

Abstract
The ability to self-renew and differentiate into multiple types of blood and immune cells renders hematopoietic stem and progenitor cells (HSPCs) valuable for clinical treatment of hematopoietic pathologies and as models of stem cell differentiation for tissue engineering applications. To study directed HSC differentiation and identify the conditions that recreate the native bone marrow environment, combinatorial biomaterials that exhibit lateral variations in chemical and mechanical properties are employed. New experimental approaches are needed to facilitate correlating cell differentiation stage with location in the culture system. We demonstrate that multivariate analysis of time-of-flight secondary ion mass spectrometry (TOF-SIMS) data can be used to identify the differentiation state of individual hematopoietic cells (HCs) isolated from mouse bone marrow. Here, we identify primary HCs from three distinct stages of B cell lymphopoiesis at the single cell level: HSPCs, common lymphoid progenitors, and mature B cells. The differentiation state of individual HCs in a test set could be identified with a partial least squares discriminant analysis (PLS-DA) model that was constructed with calibration spectra from HCs of known differentiation status. The lowest error of identification was obtained when the intra-population spectral variation between the cells in the calibration and test sets was minimized. This approach complements the traditional methods that are used to identify HC differentiation stage. Further, the ability to gather mass spectrometry data from single HSCs cultured on graded biomaterial substrates may provide significant new insight into how HSPCs respond to extrinsic cues as well as the molecular changes that occur during cell differentiation.
Keywords: Hematopoietic stem cell, differentiation, B cell, classification, time-of-flight secondary ion mass spectrometry, PLS-DA, multivariate analysis, single cell analysis
INTRODUCTION
The body’s full spectrum of blood and immune cells is generated from a small number of hematopoietic stem cells (HSCs) that are located within the bone marrow.1–4 HSCs self-renew and differentiate into increasingly less-rare and more-mature HCs.2,5–9 This renders HSCs of significant value for clinical treatment of hematopoietic pathologies and as models of stem cell differentiation.10
Presently, much research focuses on developing culture systems that mimic the bone marrow’s chemotactic and micromechanical properties to enable control over HSC differentiation. Engineered biomaterials that exhibit spatial variations in mechanical properties and ligand presentation are especially attractive because they enable screening the effects of multiple microenvironments on HSC fate using a minimal number of cells.11–13 To utilize such combinatorial systems, rigorous methodologies to aid identification of HC differentiation stage at the single cell level with location specificity are highly attractive. Currently, differentiation stage is most commonly assessed via fluorescence microscopy using cocktails of differentiation stage specific antibodies;14 however ambiguity from single-cell fluorescence analysis of small cell populations and inter-user variability of immunolabeling approaches can be a significant drawback.15
We hypothesized that information about the expression profiles of cell surface antigens as well as other differences in cell surface chemistry could be acquired with time-of-flight secondary ion mass spectrometry (TOF-SIMS), and exploited to identify the differentiation stage of individual HCs within a culture. Mass spectral maps of the molecules at the surface of an individual cell can be collected with TOF-SIMS.16–18 Because the spectra collected from biomaterials using TOF-SIMS instruments equipped with liquid metal primary ion sources are dominated by low mass (m/z<300) fragment ions that are common to multiple biomolecules, multivariate analysis is often employed to discriminate the spectra of biomolecules and cells.16,19–30 Unknown samples have also been identified with supervised multivariate models that are constructed from the TOF-SIMS data of known samples.20,27 For example, two different cell lines in a heterogeneous culture have been identified with location specificity by partial least-squares discriminant analysis (PLS-DA) of TOF-SIMS data.31 Though this work achieved the location-specific identification required for studies of HSC fate decision, the accuracy of identifying primary cells that exhibit higher intra-population heterogeneity than laboratory cells lines32 with this approach has not been quantitatively assessed.
Here, we report our efforts to classify individual primary murine HCs isolated from the bone marrow according to their stage in the B lymphocyte differentiation pathway by multivariate analysis of TOF-SIMS data. We focus on identifying three populations of primary HCs that were isolated from murine bone marrow via conventional flow cytometry (Figure 1): 1) hematopoietic stem and progenitor cells (HSPCs) that do not express lineage antigens (Lin−) but that do express Sca1 and cKit (Lin−Sca-1+c-Kit+, LSK); 2) common lymphoid progenitors (CLPs, Lin−IL-7Rα+Sca-1medc-Kitmed); and 3) mature B cells (B220+IgM+).6,33 These populations represent distinct cell phenotypes during lymphopoiesis: uncommitted stem and progenitor cells (HSPCs), lineage specified progenitor cells (CLPs), and fully-differentiated cells (B cells). We further investigated use of TOF-SIMS data to identify differences between HSPC populations isolated from young and old mice. Though isolated using identical sorting criteria, significant age-related differences in HSC functionality have been previously reported, making classification approaches that do not rely on surface antigen expression especially significant.34–36 The potential for existence of populational subfractions with improved HSPC stemness, therefore, motivated study of whether TOF-SIMS approaches could segregate HSPCs isolated via identical flow conditions from young and old mice.
