

Abstract
Ack1 (also known as ACK, TNK2, or activated Cdc42 kinase) is a structurally unique non-receptor tyrosine kinase that is expressed in diverse cell types. It integrates signals from plethora of ligand-activated receptor tyrosine kinases (RTKs), for example, MERTK, EGFR, HER2, PDGFR and insulin receptor to initiate intracellular signaling cascades. Ack1 transduces extracellular signals to cytosolic and nuclear effectors such as the protein kinase AKT/PKB and androgen receptor (AR), to promote cell survival and growth. While tyrosine phosphorylation of AR at Tyr267 regulates androgen-independent recruitment of AR to the androgen-responsive enhancers and transcription of AR target genes to drive prostate cancer progression, phosphorylation of an evolutionarily conserved Tyrosine 176 in the kinase domain of AKT is essential for mitotic progression and positively correlates with breast cancer progression. In contrast to AR and AKT, Ack1-mediated phosphorylation of the tumor suppressor Wwox at Tyr287 lead to rapid Wwox polyubiquitination followed by degradation. Thus, by its ability to promote tumor growth by negatively regulating tumor suppressor such as Wwox and positively regulating pro-survival factors such as AKT and AR, Ack1 is emerging as a critical player in cancer biology. In this review, we discuss recent advances in understanding the physiological functions of Ack1 signaling in normal cells and the consequences of its hyperactivation in various cancers.
Receptor tyrosine kinases (RTKs) respond to cues from the outside environment to activate specific programs within cells to regulate cell growth, cell proliferation, and cell differentiation. There is a wealth of information on how the ligand bound RTKs communicate specific signals by precisely activating a complex network of protein machinery through single or multiple phosphorylation events (Manning et al., 2002). Each signal may be amplified several fold and each step is tightly regulated and is kinetically controlled. Dysfunction of this signaling event is evident in the form of increased levels of effecter phosphorylation, hyperactivated pathways, enhanced cell growth, and cell proliferation. Interestingly a group of tyrosine kinases do not directly receive signals from the extracellular mileu but are rapidly activated. These proteins are referred to as the non-receptor or cytoplasmic tyrosine kinases (NRPTKs or cytTKs) (Neet and Hunter, 1996). Although NRPTKs do not bind growth factors, they appear to be critical in delivering the signals of the RTKs, as their activation is tightly regulated by the activation of the RTKs. Ack1 is one such NRPTK that was initially identified as a tyrosine kinase that specifically bound the activated form of a small G-protein, Cdcd42 (Manser et al., 1993). However recent studies have uncovered Cdc42-independent role of Ack1 in cell signaling (Mahajan et al., 2005, 2007, 2010b). This review is focused on mechanisms of Ack1 activation and its distinctive mode of regulating activities of important cellular proteins, AKT and androgen receptor (AR) in normal and cancer cells.
Ack1 Structure
Ack1 is an atypical non-receptor tyrosine kinase; its relatively large size (1,038 amino acids or ∼ 143 kDa) coupled with the presence of multiple domains distinguishes it from other nonreceptor tyrosine kinases (Fig. 1). It consists of an amino-terminal sterile α motif or SAM domain (4–70 amino acids), tyrosine kinase catalytic domain (126–385 amino acids), a SH3 domain (386–447 amino acids), a Cdc42/Rac interactive binding or CRIB domain (448–468 amino acids), a large carboxy-terminal region that contains proline-rich sequences (577–958 amino acids) and ubiquitin-association or UBA domain (963–1,026 amino acids) (Fig. 1). These multiple domains regulate various functional aspects of Ack1. While the amino-terminal SAM domain is involved in Ack1 membrane targeting, the neighboring catalytic domain possesses tyrosine kinase activity (Yokoyama and Miller, 2003; Mahajan et al., 2005, 2010b; Galisteo et al., 2006). Three-dimensional structures have been determined for the isolated Ack1 kinase domain (Lougheed et al., 2004) and the CRIB domain (Mott et al., 1999), but not for larger constructs or full-length Ack1. Crystallization of the human Ack1 kinase domain structure revealed an interesting feature; a typical kinase fold with an unusual substrate-binding cleft. The presence of the SH3 domain, carboxy-terminal to the kinase domain in Ack1 is also unusual among families of NRPTKs (Hubbard and Till, 2000; Blume-Jensen and Hunter, 2001). A point mutation in SH3 domain (W426K) resulted in enhanced tyrosine autophosphorylation, (Galisteo et al., 2006). Moreover, the isolated SH3 domain bound to Ack1 or to the isolated proline-rich region of Ack1. These results suggest that SH3 domain could play an autoinhibitory role by binding to the proline-rich region by an intramolecular mechanism, similar to the autoinhibition conferred by the other SH3 domains (Andreotti et al., 1997; Moarefi et al., 1997; Xu et al., 1997; Barila and Superti-Furga, 1998). The carboxy-terminus proline-rich domain contains a region of homology to Mig6/RALT/Gene-33 (805–879 amino acids), which acts as a generic RTK binding domain (Shen et al., 2007; Pao-Chun et al., 2009). The UBA domain binds to ubiquitin to regulate Ack1 polyubiquitination and degradation (Shen et al., 2007; Chan et al., 2009).
Fig. 1.
