown here are primarily due to Ack1 inhibition.
Tyrosine kinase inhibitors (TKIs) have emerged as a new targets for treatment of several solid and hematological malignancies [24]. Although EGFR is overexpressed in prostate cancer, clinical trials showed insignificant clinical benefit in patients with CRPC [25–28]. While Src induces AR tyrosine phosphorylation at multiple sites, AZD0530, a dual Src/Abl kinase inhibitor has shown little clinical efficacy in patients with advanced CRPC [29]. Why unlike in other tumors, are TKIs so ineffective in CRPC? Chronic treatment with bicalutamide inducing overexpression of HER2 and increased AKT activity were suggested to be the main factors responsible for observed inefficacy of EGFR inhibitor, erlotinib [30]. Inhibition of one RTK may not be sufficient for prostate tumor regression. Since Ack1 is able to integrate signals from various RTKs including EGFR and HER2, an Ack1 inhibitor could block signals from multiple RTKs. Our data indicates that Ack1 inhibitor/s could have significant antitumor effects in CRPC patients.
Supplementary Material
Acknowledgments
Grant sponsor: We thank Zena Sayegh for prostate TMA construction and Alexis Lopez for reading; Laura Hall for qPCR; Jodi Kroeger and Kate Shapeland for Flow Cytometry. This work is supported by the Molecular Biology, Flow Cytometry, Tissue Core and Microscopy Facilities at Moffitt Cancer Center. N.P.M. is a recipient of Elsa U. Pardee Foundation Award, Donald A. Adam Comprehensive Melanoma Research Center Award, and Career Development Awards in Lung Cancer.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93(22):1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- 2.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23(32):8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 3.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 4.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332(21):1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 5.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, Qiu Y. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10(4):309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Earp HS, Whang YE. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104(20):8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59(11):2511–2515. [PubMed] [Google Scholar]
- 8.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 9.Joyce R, Fenton MA, Rode P, Constantine M, Gaynes L, Kolvenbag G, DeWolf W, Balk S, Taplin ME, Bubley GJ. High dose bicalutamide for androgen independent prostate cancer: Effect of prior hormonal therapy. J Urol. 1998;159(1):149–153. doi: 10.1016/s0022-5347(01)64039-4. [DOI] [PubMed] [Google Scholar]
- 10.Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65(22):10514–10523. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- 11.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, Weber MJ. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277(32):29304–29314. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- 12.Galisteo ML, Yang Y, Urena J, Schlessinger J. Activation of the nonreceptor protein tyrosine kinase Ack by multiple extracellular stimuli. Proc Natl Acad Sci USA. 2006;103(26):9796–9801. doi: 10.1073/pnas.0603714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Riviera C, Cheng JQ, Schönbrunn E, Sebti SM, Earp SH, Mahajan NP. Ack1 mediated AKT/PKB Tyrosine 176 phosphorylation regulates its activation. PLoS ONE. 2010;5(3):e1–17. doi: 10.1371/journal.pone.0009646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modzelewska K, Newman LP, Desai R, Keely PJ. Ack1 mediates Cdc42-dependent cell migration and signaling to p130Cas. J Biol Chem. 2006;281(49):37527–37535. doi: 10.1074/jbc.M604342200. [DOI] [PubMed] [Google Scholar]
- 15.Eisenmann KM, McCarthy JB, Simpson MA, Keely PJ, Guan JL, Tachibana K, Lim L, Manser E, Furcht LT, Iida J. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat Cell Biol. 1999;1(8):507–513. doi: 10.1038/70302. [DOI] [PubMed] [Google Scholar]
- 16.van der Horst EH, Degenhardt YY, Strelow A, Slavin A, Chinn L, Orf J, Rong M, Li S, See LH, Nguyen KQ, Hoey T, Wesche H, Powers S. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc Natl Acad Sci USA. 2005;102(44):15901–15906. doi: 10.1073/pnas.0508014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7(6):591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 18.DiMauro EF, Newcomb J, Nunes JJ, Bemis JE, Boucher C, Buchanan JL, Buckner WH, Cheng A, Faust T, Hsieh F, Huang X, Lee JH, Marshall TL, Martin MW, McGowan DC, Schneider S, Turci SM, White RD, Zhu X. Discovery of 4-amino-5,6-biaryl-furo[2,3-d]pyrimidines as inhibitors of Lck: Development of an expedient and divergent synthetic route and preliminary SAR. Bioorg Med Chem Lett. 2007;17(8):2305–2309. doi: 10.1016/j.bmcl.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 19.Foloppe N, Fisher LM, Howes R, Kierstan P, Potter A, Robertson AG, Surgenor AE. Structure-based design of novel Chk1 inhibitors: Insights into hydrogen bonding and protein–ligand affinity. J Med Chem. 2005;48(13):4332–4345. doi: 10.1021/jm049022c. [DOI] [PubMed] [Google Scholar]
- 20.Mahajan NP, Earp HS. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: A mechanism for localizing guanine nucleotide-exchange factor action. J Biol Chem. 2003;278(43):42596–42603. doi: 10.1074/jbc.M305817200. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama N, Miller WT. Biochemical properties of the Cdc42-associated tyrosine kinase ACK1. Substrate specificity, authphosphorylation, and interaction with Hck. J Biol Chem. 2003;278(48):47713–47723. doi: 10.1074/jbc.M306716200. [DOI] [PubMed] [Google Scholar]
- 22.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res. 2006;66(22):11047–11054. doi: 10.1158/0008-5472.CAN-06-0596. [DOI] [PubMed] [Google Scholar]
- 24.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 25.Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN, Berry S, Ernst DS, Douglas L, Brundage M, Fisher B, McKenna A, Seymour L. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: A trial of the National Cancer Institute of Canada—Clinical Trials Group. J Clin Oncol. 2005;23(3):455–460. doi: 10.1200/JCO.2005.02.129. [DOI] [PubMed] [Google Scholar]
- 26.Small EJ, Fontana J, Tannir N, DiPaola RS, Wilding G, Rubin M, Iacona RB, Kabbinavar FF. A phase II trial of gefitinib in patients with non-metastatic hormone-refractory prostate cancer. BJU Int. 2007;100(4):765–769. doi: 10.1111/j.1464-410X.2007.07121.x. [DOI] [PubMed] [Google Scholar]
- 27.Gravis G, Bladou F, Salem N, Goncalves A, Esterni B, Walz J, Bagattini S, Marcy M, Brunelle S, Viens P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann Oncol. 2008;19(9):1624–1628. doi: 10.1093/annonc/mdn174. [DOI] [PubMed] [Google Scholar]
- 28.Gross M, Higano C, Pantuck A, Castellanos O, Green E, Nguyen K, Agus DB. A phase II trial of docetaxel and erlotinib as first-line therapy for elderly patients with androgen-independent prostate cancer. BMC Cancer. 2007;7:142. doi: 10.1186/1471-2407-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lara PN, Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, Posadas E, Stadler W, Gandara DR. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: A California Cancer Consortium Study. Anticancer Drugs. 2009;20(3):179–184. doi: 10.1097/CAD.0b013e328325a867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Festuccia C, Gravina GL, Biordi L, D'Ascenzo S, Dolo V, Ficorella C, Ricevuto E, Tombolini V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate. 2009;69(14):1529–1537. doi: 10.1002/pros.20995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.