Figure 1.
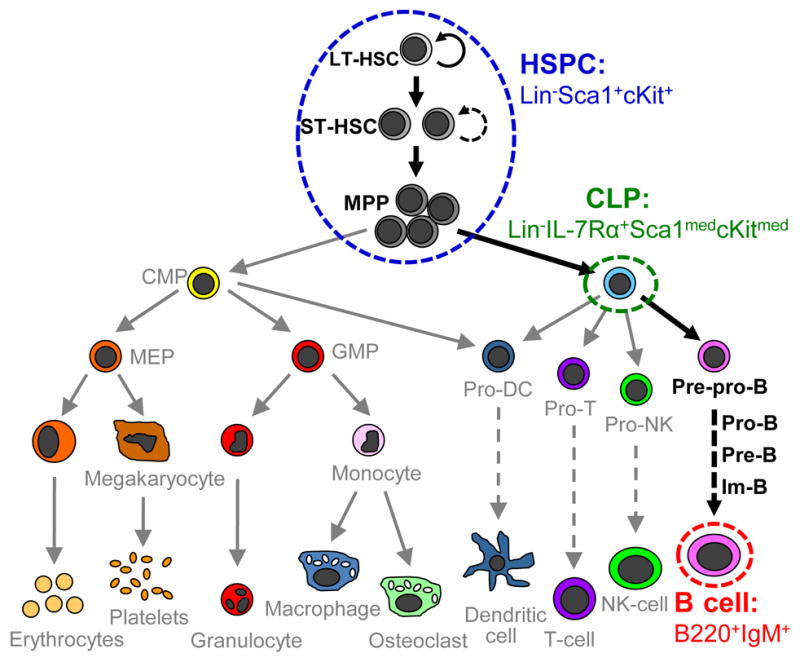
Cellular constituents of HSC-mediated hematopoiesis. The blue, green, and red envelopes indicate the cell populations used in this study. The HSPC population (Lin−Scal+cKit+) used in our study (blue envelope) contains long-term HSCs (LT-HSCs) capable of sustained hematopoietic reconstitution, short-term HSCs (ST-HSCs) capable of limited hematopoietic constitution, and multipotent progenitors (MPPs) which retain lymphoid/myeloid lineage plasticity. B cell lymphopoiesis is marked by MPP progression to a common lymphoid progenitor (CLP, green envelope) cell capable of generating all T lymphocytes, B lymphocytes, dendritic cells (DC), and natural killer (NK) cells. B lymphopoieis further progresses through a sequence of defined precursor populations: pre-pro-B cell, pro-B cell, pre-B cell, immature-B cell (Im-B), and finally mature B cell (red envelope).6 Additional cell constituents depicted: CMP, common myeloid progenitor; MEP, megakaryotic/erythroid progenitor; GMP, granulocyte/monocyte progenitor. Image modified from Passegue et al.53
Identification of the differentiation stage of individual primary HCs by multivariate analysis of TOF-SIMS data is complicated by the high degree of heterogeneity that exists within primary cell populations.37–39 Such intra-population heterogeneity can hinder the detection of the differentiation-related spectral features.19,22 We selectively captured the spectral variation between, but not within, each cell population, by employing PLS-DA models constructed using spectra from HCs of known differentiation status (calibration data set) to accurately identify the differentiation state of test HC cells. The lowest error of identification was achieved when the intra-population spectral variance that may be caused by auto-specific and age-related differences in cell surface chemistry were minimized. This approach may enable identifying cell differentiation status and its relationship to location within a colony or engineered microenvironment.
METHODS
HC isolation and preparation
The hematopoietic subpopulations were isolated from the femoral and tibial bone marrow of female C57BL/6 mice (Jackson Labs) (see the Supplemental Information for details of isolation and preparation). Distinct age ranges were used for ‘old’ (10 months old) and ‘young’ (2 – 4 months old) mice.