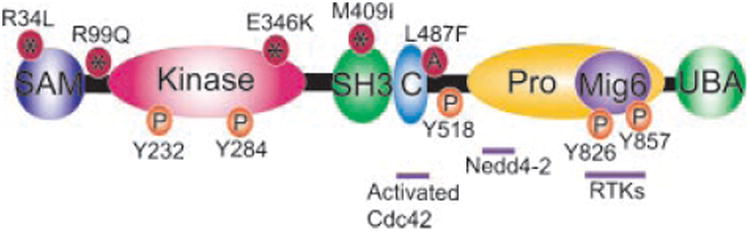
Schematic representation of Ack1 domain architecture, mutations and interaction regions. SAM, sterile alpha motif; kinase, kinase domain; SH3, Src homology domain 3; C, Cdc42/Rac interactive binding domain; Pro, proline rich domain; Mig6, Mig6 homology domain; UBA, ubiquitin association domain. Four somatic missense mutations identified in the COSMIC database are shown as red*. Tyr-phosphorylation sites are indicated in orange. Protein–protein interaction regions within Ack1 are shown.
Ack1 Interacting Proteins
Considering the important role for Ack1 in the cell signaling and its unusual peptide substrate-binding region, it is not surprising that over the past decade several Ack1 substrates have been uncovered with different physiological outcomes (Table 2). Heat shock protein 90β (Hsp90β) bound Ack1 and treatment of cells with geldanamycin, a Hsp90β inhibitor, inhibited Ack1 kinase activity and suppressed tumorigenesis (Mahajan et al., 2005), suggesting that the molecular chaperones, for example, Hsp90β are required for maintenance of optimal Ack1 tyrosine kinase activity. The carboxy-terminus of Ack1 contains a region of homology to Mig6/RALT protein, a protein known to bind and inhibit the activity of RTKs (Fiorentino et al., 2000; Zhang et al., 2007). One of the functional outcomes of Ack1–RTK interaction appears to be the modulation of levels of activated RTK levels, for example, EGFR and AXL, following stimulation with their respective ligands (Shen et al., 2007; Pao-Chun et al., 2009). Interestingly, Ack1 Mig6 homology domain possesses four tyrosine phosphorylation sites, Tyr826, Tyr857, Tyr866, and Tyr870. Mutation of Tyr358 in Mig6 abrogates binding to the EGFR kinase domain; Tyr358 corresponds to Tyr826 in Ack1. This opens up intriguing possibility that Ack1 autophosphorylation at Tyr826 (and possibly other sites) upon EGFR binding leads to transient EGFR–Ack1 complex formation. Whether EGFR directly phosphorylates Ack1 at Tyr826, Tyr857, Tyr866, and Tyr870 sites remains to be investigated. In this review three Ack1 interacting proteins AR, AKT, and Wwox have been discussed in greater detail, primarily due to the pivotal role these proteins play in the development of various cancers.
Table 2. Ack1 interacting proteins.
| Interacting proteins | Cellular function | Regulatory role | Refs. |
|---|---|---|---|
| AKT/PKB | Proto-oncogene | Phosphorylated at Tyr176 by Ack1, leading to AKT kinase activation | Mahajan et al. (2010b) |
| Androgen receptor (AR) | Steroid receptor | Phosphorylated at Tyr267 and Tyr363 leading to androgen-independent transcriptional activation | Mahajan et al. (2007, Mahajan et al. 2010a) |
| Cdc42 | GTPase | Direct binding with Ack1 lead to inhibition of Cdc42 GTPase activity | Manser et al. (1993) |
| Clathrin | Trafficking | Associates directly with heavy chain of clathrin and regulates clathrin distribution | Teo et al. (2001) |
| Dbl | Guanine nucleotide exchange factor | Association with Ack1 facilitates GEF activity | Kato-Stankiewicz et al. (2001) |
| MERTK, EGFR, PDGFR, AXL ALK, LTK | RTKs | Association with Ack1 leads to Ack1 activation | Mahajan et al. (2005), Galisteo et al. (2006), Shen et al. (2007), Howlin et al. (2008), Pao-Chun et al. (2009) |
| Grb-2 | Adapter protein | Association with proline-rich domain of Ack1 | Galisteo et al. (2006) |
| Hck | Non-receptor tyrosine kinase | Hck SH3 domain interacts with the proline-rich region of Ack1 | Yokoyama and Miller (2003) |
| HSH2 | Adaptor protein | Associates with proline-rich region of Ack1 and regulates cytokine signaling | Oda et al. (2001) |
| HSP90β | Chaperone | Binds to Ack1 and maintains Ack1 kinase conformation | Mahajan et al. (2005) |
| MCSP: melanoma chondriotin sulfate proteoglycan | Cell surface antigen | MCSP regulates cell spreading through Ack1 and p130cas | Eisenmann et al. (1999) |
| Nck | Adaptor protein | Association with Ack1 | Galisteo et al. (2006) |
| Nedd4-1 and Nedd4-2 | E3-ubiquitin ligase | Ubiquitination/degradation of Ack1 | Chan et al. (2009); Lin et al. (2010) |
| Nephrocystin | Epithelial cell organization | Partially colocalizes with Ack1 at cell to cell contacts | Eley et al. (2008) |
| P130Cas | Adaptor molecule | Ack1 associates with p130Cas via SH3 domain, phosphorylates it at Tyr165, 249, and 410, promotes cell migration | Modzelewska et al. (2006) |
| PTPN12, PTPRJ, and PTPRC | Tyrosine phosphatase | Association with Ack1 | Barr et al. (2009) |
| SNX9 | Vesicle dynamics | Binds to Ack1 (residues 920–955) | Yeow-Fong et al. (2005) |
| WASP: Wiskott-Aldrich syndrome protein | Cdc42 effecter | Phosphorylated at Ser242, Tyr256 | Yokoyama et al. (2005) |
| Wwox | Tumor suppressor | Phosphorylated at Tyr287 by Ack1 leading to polyubiquitination and degradation | Mahajan et al. (2005) |
Ack1 Signaling
Purification of tyrosine–phosphoprotein complexes from prostate cancer derived cells treated with Gas6 ligand followed by mass spectrometry first led to the identification of Ack1 as a crucial downstream target of the RTK, MERTK (Mahajan and Earp, 2003; Mahajan et al., 2005). Subsequently it was observed that Ack1 can integrate signals from a variety of RTKs in different cell types (Mahajan et al., 2005, 2007, 2010a, 2010b; Galisteo et al., 2006; Shen et al., 2007; Pao-Chun et al., 2009). Ack1 is recruited to the plasma membrane as a consequence of association with membrane bound activated RTKs which allows it to undergo autophosphorylation at Tyr284 in the activation loop and activation (Mahajan et al., 2005, 2010b). Tyr284 is highly conserved and mutation of this tyrosine to phenylalanine significantly reduces the levels of tyrosine phosphorylation in vivo and kinase activation, suggesting it to be the primary autophosphorylation site (Yokoyama and Miller, 2003; Mahajan et al., 2005).