TOF-SIMS
Mass spectral images were acquired in unbunched mode (total primary ion dose = 3 × 1013 ions/cm2) using a PHI Trift-III TOF-SIMS (Physical Electronics Incorporated, Chanhassen, MN) instrument with a 197Au+ liquid ion gun that was operated at 22 kV. The primary ion beam was raster scanned across the sample, and positive-ion spectra with a mass range of 0 to 800 amu were acquired at each pixel.
Data analysis
Multivariate analysis was performed using the PLS Toolbox (v.6.2.1, Eigenvector Research, Manson, WA) run in MATLAB (v.7.12.0 R2011a, MathWorks Inc., Natick, MA). Unit mass binned spectra of individual cells were extracted from the TOF-SIMS images and imported into the PLS toolbox. Outlier spectra that exhibit unusual variation were identified as described in the Supplemental Information and Figure S1, and excluded from further analysis. Construction of the PLS-DA and PCA models is described in the Supplemental Information, Table S1, and Figures S2 – S5.
RESULTS
The Differentiation Status of Primary HCs Isolated from Mouse Bone Marrow Can Be Identified by PLS-DA of TOF-SIMS Data
A PLS-DA model was constructed from a calibration data set consisting of the spectra acquired from 15 B cells, 13 CLPs, and 15 HSPCs isolated from five old mice. This PLS-DA model was then used to identify the differentiation stage of 15 B cells, 12 CLPs, and 15 HSPCs (test data set) also harvested from the same mice. To increase the probability that the identification was based on cell surface biomolecules and not contaminants, only the peaks that were related to amino acids, phosphocholine, and fatty acids were analyzed (cell-related peak set, Table S1).22,40 Figure 2A – 2C shows the identifications made with the PLS-DA model, where the cells that exceeded the classification threshold (red dashed lines in Fig. 2A – 2C) were identified as the indicated population. Table 1 lists the sensitivity (the fraction of cells in the specified population correctly identified as that population), specificity (the fraction of cells from other populations that were correctly identified as not in the specified population) and error (average of the false positive and false negative rates) for identifying each HC population. The differentiation stages of the calibration cells were re-identified with high sensitivity and specificity, and the errors for identifying the B cells, CLPs, and HSPCs in the calibration set were only 0%, 2%, and 10%, respectively. Likewise, the differentiation stages of the cells in the test set were also identified with high sensitivity, high selectivity, and low error (3%, 8%, and 11% identification error for the test B cells, CLPs, and HSPCs, respectively). Thus, the variance in the peaks related to amino acids, phosphocholine, and fatty acids in the spectra was characteristic of HC populations, and could be exploited to identify the differentiation stage of individual HCs.
Figure 2.

Identification plots and VIP score plots for the PLS-DA models constructed using the cell-related peaks in the calibration spectra of HCs that were harvested from the same mice as the cells in the test set. The cells that exceeded the classification threshold (red dashed line) in the prediction plots were identified as (A) B cells, (B) CLPs, and (C) HSPCs. The VIP score plots for this model show the importance of each mass peak towards the identification of the (D) B cells, (E) CLPs, and (F) HSPCs. Peaks with VIP scores greater than unity are important for identifying the indicated population.
Table 1.
The sensitivity, specificity, and class error of differentiation stage identification made with the PLS-DA model constructed using the cell-specific peak set and the complete peak set.
| Cell-Specific Peak Set | Complete Peak Set | |||||
|---|---|---|---|---|---|---|
| B Cells | CLPs | HSPCs | B Cells | CLPs | HSPCs | |
| Sensitivity of Identification of Calibration Samples | 1.0 | 1.0 | 0.87 | 1.0 | 0.92 | 1.0 |
| Specificity of Identification of Calibration Samples | 1.0 | 0.97 | 0.93 | 1.0 | 1.0 | 0.85 |
| Sensitivity of Identification of Test Samples | 0.93 | 0.83 | 1.0 | 1.0 | 0.92 | 1.0 |
| Specificity of Identification of Test Samples | 1.0 | 1.0 | 0.78 | 1.0 | 1.0 | 0.85 |
| Class Error of Identification of Calibration Samples | 0 | 0.02 | 0.10 | 0 | 0 | 0.02 |
| Class Error of Identification of Test Samples | 0.03 | 0.08 | 0.11 | 0 | 0.04 | 0.07 |
The contributions of each mass peak to the spectral variance that identifies the B cells, CLPs, and HSPCs are shown in the variable importance in projection (VIP) plots (Fig. 2D – 2F). Peaks with VIP scores greater than unity exhibit variance that is important towards identifying the indicated population.41–42 For at least two of the three cell types, peaks m/z 53, 55, 86, 130, 148, 166, 184, 190, 205, 206, 210, and 279 have high VIP scores. Although some of these peaks are related to multiple biomolecular building blocks, phosphocholine, fatty acids, glutamine, glutamic acid, leucine, and tryptophan are likely candidates for the parent components because they are related to two or more of the peaks with high VIP scores. The amino acid-related peaks with high VIP scores might reflect changes in the differentiation-specific proteins expressed on the cell surface, or the presence of the different antibodies used to isolate each HC population by flow cytometry. To investigate whether the antibodies used for cell isolation significantly contributed to cell identification, a PLS-DA model was constructed using spectra from the antibody cocktails that were used to isolate each HC population. Few of the peaks with the highest importance towards identifying the B cells, CLPs, and HSPCs (Fig. 2D – 2F) also had high importance towards identifying the antibodies used to isolate the B cells, CLPs, and HSPCs (Fig. S3A – S3C), respectively. This suggests that the mass fragments produced by the population-specific antibodies did not contribute significantly to the spectral variation that identified each HC population. Though analysis of only the peaks related to amino acids, phosphocholine, and fatty acids increases the probability that the identifications are based on biomolecules and not sample-specific contaminants, it precludes detecting differentiation-related variations in other cell surface components, such as glycans.43–44 Higher sensitivity and specificity of identifying the calibration and test cells was achieved when PLS-DA was performed using all of the peaks between 50 and 300 m/z that were not related to known contaminants (Table 1 and Fig. S4). However, the resulting model may not be applicable towards identifying the differentiation stage of other cell samples if the peaks with high VIP scores were related to sample-specific contaminants, and not cell biomolecules.
Extent of Intra-Population Variation between HCs from Mice that Differ in Age
This approach to identifying HC differentiation status would have greater applicability if the PLS-DA model could be constructed using the spectra of cells that were harvested from different mice as those in the test set. However, primary cells from different mice exhibit auto-specific and age-related differences in cell surface chemistry.32,34 Though subtle, these differences may increase the intra-population spectral variance between the calibration and test cells to a level that is detrimental to identifying cell differentiation stage. To investigate whether such changes in cell surface chemistry induce detectable spectral variance within each HC population, PCA was performed on the spectra of cells that were harvested from two sets of C57BL/6 mice that differed in age: 10-month-old (old) and 2- to 4-month-old (young) mice. These two age groups were selected because HSCs from mice of these ages exhibit identical surface antigen expression but significant functional and epigenetic differences.34–36 Note that the intra-population spectral variation detected between these old and young cells is likely larger than that exhibited by the cells that are used to study HSC fate decisions (typically < 6 months of age).38,45–48
The B cells from the old and young mice were not separated on the first principal component (PC) of the PCA model (Fig. S5). This indicates the linear combination of mass peaks whose intensities varied the most (26%) within this B cell population was not caused by auto-specific or age-related changes in cell chemistry. However, the B cell spectra from the young and old mice were separated on PC2 and PC3; the young B cells had positive scores on PC2 and PC3, whereas the old B cells had negative scores on PC2 and/or PC3 (Fig. 3A). The majority of the peaks with high negative loadings on both PC2 and PC3, and therefore, higher normalized intensities in the spectra of the B cells from the old mice, were mainly related to lipids (m/z 86, 166, 168, 182, 184, and 224). In contrast, many of the peaks with high positive loadings on PC2 and PC3, and higher normalized intensities in the spectra of B cells from the young mice, were related to amino acids (m/z 51, 130, 155, 178, 179, 205, 263, and 279). This suggests that the B cells from the young mice had a higher ratio of proteins to lipids on their surfaces than the B cells from the old mice.
Figure 3.

PC score and loadings plots were constructed using the spectra from the B cells (A), CLPs (B), and HSPCs (C) that were harvested from the old and young mice. The region within the dashed blue line on each score plot represents the border for the 95% confidence limit of the entire PC model. The loading plots for each cell type show the extent that each mass peak contributed to the variance captured by the indicated PC.