Ack1 activity is regulated by at least three different mechanisms; (a) Growth factor bound RTKs or RTKs amplification/autoactivation facilitating Ack1 activation; (b) Ack1 gene amplification causing Ack1 autoactivation, and (c) Somatic autoactivating mutations in Ack1 leading to Ack1 activation. Details of growth-factor-mediated Ack1 activation is described above. Ack1 gene amplification has been detected in lung, breast, and prostate cancers (van der Horst et al., 2005). In humans, Ack1 gene is located on chromosome 3q29; array CGH analysis combined with real-time PCR of relative DNA copy number along chromosome 3 revealed a distinct amplification of the region containing Ack1 gene (van der Horst et al., 2005). While growth factors binding to RTKs or gene amplification lead to Ack1 kinase activation have been well-studied mechanisms of Ack1 activation, somatic autoactivating mutations in Ack1 were not identified. Recently four somatic missense mutations and two nonsense mutations have been identified in Sanger COSMIC database (shown in Table 1 and Fig. 1). Of these, the E346K mutation in the kinase domain, found in endometrioid carcinoma of the Ovary autoactivates Ack1 (Mahajan et al., 2010b). E346K point mutant not only undergoes autoactivation in serum starved cells, that is, in the absence of RTK signaling, but is also able to phosphorylate Ack1 effectors, for example, AKT and AR (Mahajan et al., 2010a,b).
Table 1. Somatic mutations in Ack1.
| Mutation | Type of mutation | Tissue |
|---|---|---|
| R34L | Missense | Lung, adenocarcinoma |
| R99Q | Missense | Ovary, mucinous carcinoma |
| E346K | Missense | Ovary, endometrioid carcinoma |
| M409I | Missense | Stomach, carcinoma |
| W75* | Nonsense | Brain, glioma |
| R84* | Nonsense | Brain, glioma |
Cells with ligand activated RTKs display transient Ack1 Tyr-phosphorylation, however, almost complete loss of Ack1 Tyr-phosphorylation occurs soon after the peak phosphorylation suggesting that Ack1 activity is tightly regulated in cells by its dephosphorylation (Mahajan et al., 2005, 2007, 2010b). Three tyrosine–protein phosphatases, PTPN12 (PTP-PEST), PTPRJ (DEP1 or receptor-type tyrosine–protein phosphatase eta) and PTPRC (CD45) have recently been suggested to be involved in Ack1 Tyr518-dephosphorylation (Barr et al., 2009). Since Tyr518 is not a primary phosphorylation site, the extent by which it downregulates Ack1 activity is not known. Whether one or all of these three tyrosine–protein phosphatases could dephosphorylate Ack1 at Tyr284, a primary activation site and downregulate Ack1 kinase activity remains to be seen.
While peak Ack1 Tyr-phosphorylation is detected within 5–10 min of EGF treatment, a significant decrease in Ack1 protein levels is observed by 40 min, suggesting that Ack1 levels within cells may be regulated by its degradation in phosphorylation-dependent manner (Mahajan et al., 2010b). Investigation into the mechanism(s) of Ack1 degradation has lead to the identification of two new Ack1-binding partners, the E3 ubiquitin ligases Nedd4-2 and Nedd4-1 (Chan et al., 2009; Lin et al., 2010). A conserved PPXY-containing region in Ack1 (632–639 amino acids) binds to Nedd4-related proteins (Chan et al., 2009) and several other WW domain-containing proteins, including a well-characterized Ack1-interacting tumor suppressor, Wwox (Mahajan et al., 2005). It was observed that EGFR-mediated Ack1 activation allowed Nedd4-2 and Nedd4-1 to promote Ack1 polyubiquitination and degradation (Chan et al., 2009; Lin et al., 2010). However, the ubiquitination sites in Ack1 targeted by Nedd4-1 or Nedd4-2 ligases have not yet been identified. Further, it is imperative to determine the Ack1 tyr-phosphorylation site(s) critical for Ack1 ubiquitination and degradation.
Modulation of AR Activity by Ack1
Although, Ack1 was initially identified as a soluble cytoplasmic tyrosine kinase, its recruitment at the plasma membrane and its role in regulating membrane associated proteins or its compartmentalization within the nucleus to influence nuclear proteins has acquired considerable significance in recent time. RTKs mediated Ack1 activation results in translocation of Ack1 to the nucleus, wherein it phosphorylates AR, a nuclear hormone (Mahajan et al., 2007). AR is essential for normal as well as malignant prostate cell growth and is regulated by the binding of its ligand, androgen, or testosterone (Feldman and Feldman, 2001; Edwards and Bartlett, 2005). Ligand-bound AR forms homo-dimer, translocates to the nucleus and initiates transcription by binding to the androgen-response elements (ARE) in the promoter/enhancer regions of target genes. After binding to DNA, AR/ligand complex recruits RNA polymerase machinery, as well as coactivators to promote transcription of AR regulated genes. AR plays a crucial role in the development of advanced metastatic prostate cancer also known as androgen independent prostate cancer or AIPC (Grossmann et al., 2001; Chen et al., 2004). Since AIPC cells thrive under low circulating levels of androgen in the presence of functional AR, it was proposed that post-translational modification of AR, for example, phosphorylation of AR at specific sites may be required for androgen independence (Culig et al., 1994; Craft et al., 1999; Wen et al., 2000).