For the PCA models constructed for the CLPs and HSPCs, the first PCs captured approximately half of the spectral variance in the data set and separated the spectra of cells from the old and young mice (Fig. 3B – 3C). Thus, the major source of spectral variation in the HSPC and CLP populations could be attributed to age-related and auto-specific differences in cell surface chemistry. In both models, the CLPs and HSPCs from the young mice had positive scores on PC1 and PC2, whereas the CLPs and HSPCs from the old mice had negative scores on PC1 and/or PC2. The peaks with high negative loadings on PC1 and PC2 were mainly related to lipids, whereas peaks that were mainly related to amino acids had high positive loadings on these two PCs (Fig. 3B – 3C). Thus, like the B cells, the surfaces of the CLPs and HSPCs from the old mice have lower protein to lipid ratios than the surfaces of the CLPs and HSPCs from the young mice.
Though a more extensive study that employs a larger number of mice would be required to confirm these results, this preliminary data suggests that all three HC populations exhibit an age-related decrease in the protein to lipid ratio on the cell surface. This finding is consistent with previous work that demonstrated glycerolipid metabolism increases in the tissues of aged mice.49 However, complementary metabolic profiling analyses would be required to exclude the possibility that age-related increases in protein misfolding lead to an increase in the degradation or intracellular accumulation of protein,50 and a decrease in the protein to lipid ratio on the aged HCs.
Effects of Intra-Population Variation between the Calibration and Test Spectra on the Identification of HC Differentiation Stage
We next assessed whether the intra-population spectral variance detected with PCA was significant enough to compromise the identification of HC differentiation stage. The spectra of 30 B cells, 25 CLPs, and 29 HSPCs that were harvested from the old mice were used to construct a PLS-DA model that was used to identify the differentiation stage of the 20 B cells, 20 CLPs, and 14 HSPCs in the test data set from the young mice. Self-identification of the old B cells, CLPs, and HSPCs in the calibration set was achieved with ≤4% error (Figure 4A – 4C, Table 2), demonstrating the variance between the calibration cells was well-captured by the model. The peaks with high VIP scores (Fig. 4D – 4F) were similar to those in Fig. 2D – 2F. Identification of the differentiation stage of the test HCs from the young mice using this model was less accurate, as the errors for identifying the test B cells, CLPs, and HSPCs from the young mice were 19%, 26% and 11%, respectively (Table 2). Thus, auto-specific and age-related differences in the calibration and test spectra appeared to compromise the identification of HC differentiation stage.
Figure 4.

The differentiation stages of HCs harvested from young mice were identified using a PLS-DA model that was created using the cell-related peaks in a calibration set of spectra from HCs harvested from old mice. The cells that exceeded the threshold (red dashed line) were identified as (A) B cells, (B) CLPs, and (C) HSPCs. The VIP score plots show the importance of each mass peak towards the identification of the (D) B cells, (E) CLPs, and (F) HSPCs.
Table 2.
The sensitivity, specificity, and class error of identification of the differentiation stage of HCs harvested from old (calibration set) and young (test set) mice determined for the PLS-DA models constructed using the cell-specific peak set and the complete peak set.
| Cell-Specific Peak Set | Complete Peak Set | |||||
|---|---|---|---|---|---|---|
| B Cells | CLPs | HSPCs | B Cells | CLPs | HSPCs | |
| Sensitivity of Identification of Calibration Samples | 1.0 | 1.0 | 0.97 | 0.93 | 1.0 | 0.93 |
| Specificity of Identification of Calibration Samples | 0.98 | 0.97 | 0.96 | 0.96 | 0.98 | 0.95 |
| Sensitivity of Identification of Test Samples | 0.65 | 0.95 | 0.86 | 0.35 | 1.0 | 0.14 |
| Specificity of Identification of Test Samples | 0.97 | 0.53 | 0.93 | 0.97 | 0.47 | 0.88 |
| Class Error of Identification of Calibration Samples | 0.01 | 0.02 | 0.04 | 0.05 | 0.01 | 0.06 |
| Class Error of Identification of Test Samples | 0.19 | 0.26 | 0.11 | 0.34 | 0.26 | 0.49 |
Finally, we investigated whether use of the complete peak set improved the precision of identifying HC differentiation stage. The errors in identifying the calibration cells were ≤6% (Table 2), respectively, which is similar to that achieved with the cell-related PLS-DA model. Inclusion of the mass peaks that were related to unknown biomolecules in the analysis was detrimental to identifying the differentiation status of the test cells from the young mice; the prediction error rose to 34%, 26%, and 49% for the test B cells, CLPs, and HSPCs, respectively (Table 2, Fig. S6). Thus, the peaks not related to amino acids, phosphocholine, or fatty acids varied significantly between the spectra in the calibration and test sets. Additional work towards identifying the parent molecules that produced the unknown peaks with high VIP scores is required to determine whether the spectral variation between the calibration and test cells was due to contaminant molecules or changes in cell surface chemistry.
DISCUSSION AND CONCLUSION
Biomaterial substrates that exhibit orthogonal gradients in composition and stiffness have the potential to permit elucidation of the combination of cues that induce specific HSC fates. The rarity of these cells in the body makes quantitative single cell analysis methods particularly valuable. To realize this potential, robust methods must be developed to identify the differentiation stages of individual HSCs at distinct regions on a biomaterial. For this purpose, we have shown that TOF-SIMS data encodes for the surface chemistries exhibited by distinct HC populations, and PLS-DA can translate this chemical data into HC differentiation stage. Because PLS-DA uses numerical algorithms to quantify the probability that a HC is at the specified differentiation stage, this approach is more objective and less prone to inter-user variability than traditional immunolabeling methods. Additionally, cell surface chemistries that may be distinctive of individual organisms or aging are also encrypted in the TOF-SIMS data. Differences between the auto-specific and age-related surface chemistries exhibited by the cells in the calibration and test sets induce spectral variance that compromises the identification of HC differentiation stage. Consequently, the cells employed to construct the PLS-DA model should be from the same age group as the unknown cells in the test set to optimize precision.
Having demonstrated the feasibility of identifying the differentiation stages of individual primary HCs with location specificity by multivariate analysis of TOF-SIMS data, we expect ongoing work may enable the identification of additional HC subpopulations. To improve our capacity to detect subtle changes in HC phenotype, we are currently incorporating additional distinct HC subpopulations to construct a next generation of PLS-DA models. Most significantly, the HSPC population used in the current analyses contains a mix of stem and progenitor cells with differential long-term stem cell potential (Fig. 1). Future work that examines significantly more rare hematopoietic subpopulations (i.e. LSK CD150+CD244−CD48−)15 that are specifically enriched for the most primitive HSCs subpopulations and show significant age-related changes in stemness48 would likely enhance our ability to identify early HSC fate decisions. Overall, we expect that use of this approach to identify the differentiation stages of individual HCs within combinatorial biomaterials will greatly facilitate correlating HSC fate decisions to environmental cues, critical to the design of ex vivo culture systems in order to control HSC bioactivity.
Our data indicate that the ratio of proteins to lipids on the surfaces of B cells, CLPs, and HSPCs decreases as the age of the donor mouse increases. Though we limited discussion of this observation to comparison with current knowledge in the field, this finding demonstrates that information on the cell surface chemistries that differ between HC populations may be acquired with this approach. Presently, the amount of compositional information that can be extracted from the data presented herein is restricted by two factors; our ability to ascertain the origins of the peaks with high importance towards identifying each HC population, and the quality of the mass spectra. Published databases of TOF-SIMS peaks that are related to lipids,22 amino acids,16,29 and nucleobases51 facilitate interpreting TOF-SIMS data. Identification of the mass peaks associated with glycans, cholesterol, and other cell surface molecules would also aid this effort. The low mass range that could be detected with our instrumentation and use of unit mass binning, which reduced the mass resolution such that multiple molecules likely contributed to each spectral peak, ultimately limit the compositional information that might be extricated from our data. Use of a TOF-SIMS instrument with a cluster ion source that enhances the yields of high mass ions52 and a mass spectrometer with higher mass resolving power and sensitivity would greatly enhance interpreting the population-specific spectral variance and identifying HC differentiation stage. Alternatively, a MALDI-TOF with sufficient spatial resolution to analyze individual cells may enable the more sensitive acquisition of mass spectra with a wider mass range and higher mass resolution from individual HCs. We expect that with the aforementioned improvements in technology and databases, multivariate analysis of TOF-SIMS data may also enhance efforts to elucidate the biomolecular changes that occur during differentiation or accompany age-related deficits in HC function.
Supplementary Material
Acknowledgments
Portions of this work were carried out in the Frederick Seitz Materials Research Laboratory Central Facilities, Univ. of Illinois, which is partially supported by the U.S. Department of Energy (DOE) under grants DEFG02-07ER46453 and DE-FG02-07ER46471. MLK holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. This work was partially supported by Grants #160673 and #189782 from the American Cancer Society, Illinois Division, Inc (BACH). The authors thank Barbara Pilas and Ben Montez from the Flow Cytometry Facility, UIUC and Mr. Bhushan Mahadik (ChBE, UIUC) for assistance with flow cytometry sorting and analysis.