In spite of the widely accepted role of AR phosphorylation in androgen-independent growth of prostate cells, the kinase(s) that activate AR were unknown. Recently, we have demonstrated that Ack1 phosphorylates AR and thus unraveled the molecular basis for interplay between Ack1/AR signaling in the progression of prostate cancer (Mahajan et al., 2007). Activated Ack1 phosphorylated AR protein at Tyr-267 and Tyr-363 located within the transactivation domain and promoted AR recruitment to the AREs (Mahajan et al., 2007, Mahajan et al., 2010a). Recruitment of Ack1/AR complex at AREs resulted in androgen-inducible gene expression in the absence of androgen which promoted androgen-independent growth of prostate xenograft tumors. Tyrosine 267 appears to be the primary phosphorylation site as mutation at this residue significantly impaired Ack1-induced AR activity (Mahajan et al., 2007). Consistent with this observation, the Ack1 inhibitor, AIM-100, was able to suppress pTyr267-AR phosphorylation (Mahajan et al., 2010a). Further, in ∼40% of primary AIPC samples, pTyr-AR expression was correlated with pTyr-Ack1. Neither was elevated in androgen-dependent prostate cancer or benign prostate samples (Mahajan et al., 2007). Human prostate TMA analysis demonstrated that the expression levels of pTyr284-Ack1 and pTyr267-AR were significantly increased as disease progressed to AIPC stages (Mahajan et al., 2010a). Further, patients with high expression of pTyr284-Ack1 were at a higher risk for cancer-related deaths (Mahajan et al., 2010a,b). Collectively, these evidences suggest that Ack1 activated by surface signals or oncogenic mechanisms directly enhance AR transcriptional function and promotes progression of prostate cancer to androgen-independent stage.
AKT Activation by Ack1
Protein kinase AKT is one of the most potent pro-survival signaling molecule that is aberrantly activated in a variety of human cancers (Manning and Cantley, 2007). AKT activation occurs when ligand binding to RTK facilitates translocation of AKT to the plasma membrane (Burgering and Coffer, 1995; Franke et al., 1995; Stokoe et al., 1997; Stephens et al., 1998) where it is phosphorylated at Thr308 and at Ser473 (Dong and Liu, 2005; Sarbassov et al., 2005) leading to its kinase activation (Alessi et al., 1996). While RTK/PI3K/AKT signaling cascade is understood to a broader extent, new evidences are emerging which indicates that AKT activation can occur in PI3K-independent fashion (Sun et al., 2001; Hennessy et al., 2005; Gami et al., 2006; Zhao et al., 2006; Carpten et al., 2007; Stemke-Hale et al., 2008). For instance, about a third of the breast and prostate tumors and large majority of the pancreatic tumors that exhibit AKT activation, retain normal PTEN and PI3K activity. The molecular mechanism of AKT activation in these tumors that harbor normal PTEN and PI3K activity is not clear (Tibes et al., 2008).
Recently, we have demonstrated a new signaling mechanism wherein RTK/Ack1 signaling directly regulates AKT function. Ack1 and AKT exist as a complex even in the absence of growth factors. However, upon activation of RTKs by growth factors, Ack1 is activated which in turn directly phosphorylates AKT at evolutionary conserved Tyrosine 176. AKT Tyr176-phosphorylation is unaffected by LY294002, a PI3K inhibitor, thus opening an intriguing possibility of PI3K-independent AKT activation in tumors that is mediated by activated Ack1 (Mahajan et al., 2010b). Tyr176, located in the kinase domain, is not only evolutionarily conserved from unicellular eukaryotes to mammals but also within all the three AKT isoforms. RTKs activated by their respective ligands, for example, EGF, insulin and heregulin resulted in Tyr176-phosphorylation of AKT which facilitated AKT translocation to the plasma membrane leading to AKT activation. Depletion of Ack1 abrogated AKT Tyr176-phosphorylation, plasma membrane localization and activation suggesting that Ack1 is a key intermediate signaling entity necessary for RTKs mediated AKT Tyr176-phosphorylation, membrane localization and thus activation (Mahajan et al., 2010b). Interestingly, in contrast to AKT which bound PIP3, the Tyr176-phosphorylated AKT bound another membrane phospholipid, phosphatidic acid (PA) which explains AKT activation in the presence of a PI3K inhibitor (Mahajan et al., 2010b).
FoxO subgroup of transcription factors are phosphorylated by AKT resulting in relocalization of FoxO proteins from the nucleus to cytoplasm, preventing transactivation of FoxO-target genes (Greer and Brunet, 2005; Huang and Tindall, 2007; Manning and Cantley, 2007). FoxO proteins regulate genes involved in cell cycle arrest, cell death and DNA repair. Ack1-mediated AKT Tyr176-phosphorylation resulted in translocation of Ack1/AKT complex to the nucleus. In the nucleus, Tyr176-phosphorylated AKT phosphorylated FoxO3a and suppressed expression of apoptotic genes and promoted mitotic progression (Mahajan et al., 2010b). Consistent with this observation, Ack1 depletion by siRNA lead to 2.5-fold increase in apoptosis (MacKeigan et al., 2005). Collectively, these data indicate that Ack1 kinase is critical for cell survival.