Footnotes
Supporting information: Additional information as noted in text. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Weissman IL. Cell. 2000;100:157–168. doi: 10.1016/s0092-8674(00)81692-x. [DOI] [PubMed] [Google Scholar]
- 2.Wilson A, Trumpp A. Nature Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 3.Yin T, Li L. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 5.Cariappa A, Pillai S. Curr Opin Immunol. 2002;14:241–9. doi: 10.1016/s0952-7915(02)00328-x. [DOI] [PubMed] [Google Scholar]
- 6.Hardy RR, Hayakawa K. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 7.Yang L, Bryder D, Adolfsson J, Nygren J, Mansson R, Sigvardsson M, Jacobsen SE. Blood. 2005;105:2717–23. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- 8.Adolfsson J, Borge OJ, Bryder D, Theilgaard-Monch K, Astrand-Grundstrom I, Sitnicka E, Sasaki Y, Jacobsen SE. Immunity. 2001;15:659–69. doi: 10.1016/s1074-7613(01)00220-5. [DOI] [PubMed] [Google Scholar]
- 9.Adolfsson J, Mansson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, Anderson K, Sitnicka E, Sasaki Y, Sigvardsson M, Jacobsen SE. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Daley GQ, Scadden DT. Cell. 2008;132:544–8. doi: 10.1016/j.cell.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Martin TA, Caliari SR, Williford PD, Harley BA, Bailey RC. Biomaterials. 2011;32:3949–3957. doi: 10.1016/j.biomaterials.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mei Y, Saha K, Bogatyrev SR, Yang J, Hook AL, Kalcioglu ZI, Cho SW, Mitalipova M, Pyzocha N, Rojas F, Van Vliet KJ, Davies MC, Alexander MR, Langer R, Jaenisch R, Anderson DG. Nat Mater. 2010;9:768–778. doi: 10.1038/nmat2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo W, Yousaf MN. J Am Chem Soc. 2011;133:10780–10783. doi: 10.1021/ja204893w. [DOI] [PubMed] [Google Scholar]
- 14.Iwasaki H, Akashi K. Oncogene. 2007;26:6687–6696. doi: 10.1038/sj.onc.1210754. [DOI] [PubMed] [Google Scholar]
- 15.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 16.Kulp KS, Berman ESF, Kinze MG, Shattuck DL, Nelson EJ, Wu L, Montgomery JL, Felton JS, Wu KJ. Anal Chem. 2006;78:3651–3658. doi: 10.1021/ac060054c. [DOI] [PubMed] [Google Scholar]
- 17.Kurczy ME, Piehowski PD, Van Bell CT, Heien ML, Winograd N, Ewing AG. Proc Natl Acad Sci USA. 2010;107:2751–2756. doi: 10.1073/pnas.0908101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrowski SG, Van Bell CT, Winograd N, Ewing AG. Science. 2004;305:71–73. doi: 10.1126/science.1099791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham DJ, Wagner MS, Castner DG. Appl Surf Sci. 2006;252:6860–6868. [Google Scholar]
- 20.Berman ESF, Wu L, Fortson SL, Kulp KS, Nelson DO, Wu KJ. Surf Interface Anal. 2009;41:97–104. [Google Scholar]
- 21.Magnusson YK, Friberg P, Sjovall P, Malm J, Chen Y. Obesity. 2008;16:2745–2753. doi: 10.1038/oby.2008.424. [DOI] [PubMed] [Google Scholar]
- 22.Anderton CR, Vaezian B, Lou K, Frisz JF, Kraft ML. Surf Interface Anal. 2012;44:322–333. [Google Scholar]
- 23.Vaezian B, Anderton CR, Kraft ML. Anal Chem. 2010;82:10006–10014. doi: 10.1021/ac101640c. [DOI] [PubMed] [Google Scholar]
- 24.Berman ESF, Kulp KS, Knize MG, Wu L, Nelson EJ, Nelson DO, Wu KJ. Anal Chem. 2006;78:6497–6503. doi: 10.1021/ac060865g. [DOI] [PubMed] [Google Scholar]
- 25.Baker MJ, Brown MD, Gazi E, Clarke NW, Vickerman JC, Lockyer NP. Analyst. 2008;113:175–179. doi: 10.1039/b712853c. [DOI] [PubMed] [Google Scholar]
- 26.Jungnickel H, Jones EA, Lockyer NP, Oliver SG, Stephens GM, Vickerman JC. Anal Chem. 2005;77:1740–1745. doi: 10.1021/ac048792t. [DOI] [PubMed] [Google Scholar]
- 27.Thompson CE, Fletcher JS, Goodacre R, Henderson A, Lockyer NP, Vickerman JC. Appl Surf Sci. 2006;252:6719–6722. [Google Scholar]
- 28.Thompson CE, Jungnickel H, Lockyer NP, Stephens GM, Vickerman JC. Appl Surf Sci. 2004;231–232:420–423. [Google Scholar]
- 29.May CJ, Canavan HE, Castner DG. Anal Chem. 2004;76:1114–1122. doi: 10.1021/ac034874q. [DOI] [PubMed] [Google Scholar]
- 30.Wagner MS, Castner DG. Langmuir. 2001;17:4649–4660. [Google Scholar]
- 31.Barnes CA, Brison J, Robinson M, Graham DJ, Castner DG, Ratner BD. Anal Chem. 2012;84:893–900. doi: 10.1021/ac201179t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freshney RI. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications. 6. John Wiley & Sons, Inc; Hoboken, New Jersey: 2010. pp. 1–10. [Google Scholar]
- 33.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Immunity. 2004;20:707–18. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayack SR, Shadrach JL, Kim FS, Wagers AJ. Nature. 2010;463:495–500. doi: 10.1038/nature08749. [DOI] [PubMed] [Google Scholar]
- 36.Warren LA, Rossi DJ. Mech Ageing Dev. 2009;130:46–53. doi: 10.1016/j.mad.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YW, Lin MS, Vora KA. Int Immunol. 1992;4:1293–1302. doi: 10.1093/intimm/4.11.1293. [DOI] [PubMed] [Google Scholar]
- 38.Schroeder T. Cell Stem Cell. 2010;6:203–207. doi: 10.1016/j.stem.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 39.das Neves RP, Jones NS, Andreu L, Gupta R, Enver T, Iborra FJ. PLoS Biol. 2010;8:e1000560. doi: 10.1371/journal.pbio.1000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oran U, Unveren E, Wirth T, Unger WES. Appl Sur Sci. 2004;227:318–324. [Google Scholar]
- 41.Chong IG, Jun CH. Chemo Intell Lab Sys. 2005;78:103–112. [Google Scholar]
- 42.Musumarra G, Barresi V, Condorelli DF, Fortuna CG, Scirè S. J Chemometrics. 2004;18:125–132. [Google Scholar]
- 43.Brown J, Greaves MF, Molgaard HV. Int Immunol. 1991;3:175–184. doi: 10.1093/intimm/3.2.175. [DOI] [PubMed] [Google Scholar]
- 44.Tang C, Lee AS, Volkmer JP, Sahoo D, Nag D, Mosley AR, Inlay MA, Ardehali R, Chavez SL, Pera RR, Behr B, Wu JC, Weissman IL, Drukker M. Nat Biotech. 2011;29:829–834. doi: 10.1038/nbt.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holst J, Watson S, Lord MS, Eamegdool SS, Bax DV, Nivison-Smith LB, Kondyurin A, Ma L, Oberhauser AF, Weiss AS, Rasko JEJ. Nat Biotech. 2010;28:1123–1128. doi: 10.1038/nbt.1687. [DOI] [PubMed] [Google Scholar]
- 46.Lutolf MP, Doyonnas R, Havenstrite K, Koleckar K, Blau HM. Integr Biol. 2009;1:59–69. doi: 10.1039/b815718a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rieger MA, Schroeder T. Cells Tissues Organs. 2008;188:139–49. doi: 10.1159/000114540. [DOI] [PubMed] [Google Scholar]
- 48.Yilmaz OH, Kiel MJ, Morrison SJ. Blood. 2006;107:924–930. doi: 10.1182/blood-2005-05-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, Auwerx J. Sci Rep. 2011:1. doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basaiawmoit RV, Rattan SIS. In: Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols. Bross P, Gregersen N, editors. Humana Press; New York: 2010. pp. 107–117. [Google Scholar]
- 51.Lhoest JB, Wagner MS, Tidwell CD, Castner DG. J Biomed Mat Res. 2001;57:432–440. doi: 10.1002/1097-4636(20011205)57:3<432::aid-jbm1186>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 52.Touboul D, Kollmer F, Niehuis E, Brunelle A, Laprévote O. J Am Soc Mass Spectrom. 2005;16:1608–1618. doi: 10.1016/j.jasms.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Passegue E, Jamieson CH, Ailles LE, Weissman IL. Proc Natl Acad Sci USA. 2003;100(Suppl 1):11842–9. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.