Ack1 as “PDK3”
Ack1/AKT signaling nexus has became highly relevant in breast cancers (Mahajan et al., 2010b). The expression levels of pTyr284-Ack1 and pTyr176-AKT were significantly increased as breast cancer progressed to metastatic stage. The expression levels of pTyr284-Ack1 were also significantly increased as prostate cancer progressed to metastatic stage (Mahajan et al., 2010a). These tumors exhibit significant correlation in their expression suggesting that upon activation of Ack1, AKT was Tyr176-phosphorylated in situ contributing in tumor progression. A significant outcome of these studies was the observation that those patients that exhibited higher expressions of pTyr284-Ack1 and pTyr176-AKT were at a higher risk for breast and prostate cancer-related deaths (Mahajan et al., 2010a,b). AKT is frequently activated in pancreatic cancer which has been shown to be highly correlated with HER-2/neu overexpression (Schlieman et al., 2003). We noticed that breast tumors of MMTV-neu mice exhibit significantly higher levels of pTyr284-Ack1 and pTyr176-AKT but have normal PTEN levels (unpublished data). Thus, tumors that possess somatic autoactivating mutations or amplification in Ack1 kinases could use PI3K/PTEN-independent mechanisms for AKT activation to promote cell growth and proliferation (Fig. 2). Further, Tyr176-phosphorylation is sufficient for AKT membrane localization followed by PDK1/PDK2-mediated activation, which defines Ack1 kinase activity as “PDK3” (Fig. 2). Collectively, these evidences points out that cells have devised multiple and possibly mutually exclusive mechanisms to activate AKT. Since a fraction of AKT employs this alternative mode of activation in normal and prominently in cancerous cells, these data would shed new light on the quest for understanding the mechanism of AKT inhibition in cancer cells by small molecule inhibitors.
Fig. 2.
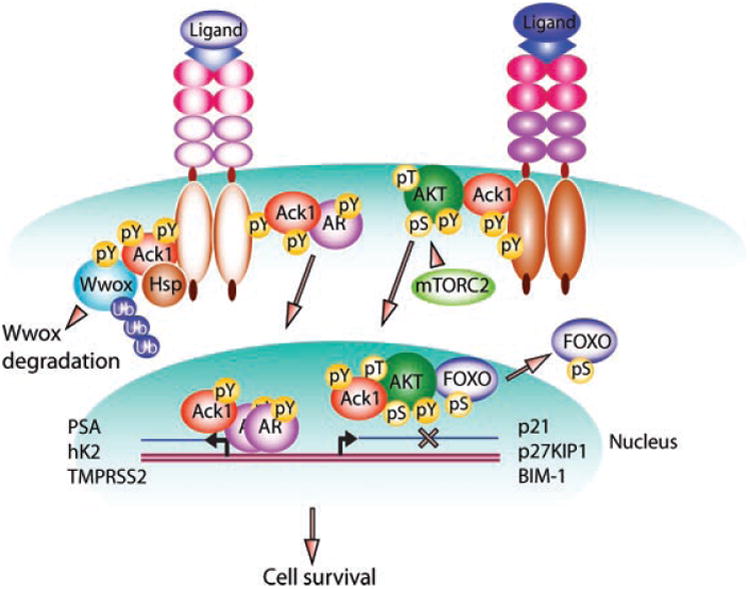
Ack1 signaling pathway in normal and cancerous cells. Ack1 is rapidly activated in normal and cancerous cells following RTK activation. In tumors, Ack1 gene amplification and autoactivating somatic mutations, E346K, can also lead to its activation. Ack1 phosphorylates AKT, at Tyr176 which facilitates AKT kinase activation and transcriptional suppression of FoxO-target genes, involved in cell cycle arrest, cell death, and DNA repair. Ack1-mediated AR activation is shown in the middle. Activated Ack1 phosphorylates AR at Tyr267 and Tyr363. Tyrosine phosphorylated AR translocates to the nucleus and transcribes AR-regulated genes in the absence of androgen. Regulation of the tumor suppressor, Wwox is shown to left. Activated Ack1 phosphorylates Wwox at Tyr287; Tyr287-phosphorylated Wwox is polyubiquitinated and degraded. Combined effect of Ack1 activation is cell survival and neoplasia, as seen in Ack1 transgenic mouse.
Identification of Ack1/AKT signaling pathway could be of considerable importance to researchers working in the field of cancer biology, cell metabolism, lipid signaling and drug discovery. Large numbers of tumors are reliant upon AKT activation for survival and growth making it an attractive target for molecular therapeutics (Cheng et al., 2005). The assay that was used routinely during the development of AKT inhibitors was primarily based on AKT Ser473-phosphorylation. Identification of RTK/Ack1/AKT signaling nexus and availability of pTyr176-AKT antibodies will allow researchers to identify a new class of AKT inhibitors based on AKT Tyr176-phosphorylation and activation. These novel inhibitors that block AKT membrane localization and activation could have major implications in various cancers and cell metabolism related disorders, for example, diabetes and obesity.
Mouse Models for Ack1
A transgenic mouse model expressing activated Ack1 (L487F autoactivating mutation) under the control of prostate-specific Probasin promoter has recently been developed (Mahajan et al., 2010b). Ack1 transgenic mice that harbor normal PTEN exhibit Ack1/AKT activation resulting in FoxO3a phosphorylation specifically in the prostate gland. These transgenic mice developed prostatic intraepithelial hyperplasia by 22 weeks and prostatic intraepithelial neoplasia or mPINs by ∼44 weeks. Formation of PINs indicates that Ack1-mediated AKT activation could be an earlier event in this neoplastic progression as an alternative to PTEN loss observed in mouse models of prostate cancer (Blanco-Aparicio et al., 2007). It would be interesting to investigate whether Ack1 transgenic mice develop PINs upon castration, that is, testosterone depletion. Further, these mice would be an excellent model to test whether Ack1 specific inhibitors can mitigate neoplasia of the castration-resistant prostate cells.
Regulation of Tumor Suppressor Wwox by Ack1
High frequency of loss of heterozygosity of chromosome 16q23 is observed in prostate, breast, ovarian, and other cancers (Carter et al., 1990; Bednarek et al., 2000). Wwox gene, composed of nine exons, has been mapped to 16q23 region (Bednarek et al., 2000). Wwox, a 46-kDa protein, possessing two NH2-terminal WW domains and a COOH-terminal short chain alcohol dehydrogenase (ADH) domain, is found in adult hormonally regulated tissues, such as testis, ovary, and prostate (Bednarek et al., 2000). The homozygous knockout mice spontaneously developed osteosarcomas in juvenile while adult heterozygotes developed lung papillary carcinoma, suggesting that Wwox is a tumor suppressor (Aqeilan et al., 2007). In a high-throughput screen in mice for mutations collaborating with either p53 or p19ARF deficiency, Wwox was found to be disrupted by seven intragenic insertions and deleted within 10 of the human cell lines (Uren et al., 2008).
In addition to deletion or rearrangement of tumor suppressor genes, loss of protein expression may also play a role in cancer development. Wwox protein expression is lost or reduced in 59–63% of invasive breast carcinomas (Guler et al., 2004; Nunez et al., 2005). Similarly, loss of Wwox protein was observed in 65% of gastric adenocarcinoma and 33% of gastric cancer cell lines, indicating that high-grade tumors were likely to exhibit lower levels of Wwox protein or lack the expression entirely. The mechanistic details of loss of Wwox expression became first evident when Wwox was identified to be an Ack1 interacting protein (Mahajan et al., 2005). Ack1 phosphorylated Wwox at Tyr287 which was shown to be prerequisite for Wwox polyubiquitination and degradation (Mahajan et al., 2005). Moreover, AIPC tumors display increased tyrosine phosphorylated Ack1 and decreased Wwox, suggesting that activated Ack1 regulates Wwox by tyrosine phosphorylation, polyubiquitination, and degradation (Fig. 2). Tyr287-phosphorylation site in Wwox was later confirmed in a global survey of tyrosine kinase signaling performed using phosphoproteomics approach (Rikova et al., 2007). This approach also identified a second phosphorylation site at Tyr293. Earlier mutation analysis had indicated that Y293F mutant does not exhibit dramatic decrease in Wwox tyrosine phosphorylation as seen in the case of Y287F mutant (Mahajan et al., 2005). Taken together, these data suggests that Tyr287 is the primary and Tyr293 is the secondary phosphorylation site in Wwox. More studies are needed to fully understand the role of Ack1-mediated Wwox phosphorylation and subsequent degradation. For example, the site(s) at which Wwox is ubiquitinated is not yet known and the identity of E3 ubiquitin ligase needed for Wwox ubiquitination is also not clear.
Ack1 Inhibitors
While tyrosine kinase inhibitors or TKIs have emerged as new drugs for treatment of variety of tumors, TKIs display limited efficacy in certain cancers, including prostate cancer (Canil et al., 2005; Krause and Van Etten, 2005; Gross et al., 2007; Small et al., 2007; Gravis et al., 2008). Long-term treatment with AR antagonist, bicalutamide, induced overexpression of HER2 and increased AKT activity which was suggested to be the main factor responsible for EGFR inhibitor erlotinib inefficacy (Festuccia et al., 2009). These data indicate that inhibition of one RTK may not be sufficient for the most tumor regression. Since Ack1 is able to integrate signals from various RTKs, Ack1 inhibitors could effectively block signal from multiple RTKs and thus would have significant antiproliferative effects in breast, ovarian, pancreatic, lung, and prostate cancer patients.
Elucidation of Ack1's role in various cancers lead to screening efforts that resulted in identification of a small molecule inhibitor of Ack1, 4-amino-5,6-biaryl-furo[2,3-d]pyrimidine (DiMauro et al., 2007). Although, 4-amino-5,6-biaryl-furo[2,3-d]pyrimidine was shown to inhibit Ack1 kinase activity selectively over Lck (DiMauro et al., 2007), whether this inhibitor could suppresses activity of Ack1 kinase and its physiological substrate in vivo was not clear. Recently, this inhibitor, renamed as AIM-100, was shown to suppress both Ack1 Tyr284- and AR Tyr267-phosphorylations (Mahajan et al., 2010a). Significant downregulation of AR Tyr267-phosphorylation blocked its recruitment to promoters/enhancers of AR-target genes, for example, PSA, TMPRSS2, and NKX3.1. Further, AIM-100 inhibited the growth of prostate cells by causing cell cycle arrest in G0/G1 phase suggesting that targeting Ack1 may be an unique therapeutic strategy to inhibit androgen-independent AR activity (Mahajan et al., 2010a). Ability of AIM-100 to inhibit autoactivated Ack1 (E346K mutant) further indicates that it is effective in repressing both oncogene induced or ligand modulated Ack1 Tyr284- and AR Tyr267-phosphorylations (Mahajan et al., 2010a).
Conclusion
Ack1 is emerging as a major non-receptor tyrosine kinase that is activated in a variety of cancers. It regulates activity of a number of proteins by tyrosine phosphorylation especially proteins critical for cell survival, cell growth, and proliferation. Its downstream effecters include many important cell survival proteins, for example, AKT, AR, and Wwox. To date Ack1 inhibitors have not been tested in clinical trials. The development of mouse model of activated Ack1 will facilitate the rapid screening of these compounds before they can be tested in cancer patients.
Acknowledgments
This work is supported by Donald A. Adam Comprehensive Melanoma Research Center Award, Career Development Awards by Moffitt Lung Cancer SPORE and Miles for Moffitt award to NPM.
Contract grant sponsor: Moffitt Lung Cancer Spore; Contract grant number: 10-15493-02-11.
Literature Cited
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS, Lian JB, Croce CM. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci USA. 2007;104:3949–3954. doi: 10.1073/pnas.0609783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barila D, Superti-Furga G. An intramolecular SH3-domain interaction regulates c-Abl activity. Nat Genet. 1998;18:280–282. doi: 10.1038/ng0398-280. [DOI] [PubMed] [Google Scholar]
- Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000;60:2140–2145. [PubMed] [Google Scholar]
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN, Berry S, Ernst DS, Douglas L, Brundage M, Fisher B, McKenna A, Seymour L. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: A trial of the National Cancer Institute of Canada-Clinical Trials Group. J Clin Oncol. 2005;23:455–460. doi: 10.1200/JCO.2005.02.129. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Carter BS, Ewing CM, Ward WS, Treiger BF, Aalders TW, Schalken JA, Epstein JI, Isaacs WB. Allelic loss of chromosomes 16q and 10q in human prostate cancer. Proc Natl Acad Sci USA. 1990;87:8751–8755. doi: 10.1073/pnas.87.22.8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W, Tian R, Lee YF, Sit ST, Lim L, Manser E. Down-regulation of active ACK1 is mediated by association with the E3 ubiquitin ligase Nedd4-2. J Biol Chem. 2009;284:8185–8194. doi: 10.1074/jbc.M806877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- DiMauro EF, Newcomb J, Nunes JJ, Bemis JE, Boucher C, Buchanan JL, Buckner WH, Cheng A, Faust T, Hsieh F, Huang X, Lee JH, Marshall TL, Martin MW, McGowan DC, Schneider S, Turci SM, White RD, Zhu X. Discovery of 4-amino-5,6-biaryl-furo[2,3-d]pyrimidines as inhibitors of Lck: Development of an expedient and divergent synthetic route and preliminary SAR. Bioorg Med Chem Lett. 2007;17:2305–2309. doi: 10.1016/j.bmcl.2007.01.057. [DOI] [PubMed] [Google Scholar]
- Dong LQ, Liu F. PDK2: The missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am J Physiol Endocrinol Metab. 2005;289:E187–E196. doi: 10.1152/ajpendo.00011.2005. [DOI] [PubMed] [Google Scholar]
- Edwards J, Bartlett JM. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 1: Modifications to the androgen receptor. BJU Int. 2005;95:1320–1326. doi: 10.1111/j.1464-410X.2005.05526.x. [DOI] [PubMed] [Google Scholar]
- Eisenmann KM, McCarthy JB, Simpson MA, Keely PJ, Guan JL, Tachibana K, Lim L, Manser E, Furcht LT, Iida J. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat Cell Biol. 1999;1:507–513. doi: 10.1038/70302. [DOI] [PubMed] [Google Scholar]
- Eley L, Moochhala SH, Simms R, Hildebrandt F, Sayer JA. Nephrocystin-1 interacts directly with Ack1 and is expressed in human collecting duct. Biochem Biophys Res Commun. 2008;371:877–882. doi: 10.1016/j.bbrc.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Festuccia C, Gravina GL, Biordi L, D'Ascenzo S, Dolo V, Ficorella C, Ricevuto E, Tombolini V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate. 2009;69:1529–1537. doi: 10.1002/pros.20995. [DOI] [PubMed] [Google Scholar]
- Fiorentino L, Pertica C, Fiorini M, Talora C, Crescenzi M, Castellani L, Alema S, Benedetti P, Segatto O. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Mol Cell Biol. 2000;20:7735–7750. doi: 10.1128/mcb.20.20.7735-7750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Galisteo ML, Yang Y, Urena J, Schlessinger J. Activation of the nonreceptor protein tyrosine kinase Ack by multiple extracellular stimuli. Proc Natl Acad Sci USA. 2006;103:9796–9801. doi: 10.1073/pnas.0603714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gami MS, Iser WB, Hanselman KB, Wolkow CA. Activated AKT/PKB signaling in C. elegans uncouples temporally distinct outputs of DAF-2/insulin-like signaling. BMC Dev Biol. 2006;6:45. doi: 10.1186/1471-213X-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravis G, Bladou F, Salem N, Goncalves A, Esterni B, Walz J, Bagattini S, Marcy M, Brunelle S, Viens P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann Oncol. 2008;19:1624–1628. doi: 10.1093/annonc/mdn174. [DOI] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Gross M, Higano C, Pantuck A, Castellanos O, Green E, Nguyen K, Agus DB. A phase II trial of docetaxel and erlotinib as first-line therapy for elderly patients with androgen-independent prostate cancer. BMC Cancer. 2007;7:142. doi: 10.1186/1471-2407-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93:1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, McCue P, Huebner K. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100:1605–1614. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Howlin J, Rosenkvist J, Andersson T. TNK2 preserves epidermal growth factor receptor expression on the cell surface and enhances migration and invasion of human breast cancer cells. Breast Cancer Res. 2008;10:R36. doi: 10.1186/bcr2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Kato-Stankiewicz J, Ueda S, Kataoka T, Kaziro Y, Satoh T. Epidermal growth factor stimulation of the ACK1/Dbl pathway in a Cdc42 and Grb2-dependent manner. Biochem Biophys Res Commun. 2001;284:470–477. doi: 10.1006/bbrc.2001.5004. [DOI] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- Lin Q, Wang J, Childress C, Sudol M, Carey DJ, Yang W. HECT E3 ubiquitin ligase Nedd4-1 ubiquitinates ACK and regulates EGF-induced degradation of EGFR and ACK. Mol Cell Biol. 2010;30:1541–1554. doi: 10.1128/MCB.00013-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lougheed JC, Chen RH, Mak P, Stout TJ. Crystal structures of the phosphorylated and unphosphorylated kinase domains of the Cdc42-associated tyrosine kinase ACK1. J Biol Chem. 2004;279:44039–44045. doi: 10.1074/jbc.M406703200. [DOI] [PubMed] [Google Scholar]
- MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Earp HS. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: A mechanism for localizing guanine nucleotide-exchange factor action. J Biol Chem. 2003;278:42596–42603. doi: 10.1074/jbc.M305817200. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65:10514–10523. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Earp HS, Whang YE. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104:8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Challa S, Coppola D, Lawrence H, Luo Y, Gevariya H, Zhu W, Chen YA, Lawrence NJ, Mahajan NP. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate. 2010a doi: 10.1002/pros.21163. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan KN, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, Rebecca S, Muraoka-Cook RS, Cheng JQ, Schönbrunn E, Sebti SM, Earp SH, Mahajan NP. Ack1 mediated AKT/PKB Tyrosine 176 Phosphorylation Regulates its Activation. PLoS ONE. 2010b;5:e9646. doi: 10.1371/journal.pone.0009646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Tan L, Lim L. A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature. 1993;363:364–367. doi: 10.1038/363364a0. [DOI] [PubMed] [Google Scholar]
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Modzelewska K, Newman LP, Desai R, Keely PJ. Ack1 mediates Cdc42-dependent cell migration and signaling to p130Cas. J Biol Chem. 2006;281:37527–37535. doi: 10.1074/jbc.M604342200. [DOI] [PubMed] [Google Scholar]
- Mott HR, Owen D, Nietlispach D, Lowe PN, Manser E, Lim L, Laue ED. Structure of the small G protein Cdc42 bound to the GTPase-binding domain of ACK. Nature. 1999;399:384–388. doi: 10.1038/20732. [DOI] [PubMed] [Google Scholar]
- Neet K, Hunter T. Vertebrate non-receptor protein-tyrosine kinase families. Genes Cells. 1996;1:147–169. doi: 10.1046/j.1365-2443.1996.d01-234.x. [DOI] [PubMed] [Google Scholar]
- Nunez MI, Ludes-Meyers J, Abba MC, Kil H, Abbey NW, Page RE, Sahin A, Klein-Szanto AJ, Aldaz CM. Frequent loss of WWOX expression in breast cancer Correlation with estrogen receptor status. Breast Cancer Res Treat. 2005;89:99–105. doi: 10.1007/s10549-004-1474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Muramatsu MA, Isogai T, Masuho Y, Asano S, Yamashita T. HSH2: A novel SH2 domain-containing adapter protein involved in tyrosine kinase signaling in hematopoietic cells. Biochem Biophys Res Commun. 2001;288:1078–1086. doi: 10.1006/bbrc.2001.5890. [DOI] [PubMed] [Google Scholar]
- Pao-Chun L, Chan PM, Chan W, Manser E. Cytoplasmic ACK1 interaction with multiple receptor tyrosine kinases is mediated by Grb2: An analysis of ACK1 effects on Axl signaling. J Biol Chem. 2009;284:34954–34963. doi: 10.1074/jbc.M109.072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Lin Q, Gu Y, Childress C, Yang W. Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol Biol Cell. 2007;18:732–742. doi: 10.1091/mbc.E06-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small EJ, Fontana J, Tannir N, DiPaola RS, Wilding G, Rubin M, Iacona RB, Kabbinavar FF. A phase II trial of gefitinib in patients with non-metastatic hormone-refractory prostate cancer. BJU Int. 2007;100:765–769. doi: 10.1111/j.1464-410X.2007.07121.x. [DOI] [PubMed] [Google Scholar]
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo M, Tan L, Lim L, Manser E. The tyrosine kinase ACK1 associates with clathrin-coated vesicles through a binding motif shared by arrestin and other adaptors. J Biol Chem. 2001;276:18392–18398. doi: 10.1074/jbc.M008795200. [DOI] [PubMed] [Google Scholar]
- Tibes R, Kornblau SM, Qiu Y, Mousses SM, Robbins C, Moses T, Carpten JD. PI3K/AKT pathway activation in acute myeloid leukaemias is not associated with AKT1 pleckstrin homology domain mutation. Br J Haematol. 2008;140:344–347. doi: 10.1111/j.1365-2141.2007.06920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, Lagcher W, Sie D, Tanger E, Cox T, Reinders M, Hubbard TJ, Rogers J, Jonkers J, Wessels L, Adams DJ, van Lohuizen M, Berns A. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133:727–741. doi: 10.1016/j.cell.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst EH, Degenhardt YY, Strelow A, Slavin A, Chinn L, Orf J, Rong M, Li S, See LH, Nguyen KQ, Hoey T, Wesche H, Powers S. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc Natl Acad Sci USA. 2005;102:15901–15906. doi: 10.1073/pnas.0508014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, Hung MC. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60:6841–6845. [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yeow-Fong L, Lim L, Manser E. SNX9 as an adaptor for linking synaptojanin-1 to the Cdc42 effector ACK1. FEBS Lett. 2005;579:5040–5048. doi: 10.1016/j.febslet.2005.07.093. [DOI] [PubMed] [Google Scholar]
- Yokoyama N, Miller WT. Biochemical properties of the Cdc42-associated tyrosine kinase ACK1. Substrate specificity, authphosphorylation, and interaction with Hck. J Biol Chem. 2003;278:47713–47723. doi: 10.1074/jbc.M306716200. [DOI] [PubMed] [Google Scholar]
- Yokoyama N, Lougheed J, Miller WT. Phosphorylation of WASP by the Cdc42-associated kinase ACK1: Dual hydroxyamino acid specificity in a tyrosine kinase. J Biol Chem. 2005;280:42219–42226. doi: 10.1074/jbc.M506996200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–744. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci USA. 2006;103:16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]